
Abstract
Atherosclerosis is a disease of large- and medium-sized arteries that is caused by cholesterol accumulation in arterial intimal cells, including macrophages and smooth muscle cells (SMC). Cholesterol accumulation in these cells can be prevented or reversed in preclinical models—and atherosclerosis reduced—by transgenesis that increases expression of molecules that control cholesterol efflux, including apolipoprotein AI (apoAI) and ATP-binding cassette subfamily A, member 1 (ABCA1). In a previous work, we showed that transduction of arterial endothelial cells (EC)—with a helper-dependent adenovirus (HDAd) expressing apoAI—enhanced EC cholesterol efflux in vitro and decreased atherosclerosis in vivo. Similarly, overexpression of ABCA1 in cultured EC increased cholesterol efflux and decreased inflammatory gene expression. These EC-targeted gene-therapy strategies might be improved by concurrent upregulation of cholesterol-efflux pathways in other intimal cell types. Here, we report modification of this strategy to enable delivery of therapeutic nucleic acids to cells of the sub-endothelium. We constructed an HDAd (HDAdXMoAntimiR33a5p) that expresses an antagomiR directed at miR-33a-5p (a microRNA that suppresses cholesterol efflux by silencing ABCA1). HDAdXMoAntimiR33a5p contains a sequence motif that enhances uptake of anti-miR-33a-5p into exosomes. Cultured EC release exosomes containing small RNA, including miR-33a-5p. After transduction with HDAdXMoAntimiR33a5p, EC-derived exosomes containing anti-miR-33a-5p accumulate in conditioned medium (CM). When this CM is added to macrophages or SMC, anti-miR-33a-5p is detected in these target cells. Exosome-mediated transfer of anti-miR-33a-5p reduces miR-33a-5p by ∼65–80%, increases ABCA1 protein by 1.6–2.2-fold, and increases apoAI-mediated cholesterol efflux by 1.4–1.6-fold (all p ≤ 0.01). These effects were absent in macrophages and SMC incubated in exosome-depleted CM. EC transduced with HDAdXMoAntimiR33a5p release exosomes that can transfer anti-miR-33a-5p to other intimal cell types, upregulating cholesterol efflux from these cells. This strategy provides a platform for genetic modification of intimal and medial cells, using a vector that transduces only EC.
Introduction
Cardiovascular disease, primarily due to atherosclerosis, is the world's leading cause of death. 1 Therefore, improved treatments for atherosclerosis would significantly improve public health. Lipid-lowering therapy with statin drugs and inhibitors of proprotein convertase subtilisin/kexin 9 (PCSK9) can prevent and treat atherosclerosis; however, the potential benefits of statin therapy are limited by poor patient adherence, 2 PCSK9 inhibitors are prohibitively expensive, 3 and both drugs leave substantial residual cardiovascular disease risk. 4
Others developed gene therapy as a treatment for atherosclerosis, primarily as a liver-targeted intervention aimed at altering plasma lipid and lipoprotein levels (i.e., lowering low-density lipoprotein cholesterol or raising high-density lipoprotein [HDL] cholesterol). 5,6 However, large viral doses are needed to efficiently transduce the liver, and these doses are often toxic. 7,8 Our group and others developed gene therapy, delivered directly to segments of the blood vessel wall, as an alternative means of preventing and reversing atherosclerosis. This approach is effective in animal models, 9 but until recently it was limited by vector-induced vascular inflammation and brevity of transgene expression. 10,11
We overcame both of these limitations by using helper-dependent adenovirus (HDAd). 12 HDAd-mediated transgene expression persists stably for ≥48 weeks in rabbit carotid arteries and is associated with minimal vascular inflammation. 13,14 HDAd-mediated expression of apolipoprotein AI (apoAI) in rabbit carotids reduces development of atherosclerosis 13,15 and stimulates regression of small-to-medium-sized lesions. 16 Nevertheless, HDAd-mediated gene therapy is only partially successful in reducing atherosclerosis (∼30% reduction) and does not stimulate regression of larger atherosclerotic lesions. 15,16
