
Abstract
Glycogen storage diseases (GSDs) type I (GSDI) and type III (GSDIII), the most frequent hepatic GSDs, are due to defects in glycogen metabolism, mainly in the liver. In addition to hypoglycemia and liver pathology, renal, myeloid, or muscle complications affect GSDI and GSDIII patients. Currently, patient management is based on dietary treatment preventing severe hypoglycemia and increasing the lifespan of patients. However, most of the patients develop long-term pathologies. In the past years, gene therapy for GSDI has generated proof of concept for hepatic GSDs. This resulted in a recent clinical trial of adeno-associated virus (AAV)-based gene replacement for GSDIa. However, the current limitations of AAV-mediated gene transfer still represent a challenge for successful gene therapy in GSDI and GSDIII. Indeed, transgene loss over time was observed in GSDI liver, possibly due to the degeneration of hepatocytes underlying the physiopathology of both GSDI and GSDIII and leading to hepatic tumor development. Moreover, multitissue targeting requires high vector doses to target nonpermissive tissues such as muscle and kidney. Interestingly, recent pharmacological interventions or dietary regimen aiming at the amelioration of the hepatocyte abnormalities before the administration of gene therapy demonstrated improved efficacy in GSDs. In this review, we describe the advances in gene therapy and the limitations to be overcome to achieve efficient and safe gene transfer in GSDs.
Glycogen Storage Diseases are Rare Genetic Diseases Characterized By a Disturbance of Glycogen Metabolism
Glycogen is a highly branched polysaccharide that is the storage form of glucose in mammals. Although most tissues store glycogen, it is enriched in liver and skeletal muscle. In the liver, glycogen serves as a source of glucose to maintain normal blood glucose levels. Accumulation and glycogen degradation depend on the levels of glucose in circulation. During the absorptive state, when the gastrointestinal tract is full, the glucose extracted from the food reaches the circulation and it is stored in the liver as glycogen. In the postabsorptive state, when blood glucose levels fall, glycogen is broken down to glucose (glycogenolysis) to maintain normal values of glycemia. This process requires the intervention of two different enzymes, glycogen phosphorylase that degrades the linear tract of glycogen and glycogen debranching enzyme (GDE) that transforms branched glycogen in a linear form. Glycogen degradation releases glucose-1 phosphate that after isomerization to glucose-6 phosphate is transformed into glucose by the glucose-6 phosphatase enzyme (G6Pase). When hepatic glycogen stores are depleted, gluconeogenesis can occur in liver, kidney, and intestine, allowing for de novo synthesis of glucose with noncarbohydrates substrates, for example, amino acids, pyruvate, and glycerol. 1 In muscles, G6Pase is not expressed and the glucose-6 phosphate derived from glycogen is used to produce ATP through the glycolytic pathway as a source of energy for muscle contraction.
Glycogen storage diseases (GSDs) are a group of rare inherited metabolic disorders (incidence from 1 in 20,000 to 1 in 100,000 newborns) characterized by abnormal storage or degradation of glycogen. 2,3 GSDs manifest with an abnormal quantity or structure of glycogen in liver, muscles, or both. To date, there are 15 known GSDs caused by genetic deficiencies of different proteins involved in glycogen synthesis or degradation, glycolysis, and glucose release in the bloodstream 2,3 (Fig. 1). Episodes of hypoglycemia are the primary manifestation of hepatic GSDs (GSD0, GSDI, GSDIII, GSDIV, GSDVI, and GSDIX), especially during short fasting since the liver is the main glucose-producing tissue in postabsorptive period. 4 In contrast, intolerance to exercise with muscle pain and rhabdomyolysis, or muscle weakness and cardiomyopathy are signs of GSDs affecting muscles (GSDII to GSDV and GSDVII to GSDXV). 2,5
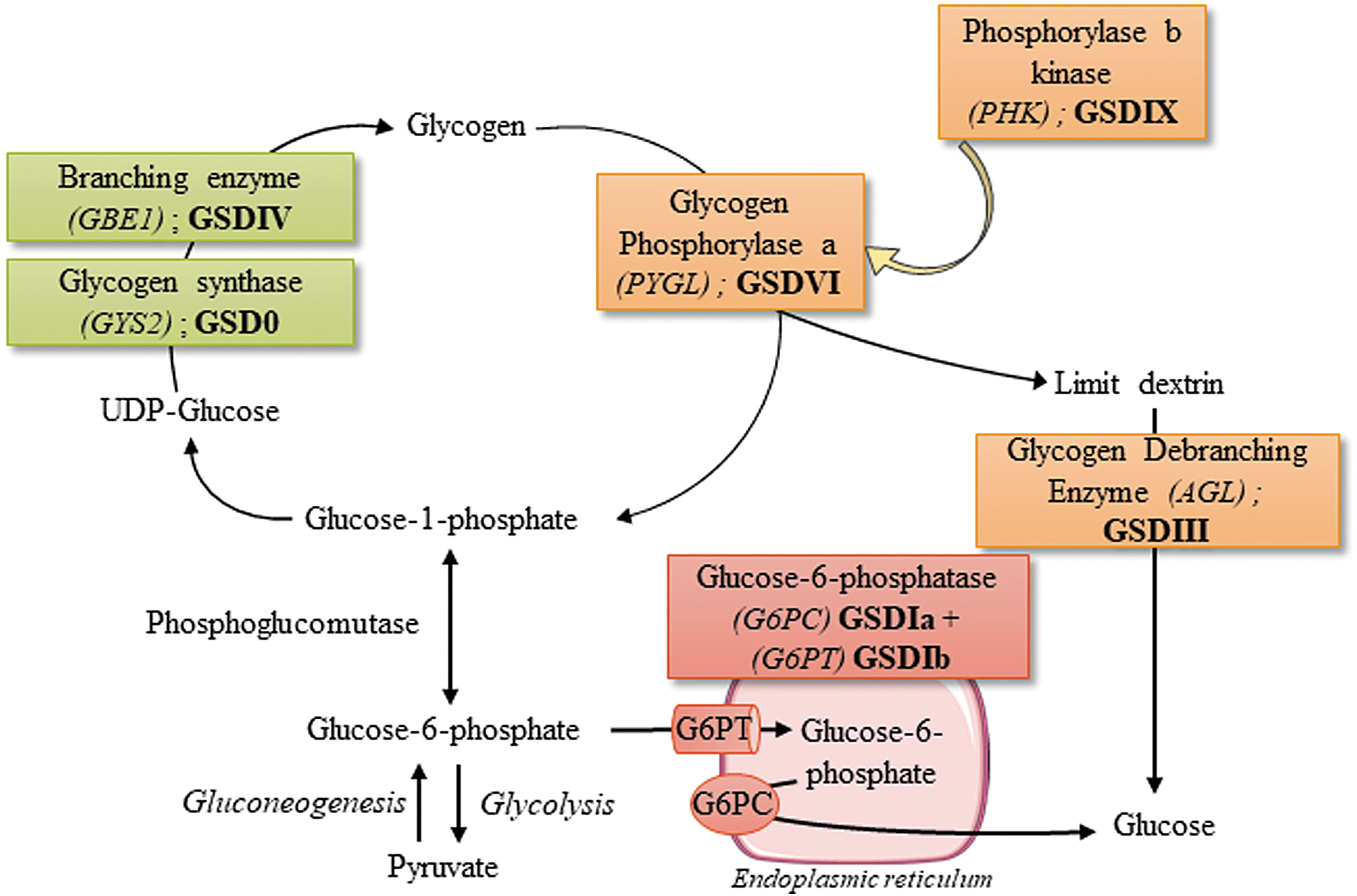
Glycogen metabolism and hepatic glycogen storage diseases. Hepatic glycogen storage disorders are indicated in roman numbers next to the defective genes. UPD-glucose, uridine diphosphate glucose.