These limitations might be explained—at least in part—by the inability of adenoviral vectors to cross intact endothelium in large arteries. 11,17 When HDAd is delivered via the lumen of a large artery, only the luminal endothelial cells (EC) are transduced. 18 However, the cellular cholesterol accumulation that drives atherosclerotic lesion growth occurs primarily in cells below the endothelium, including macrophages and smooth muscle cells (SMC). 19 ApoAI secreted from transduced EC can enter the artery wall and stimulate cholesterol efflux from underlying cells; however, this activity of apoAI depends on target-cell expression of cholesterol-efflux proteins such as ATP-binding cassette subfamily A, member 1 (ABCA1). 20,21
We considered whether we could combine EC-targeted apoAI gene therapy with a gene-based intervention that upregulates expression of cholesterol-efflux proteins in underlying macrophages and SMC. We identified anti-miR-33a therapy as a promising approach. Anti-miR-33a therapy reliably increases cellular cholesterol efflux, primarily by upregulating expression of the miR-33a target ABCA1. 22
We next identified exosome-packaged antagomiRs as an approach whereby anti-miR-33a could potentially be expressed in EC and delivered, via cell
Materials and Methods
Cell culture and transduction
293-Cre cells (Microbix, Toronto, ON, Canada) 26 were maintained in high-glucose Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 1% Pen-Strep, and 400 μg/mL G418 (Thermo Fisher Scientific, Waltham, MA). Bovine aortic EC (BAEC) (Cell Applications, San Diego, CA) and bovine aortic SMC (BASMC) (Cell Applications) were maintained in high-glucose DMEM (GIBCO, Grand Island, NY), 10% FBS (VWR Life Science Seradigm, Radnor, PA), and 1% Pen-Strep (GIBCO) and they were used between passages 3 and 7. Human THP-1 monocytes (ATCC, Manassas, VA) were maintained in high-glucose RPMI-1640 (ATCC) containing 10% FBS and 1% Pen-Strep and were differentiated into macrophages by treatment with phorbol myristate acetate (100 ng/mL) for 72 h. All cells were cultured at 37°C with 5% CO2.
We used BAEC because they are easily propagated, which is necessary to produce the large amount of exosome-containing medium that we anticipated would be needed for these experiments. We used BASMC, because they are a useful experimental model of cellular cholesterol loading and inducible cholesterol efflux. 27,28 We used THP-1 cells because they are also a useful model of cholesterol loading and inducible cholesterol efflux, and they express both miR-33a-5p and ABCA1 (useful as positive controls). 22
For transduction, BAEC were grown to 80–90% confluency; then, they were exposed to viral vectors (1 × 1010 viral particles (vp)/mL in growth medium) for 6 h. The vector-containing medium was then removed; cells were washed with phosphate-buffered saline (PBS) and fed with either growth medium or serum-free DMEM. This BAEC-conditioned medium (CM) was collected 48 h later and either used immediately or stored at −80°C.
Isolation of extracellular vesicles from CM of untransduced BAEC
BAEC were grown to 80–90% confluency in 150-mm dishes, washed with PBS, and fed with serum-free DMEM (8 mL per 150-mm dish). Forty-eight hours later, CM was collected and extracellular vesicles (EV) were isolated, essentially as described. 29 Briefly, cells and cell debris were removed by serial centrifugation of CM and decantation of EV-containing supernatant: 300 g for 10 min, 2,000 g for 20 min, and 10,000 × g for 30 min (all at 4°C). EV were then pelleted from the CM by ultracentrifugation (100,000 g for 90 min at 4°C). The post-ultracentrifugation CM supernatant (termed “EV-depleted CM”) was either discarded or stored at −80°C for future use, as a control. The pelleted EV were gently resuspended in PBS and re-pelleted by ultracentrifugation, as described earlier. The post-ultracentrifugation PBS supernatant was then either discarded or stored at −80°C for future use, as a control. The pelleted EV were either used immediately (for nanoparticle tracking analysis, imaging, or RNA/protein analyses) or gently resuspended in ∼50 μL PBS and stored at −80°C.
EV size and appearance
Freshly isolated EV, EV-depleted CM, and post-ultracentrifugation PBS supernatant were evaluated with nanoparticle tracking analysis (NanoSight NS300 instrument; Malvern Instruments, Malvern, UK), which measures particle concentration and diameter. 30 Briefly, pelleted EV samples were vortexed and serially diluted at a ratio of 1:6,000–1:15,000 with molecular-grade double-distilled water. This diluent also served as a negative control. Three freshly isolated EV samples were measured with nanoparticle tracking, and mean values were calculated.