The monogenic origin of hepatic GSDs and their liver involvement make these diseases an ideal target for gene replacement strategies. Gene therapy research in animal models has generated proof of concept for the treatment of GSDI and GSDIII. The extensive preclinical experience accumulated in GSDIa lately resulted in a clinical trial (
GSDI and GSDIII
GSDI, also referred as von Gierke disease, is characterized by the absence of endogenous glucose production due to deficient G6Pase activity. More precisely, GSDIa and GSDIb are caused by mutations in the genes encoding for the catalytic subunit of the G6Pase complex (G6PC1) or the glucose-6 phosphate transporter (SLC37A4 or G6PT), respectively. 6 –9 Loss-of-function mutations in these two subunits result in severe hypoglycemia after a short fasting period that can be fatal if untreated immediately. Patients with GSDI also displayed hepatomegaly and nephromegaly due to the abnormal storage of glycogen in liver and kidneys. Indeed, the accumulation of glucose-6 phosphate, resulting from reduced hydrolysis to glucose, leads to a complete hepatic and renal metabolism reprogramming, characterized by increased glycogen turnover, and activation of de novo lipogenesis and pentose phosphate pathway. 10 –12 As a consequence, GSDI patients exhibit swollen hepatocytes and renal epithelial cells due to the accumulation of high levels of glycogen and lipids leading to fatty liver, kidney injuries, and secondary biochemical abnormalities, including hyperlactatemia, hyperuricemia, and hyperlipidemia. 8,9 Hepatocellular adenomas (HCAs) occur in 70–80% of patients >25 years, with a high probability to transform into hepatocellular carcinomas (HCCs), which may require liver transplantation. 3,9 Indeed, we showed similarity in the metabolism of GSDI hepatocytes and tumor cells, both characterized by increased cell proliferation, and loss of several tumor suppressors. 10 Most of the GSDI patients older than 25 years have kidney disease. 9 Studies in a murine model of the disease indicate that the accumulation of lipids is responsible for renal injuries leading to a chronic kidney disease (CKD), 11,13,14 which may require dialysis or kidney transplantation in patients. 8,9 GSDI patients also develop intestinal symptoms that appear more serious in GSDIb. 9 In GSDIb, differently from GSDIa, patients present severe infectious complications, due to neutropenia and neutrophil and monocyte functional defects. 9 In GSDI, normoglycemia is maintained by frequent meals enriched in uncooked cornstarch even during the night or continuous nasogastric feeding. 9,15 To limit glycogen and fat accumulation, lactose, fructose, and products containing free sugar should be avoided. Thus, frequent daytime feeding, intragastric continuous infusions, and a strict dietary regimen allow for the control of hypoglycemia and reduce the metabolic impairment. Although a strict metabolic control may delay or, in some cases, lead to the regression of long-term complications in GSDI, poor compliance with the dietary treatment makes liver and/or kidney transplantation the only curative treatment for this disease. 9,16 –20
GSDIII or Cori disease is caused by mutations in AGL resulting in reduced activity of GDE that degrades branched glycogen. 21 GDE deficiency leads to the accumulation of abnormal glycogen (called limit dextrin) virtually in all tissues. GSDIII has variable features involving generally both liver and muscle (type IIIa, representing 85% of cases) or liver only (type IIIb; in ∼15% of cases). The disease manifests during childhood with hepatomegaly, hypoglycemia, and hyperlipidemia. Dietary management is based on frequent meals enriched in uncooked cornstarch, as in GSDI, to prevent hypoglycemia and hyperketonemia. 21,22 Differently from GSDI patients, the activation of gluconeogenesis triggered by protein- and fat-enriched diet permits better control of blood glucose levels in GSDIII patients. 21 –26 Nevertheless, during childhood, the control of glycemia in GSDIII patients is quite variable and they experience episodes of life-threatening severe hypoglycemia. In most GSDIII patients, hepatomegaly and the control of glycemia improve with aging. However, the decreased liver size can be misleading as progressive liver cirrhosis and hepatic failure occur. 21,27,28 Recent results showed that proliferation and differentiation of stellate cells lead to the development of fibrosis, which plays a central role in the progression of liver disease. 29 In rare cases, HCA and HCC were reported. 27,28 In addition, plasmatic transaminases are generally elevated in GSDIII patients, suggesting severe hepatic damages. 21 At the muscle level, creatine kinase elevation, muscular pain after exercise, and both distal and proximal myopathy were reported in GSDIII patients. 21,30 –32 Intolerance to exercise seems to be only marginally related to the low blood glucose. 33 A careful characterization of the muscle function indicated a progressive degeneration starting in the third decade. 30,31 In general, hypotonia and muscle weakness leads to intolerance to exercise and, in some cases, to the loss of independent walking. Cardiac involvement with left ventricular hypertrophy or other forms of hypertrophy were reported in 58% of a young patient's population (median age: 20.6 years range 1–64.1). 21 In the same cohort, 15% of the patients had cardiomyopathy defined as severe intolerance to exercise and/or the use of pharmacological treatment for symptoms of heart failure. Importantly, high-fat, high-protein, and low-carbohydrate diet was associated with an improvement of the cardiac phenotype. 23
To resume, both GSDI and GSDIII are characterized by glycogen accumulation in the liver that directly or indirectly causes stress virtually in all the hepatocytes, leading to abnormal liver parenchyma and hepatic complications. Interestingly, increased hepatocyte proliferation was observed in GSDIa and GSDIII hepatocytes long before hepatic tumor formation. 10,29 In addition, hepatocyte metabolism characterized in GSDIa mouse was very similar to that of tumor cells, 10,12 indicating a high potential for transformation of GSDI hepatocytes. Given the risk of tumor occurrence in GSDI and GSDIII patients, it is important to define the residual level of activity needed to avoid the development of tumors at long term.
Another important feature shared by the two diseases is the multiorgan manifestation. In addition to hypoglycemia and liver pathology, renal, myeloid, or muscle complications affect GSDI and GSDIII patients and need to be addressed. In GSDI, the short-term follow-up of the transplanted patients and the simultaneous liver and kidney transplantation make the prevention of renal disease by liver transplantation still an open question. 34,35 In animal models of GSDI, liver-directed gene therapy did not prevent kidney degeneration and CKD at long term. 36,37 In addition, targeted deletion of G6Pase in the kidney is sufficient to induce the characteristic GSDI nephropathy in mice. 13 Similarly, in GSDIII patients, liver transplantation did not rescue the cardiac and muscle function impairment, 38,39 and both liver- and muscle-targeted gene therapies were required to rescue the phenotype of a mouse model of GSDIII. 40 Thus, a key challenge for successful gene therapy in GSDI and GSDIII is to develop gene therapy tools to efficiently target kidney and muscle in addition to the liver, to prevent both hypoglycemia and long-term pathologies.
Gene Therapy Approaches in GSDI and GSDIII
Recent clinical successes in hemophilia A and B, in retinitis pigmentosa, and in spinal muscular atrophy indicate adeno-associated virus (AAV) as the vector of choice for in vivo gene therapy. 41 –43 Today, engineering of recombinant AAV vectors has made remarkable strides permitting efficient liver-directed gene therapy. 44 Moreover, the liver is a preferred target for AAV vectors, thanks to its unique anatomic properties with fenestrated endothelium and a high blood supply, leading to efficient accumulation of AAV vectors within the liver after systemic administration. In addition, AAV genomes are relatively stable in quiescent hepatocytes, generally found in episomal form. 45,46
Starting from the identification of the molecular basis of GSDs, different approaches were tested to restore the expression of the mutated proteins and rescue the disease phenotype in animal models of hepatic GSDs. Vectors used to deliver a corrected copy of the transgene included adenovirus, 47 –50 lentivirus, 51,52 and naked DNA combined with ultrasound-mediated delivery. 53 Starting from 2006, AAV became the vectors of choice for gene replacement in hepatic GSDs. More recently, mRNA lipid nanoparticles were used to target rare genetic metabolic diseases 54 and GSD. 55 The advantages and limitations of these therapeutic strategies for DNA or RNA delivery into the cell were recently discussed. 56,57
AAV gene therapy for GSDI
More than 20 years ago, the availability of murine and canine models of GSDIa and GSDIb stimulated the development of gene replacement strategies for those diseases. Unfortunately, these animal models suffer from severe hypoglycemia and have a very low life expectancy, which makes them unsuitable for studying the effects of gene therapy on the GSDI long-term complications. In total G6pc−/− mice or dogs, to avoid premature deaths, glucose therapy, that is, glucose injections, glucose-fortified water, or food supplementation was maintained before AAV injections. More recently, tissue-specific knockout mouse models were obtained with extended life expectancy. 58 The liver-specific G6pc−/− mice recapitulated the human hepatic disease, including intolerance to fasting, metabolic disturbance, and tumor development, 59 whereas the kidney-specific G6pc−/− mice exhibited CKD, as reported in GSDIa patients. 11,13
Another important advantage of GSDI regarding AAV gene therapy is that the G6PC or the G6PT transgenes are relatively short, and this provides a relative freedom in the choice of the elements constituting the transgene expression cassette (Table 1).
Adeno-associated viral vector-based gene therapies in hepatic glycogen storage diseases type I and type III
Neonates: 1–2 day-old mice.
AAV, adeno-associated virus; CAG, CMV early enhancer/chicken beta actin promoter; CMV, cytomegalovirus promoter; ds, double stranded; GSD, glycogen storage disease; hAAT, human α-1–antitrypsin promoter; hG6PC, human glucose-6 phosphatase; hGAA, human acid alpha-glucosidase; hGDE, human glycogen debranching enzyme; hGPE, human G6PC promoter (−2,864 to −1); mG6PC, murine glucose-6 phosphatase; mini hGPE, minimal human G6PC promoter (−298 to +128); mini hGPTE, minimal human G6PT promoter (610 bp); ss, single stranded; vg, vector genome.
Finally, restoration of a low hepatic G6Pase activity is sufficient to control glycemia. Indeed, the first data obtained with AAV gene therapy in canine and murine models of GSDIa confirmed that low levels of G6PC transgene expression, that is, 3–5% of wild-type (WT) activity, were sufficient to prevent hypoglycemia. 60 –62 Similarly, in GSDIb, gene therapy with AAV vectors resulted in improved survival and long-term correction of the hypoglycemia in G6pt−/− mice, at doses comparable with those used in GSDIa. 63,64 As expected, due to the very low integration rates of AAV vectors, gene therapy resulted in the transient correction of myeloid abnormalities in G6pt−/− mice. 63
However, those pioneer studies highlighted some of the limitations of AAV gene therapy for GSDI. First, the treatment of 2-week–old mice resulted in vector genome loss possibly due to the liver and kidney degeneration underlying the disease pathology and/or liver growth. 37 Despite vector dilution, the correction of liver impairment was stable up to 6 months in mice and 1 year in dogs after administration of an AAV8 vector expressing the G6PC transgene. 65 Long-term studies conducted in dogs reported a progressive transgene loss, requiring up to three repeated administrations of AAV vectors pseudotyped in different serotypes to avoid IgG-mediated neutralization and achieve sustained efficacy (Table 1). 36,66 –68 The need of simultaneous targeting of liver and kidney constituted a second technical barrier for the correction of the disease at a systemic level with some AAV serotypes that were very inefficient in kidney targeting. 37,65 Despite this limitation, correction of kidney and liver disease was partially achieved with single-stranded AAV1 vectors. 69 Differently from WT AAV that has a single-stranded (ssAAV) genome, recombinant AAV vectors can have both single and double-stranded genomes. The use of double-stranded AAV (dsAAV) vectors may improve transduction efficacy and AAV vector genome stability. 70,71 Accordingly, in GSDIa, dsAAV9 vectors allowed for the efficient expression of G6PC in both liver and kidney and the rescue of glycogen accumulation, glycemia, and kidney function. 72 However, low transduction efficacy was obtained and the loss of transgene expression was reported in the kidney at long term (∼1 year after vector injection), suggesting that the tissue degeneration associated with the diseased state was not fully corrected by gene therapy. 72 Another possible cause of the low persistence of the G6PC transgene may be due to the formation of an antitransgene immune response, leading to the clearance of the cells expressing the transgene. Early finding indicated that the use of a constitutive promoter (cytomegalovirus early enhancer/chicken beta actin promoter [CAG]) to express G6PC was associated with higher CD8 positive infiltrates in the liver and lower transgene persistence. 73 However, a similar transgene expression cassette was used to transduce hepatocytes in GSDIa dogs without any evident immune response, even after readministration. 68 This suggests that antitransgene immune responses may have a marginal role in the low transgene persistence observed in the different animal models.
The second limitation concerns the prevention of long-term hepatic and renal complications, very prevalent in the GSDI population, 9 by AAV gene therapy. Preclinical studies performed in total G6pc−/− mice showed that the long-term expression of the G6PC transgene under transcriptional control of the human G6PC promoter (hGPE) prevented the formation of liver tumors (Table 1). 61,62,74,75 Results indicated that a low percentage of G6Pase activity (>2% of normal hepatic G6Pase activity) could be sufficient to avoid the formation of HCA in GSDIa mice. 61 In GSDIb mouse model, a higher percentage (6–8%) of the WT G6PT activity was required to prevent the formation of HCA in 60–78-week–old animals. 63 The prevention of tumors formation by AAV gene therapy in GSDIa dogs is less clear and controversial. Indeed, in one study, despite high G6Pase activity (21% to 52% of normal hepatic G6Pase activity), four out of five treated dogs developed HCA and HCC. 36 Moreover, three of the five AAV-treated dogs developed CKD due to a very low kidney transduction. 36 In a second study, none of the dogs developed long-term complications. 67 This difference, attributed to a strict dietary management, allowing for a better control of liver degeneration, may account for the improved efficacy of gene therapy 76,77 and supports the use of combination therapies in gene transfer. 78 However, it should be noted that in the second study, the treated GSDIa dogs showed a time-dependent decrease in blood glucose and died at a mean age of 5.9 years, despite the strict metabolic control and the sequential administration of two AAV vectors. 67
The experience accumulated in >20 years of practice with the animal models and gene transfer in GSDIa resulted, this year, in a phase 1/2 open-label clinical trial (NCT03517085; Ultragenyx Pharmaceutical, Inc.). Preliminary results obtained from three patients in the low dose cohort (2.0 × 1012 vg/kg) demonstrated efficacy with an average 80% of improvement in the time to hypoglycemic event, together with a 38% reduction in the total daily cornstarch consumption. 79 The administration of the AAV8 vectors expressing human G6PC at the low dose was associated with alanine aminotransferase (ALT) elevation in two patients out of three. 79 Based on the results obtained in clinical trials on the use of AAV8 for hemophilia B, the ALT elevation observed in the GSDIa trial is likely associated with an immune response to the AAV vector capsid and can be controlled by a short course of corticosteroids. 80 However, the kinetic of the formation of the immune response in patient 1, which required two corticosteroid treatments at week 8 and week 20, may suggest a distinct trait when compared with the hemophilia B trial. 80 So far, the sponsor has provided no interpretation of these results based on immune-monitoring data. Given the demonstration of efficacy and the acceptable safety profile, the Data Monitoring Committee positively advised to continue the trial with the enrollment of cohort 2 at the dose of 6.0 × 1012 vg/kg.
AAV gene therapy for GSDIII
In hepatic GSD other than GSDI, the experience with gene transfer in general, and with AAV in particular, is limited. In GSDIII, the development of an AAV-based gene therapy approach was delayed by (1) the complexity of the disease, which requires simultaneous liver and muscle targeting, (2) the lack of small animal models, and (3) the size of the transgene that is close to the AAV genome packaging limit (∼5 kb).
A dog model of GSDIII was described in 2012. This model showed extensive accumulation of glycogen in liver and muscle, together with hepatic cirrhosis and muscle fibrosis at long term. 81,82 Starting from 2014, four mouse models of GSDIII were described and all recapitulated the characteristics of the human GSDIII disease with glycogen accumulation in the liver and muscle, hypoglycemia, muscle strength impairment, and intolerance to exercise. 29,40,83,84 The availability of these small animal models allowed for the development of an AAV vector-based gene therapy approach for GSDIII. The use of recombinant human acid alpha-glucosidase (GAA) was suggested for the clearance of cytosolic glycogen accumulated in GSDIII primary myoblasts. 85 Similarly, the treatment of GSDIII mice with an optimized vector expressing a liver-secreted GAA 86 was able to reduce glycogen accumulation in liver. 40 Surprisingly, despite the high levels of GAA enzyme taken up in muscle, no reduction of glycogen accumulation was observed, 40 suggesting different mechanisms underlying glycogen degradation in liver and muscle.
One of the biggest obstacles for the development of an AAV gene therapy approach for GSDIII is the size of the transgene, which is larger than the AAV vector encapsidation capacity. A dual vector strategy was developed to express GDE in muscle and liver. Following this strategy, the GDE transgene sequence was split into two different vectors. The recombination relied on an overlapping sequence, derived from the GDE transgene and present in both vectors. The use of a constitutive cytomegalovirus promoter permitted for the efficient reconstitution of the full size GDE sequence and the consequent expression of the transgene in muscle. This was associated with reduced glycogen accumulation in different muscles and the rescue of muscle strength. Interestingly, when a liver-specific promoter was used, glycemia was only partially rescued and a high variability in the expression of the GDE transgene was reported. Hepatocyte proliferation and degeneration due to the development of fibrosis in GSDIII 29 may explain the partial rescue of glycemia despite the high doses of AAV vectors used to target the liver. 29,40
Alternative Gene Therapy Approaches Addressing the Limitations of AAV Gene Therapy
Liver gene therapy with AAV vectors in neonate animals results in vector genome dilution and transgene loss after liver growth since the AAV vector genomes remain for the vast majority in the nonintegrated episomal form. 45,46 In adult hemophilia B dogs, canine coagulation Factor IX was stably expressed for more than 10 years. 87 Accordingly, in a pioneer clinical trial for hemophilia B, Nathwani et al. demonstrated stable expression of the human coagulation Factor IX for a period of 6 years, indicating the lack of transgene expression loss in hepatocytes. 88 One important advantage of hemophilia B is that the underlying mutations slightly affect liver function and hepatocytes can be considered as normal.
In GSDI and GSDIII, the impaired enzymatic function results in the modification of hepatocyte metabolism that ultimately leads to enhanced proliferation, apoptosis, and/or transformation and cell death. 10,29 Increased hepatocytes proliferation and wasting are likely to result in AAV vector genome dilution and transgene expression loss. Indeed, repeated administrations of AAV vectors were required to achieve stable correction of the hypoglycemia in GSDIa dogs (Table 1) and glycemia correction was reduced over time when the vector was administered early in life (up to 6 months of age). 67 Integrative approaches, based on lentivirus 51,52 or zinc finger (ZFN)-mediated targeted integration, 89,90 were developed in GSDIa mice. Interestingly, integration of the G6PC transgene by lentivirus resulted in an increased transduction of hepatocytes between neonatal state and 9 months postinjection, leading to ∼20% of G6PC-expressing hepatocytes and resulting in the prevention of tumor formation in liver-specific G6pc −/− mice. 51 However, the potential genotoxicity of the integrative approaches in vivo is still questioned and it is particularly relevant in hepatic GSDs that are associated with hepatocytes degeneration and HCA and HCC formation. Recently, data obtained in vivo by genome editing with ZFN or CRISPR/Cas9 targeted to the ROSA26 safe harbor locus demonstrated specific integration of the G6PC transgene together with complete rescue of hypoglycemia in G6pc −/− mice. 89 –91
Another potential approach, circumventing both the AAV persistence and the potential genotoxicity of integrative approaches, is the use of synthetic mRNA delivered by lipid nanoparticles. 92 Proof-of-concept studies performed in liver-specific G6pc−/− mice showed a marked improvement of glycemia and hepatomegaly after single administration of the mRNA nanoparticle complex allowing for the efficient expression of G6Pase. 55,93
Finally, it was suggested that combination therapy might improve liver function and decrease apoptosis, leading to a better efficacy of gene therapy. In principle, the rescue of hepatocytes degeneration may improve transgene persistence and ultimately lead to long-term correction of the hepatic manifestation in GSDI and GSDIII by a single dose of AAV vector. For example, substrate reduction strategies were attempted to rescue the metabolic phenotype in GSDIa and GSDIII. A small interfering RNA (siRNA) specific for the liver isoform of glycogen synthase (Gys2) was developed to decrease glycogen synthesis and accumulation in the liver. Long-term treatment with Gys2-specific siRNA prevented glycogen accumulation in the liver and rescued hepatomegaly in a mouse model of GSDIII. 29 Similarly, in liver-specific G6pc−/− mice, decreased glycogen accumulation and normalization of the liver morphology were observed. 29 Thus, although the siRNA-mediated inhibition of Gys2 did not rescue glycemia, this approach can be considered in combination with gene therapy. Bezafibrate, a pan-peroxisome proliferator-activated receptor agonist, was proposed in combination with ZFN-mediated targeted integration to improve treatment efficacy. 89 Bezafibrate treatment in combination with AAV gene therapy improved survival, glycemia, and hepatomegaly in G6pc−/− mice. The beneficial effect observed after bezafibrate treatment, possibly due to autophagy induction, 94 was associated with improved vector genome copy number and integration in hepatocytes. 89 This could be linked to the global amelioration of the metabolism of targeted organs, since fenofibrate, a bezafibrate analog, has recently been reported to prevent the deleterious evolution of both liver and kidney diseases in liver- and kidney-specific deficient G6pc−/− mice, respectively. 14
Conclusions
In the past years, gene therapy reached maturity, moving from proof of concept to preclinical studies and clinical trials. New therapeutic approaches are being developed for a growing number of indications. However, current limitations of AAV-mediated gene transfer, in addition to those related to the immune response to the AAV product, include (1) the reduced persistence of vector genomes, in particular, in diseases with an underlying liver degeneration, (2) the potential genotoxicity of integrated genomes, and (3) the need of high vector doses to target nonpermissive tissues (Fig. 2). The hepatocyte degeneration observed in GSDI and GSDIII makes them an ideal test bench to develop novel approaches to overcome the actual gene therapy limitations in liver gene transfer. In addition to that, the need to treat a second tissue other than liver and the transgene size in GSDIII represents technical challenges for the available gene therapy tools. Hybrid promoters with dual tissue specificity, 95 new capsids with enhanced targeting of both liver and muscle or kidney, and new strategies to increase dual vectors recombination 96 may represent a potential solution.
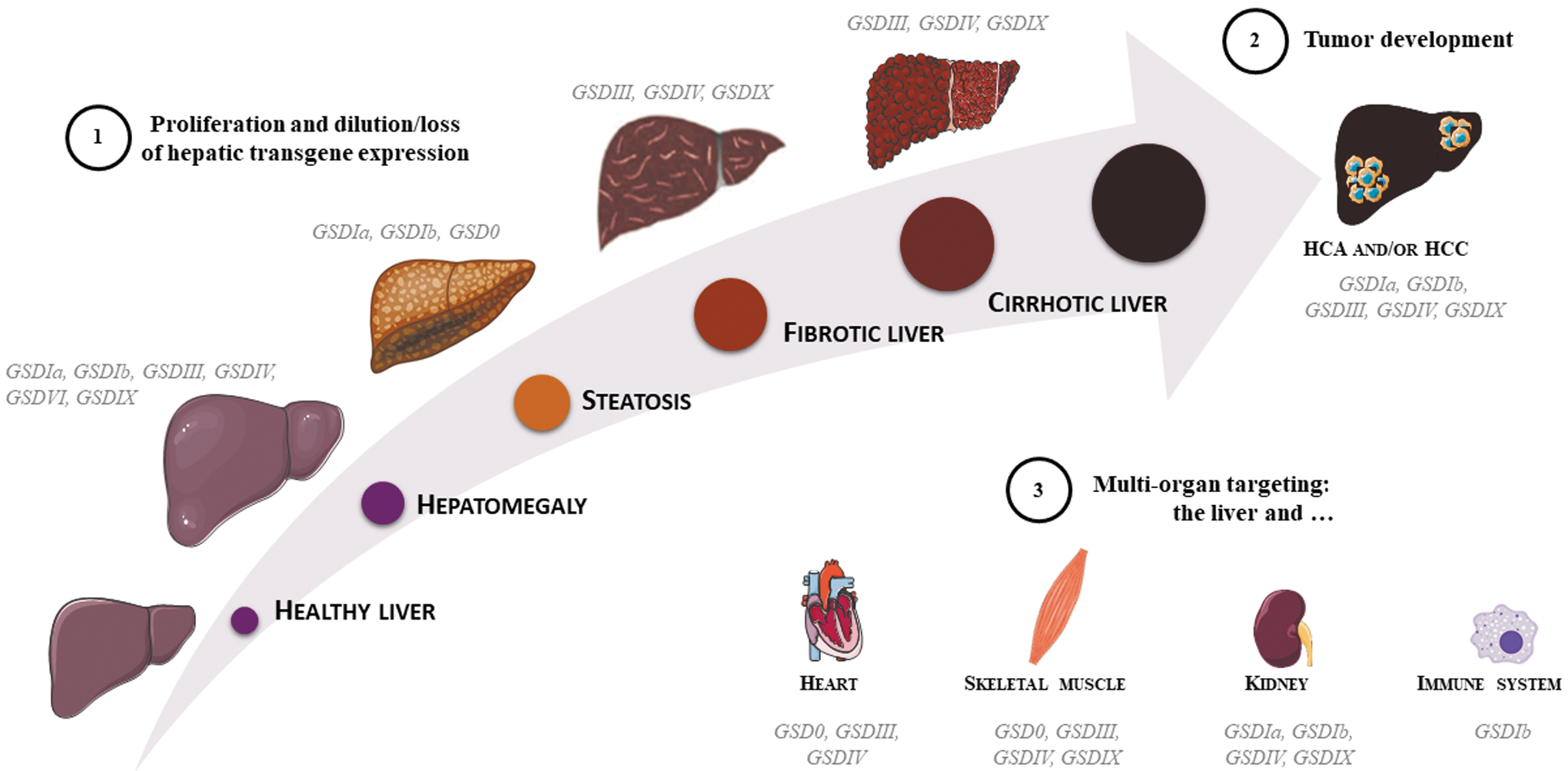
AAV gene therapy challenges in hepatic glycogen storage diseases. Increased liver size (i.e., hepatomegaly) is a hallmark of hepatic GSDs. The liver function impairment may evolve in steatosis or fibrosis/cirrhosis and ultimately in the formation of HCA or HCC.
An emerging concept particularly adapted to the treatment of metabolic diseases is the combination of gene therapy with other therapeutic strategies. The 20-year–long experiences in gene therapy for GSDIa clearly indicate that the degeneration of hepatocytes affects the long-term efficacy of gene therapy. Pharmacological intervention or dietary regimen aimed at the amelioration of the liver impairment before the administration of gene therapy demonstrated improved efficacy in GSDI. The combination of specific dietary regimen with gene therapy has the potential to improve the outcome of the treatment by improving vector persistence and by delaying the long-term complications onset.
In conclusion, the experience accumulated with both small and large animal models of GSDI and GSDIII hints at new therapeutic strategies for metabolic diseases. Those models represent a unique opportunity to identify molecular targets to improve the efficacy of gene transfer in the liver. Although we cannot exclude the existence of disease-specific pathways, it is likely that at least some parts of the underlying mechanisms are shared among the different diseases with hepatocyte degeneration. Therefore, the improvement of gene therapy for GSDI and GSDIII represents a unique opportunity to devise novel approaches and ultimately provide safe and efficacious treatments for genetic metabolic diseases.
Footnotes
Acknowledgments
GSDI and GSDIII researchers at Genethon and Inserm U1213/Université Lyon I have been supported by Genethon, the “Association Française contre la Myopathie”, the “Association Francophone des Glycogénoses,” and the National Research Agency (ANR16-CE14-0022-02 and ANR-17-CE18-0014).
Author Disclosure
G.R. is inventor in patents related to the development of gene therapy strategies for GSDIII.