We used an established protocol to image EV by transmission electron microscopy. 29 Briefly, the remainder of the three EV samples used for nanoparticle tracking analysis were frozen, then thawed, and pooled (∼100 μL total volume). We added an equal volume of 4% paraformaldehyde; then, we deposited the mixture by airfuge on Formvar-carbon coated transmission electron microscopy grids (Ted Pella, Redding, CA). Samples were embedded and contrasted by treatment with uranyl-oxalate solution (pH 7; Electron Microscopy Sciences, Hatfield, PA) for 5 min, followed by treatment with methyl-cellulose-uranyl-acetate (Sigma-Aldrich, St. Louis, MO) on ice for 10 min. Transmission electron micrographs were acquired by using a JEM-1400 transmission electron microscope (JEOL, Tokyo, Japan) at 120kV along with an Ultrascan 1000XP digital camera (Gatan, Pleasanton, CA).
Molecular cloning and HDAd generation, amplification, and characterization
We purchased two plasmids (MZIP33a-PA-1 and XMIRXP-NT; SBI, Palo Alto, CA). MZIP33a-PA-1 contains an expression cassette in which the H1 promoter 31 drives transcription of an antagomiR targeted at miR-33a-5p. 22 XMIRXP-NT contains an expression cassette in which the H1 promoter drives transcription of a non-targeted “scrambled” control shRNA. 22 XMIRXP-NT also includes an “X-motif” DNA sequence that, when transcribed, increases uploading of shRNA transcripts into exosomes. 32 Both expression cassettes also contain a “5T” (TTTTT) termination signal. 33
We used these plasmids as templates for PCR (Q5® High-Fidelity DNA Polymerase; New England Biolabs, Ipswich, MA; see Supplementary Table S1 for primer sequences) that generated three expression cassettes (Supplementary Fig. S1), all flanked by AscI sites for use in ligating the cassettes into an HDAd backbone plasmid. For the first reaction, we used the MZIP33a-PA-1 plasmid as template, and primers to amplify the anti-miR-33a-5p expression cassette. For the second reaction, we used XMIRXP-NT as a template for amplification of the scrambled control expression cassette. For the third reaction, we again used MZIP33a-PA-1 as a template, but we incorporated nucleotides representing the X-motif into the reverse PCR primer. The three amplicons were digested with AscI and ligated into AscI-digested/phosphatased pC4HSU, an HDAd backbone plasmid. 12 We named the ligation products pC4HSU-AntimiR33a5p, pC4HSU-XMoScr, and pC4HSU-XMoAntimiR33a5p.
These plasmids were transformed into XL10-Gold® ultracompetent cells (Stratagene, La Jolla, CA), followed by plating onto LB Amp-100 agar plates and colony PCR to detect the inserted sequences. Plasmid DNA was extracted from positive clones and transformed into SURE 2 supercompetent cells (Agilent, Santa Clara, CA), followed by screening with restriction digests. DNA from positive clones was sequenced (Sequetech, Mountain View, CA), and in all cases the expression cassettes were intact. The HDAd plasmids were then digested with PmeI, transfected into 293-Cre cells, 26 followed by infection with H14 helper virus (Microbix) 12 to produce HDAdAntimiR33a5p, HDAdXMoScr, and HDAdXMoAntimiR33a5p. HDAds were amplified, purified, and characterized as described. 34 All HDAd preparations contained <1 in 106 E1A-positive genomes and <1% helper-virus contamination.
RNA and DNA analyses
Cells or EV (the latter purified by ultracentrifugation) were lysed with TRIzol (Thermo Fisher Scientific). Total RNA (including miRNA) was purified by using Direct-Zol columns (Zymo Research, Irvine, CA) and quantified with NanoDrop (Thermo Fisher Scientific). We measured the size of cellular and EV RNA with a 2100 BioAnalyzer Pico Chip (Agilent), using a sample concentration of 1.2 ng/μL.
To measure specific RNA transcript levels, 100 ng of cellular or EV RNA was used as a substrate for polyadenylation and cDNA synthesis with an oligo-dT primer that includes a 3′ universal tag (miScript kit; Qiagen, Hilden, Germany). The product of this reaction was used as a substrate for either end-point PCR or quantitative PCR. PCR primers for these reactions (Qiagen) included: a reverse primer that binds the universal tag for all reactions (miScript kit), and a gene-specific forward primer that binds to either: (1) the reference gene RNU6B (miScript kit); (2) miR-33a-5p (MS00003304); (3) anti-miR-33a-5p (MSC0075705); or (4) the scrambled control shRNA (MSC0075764). For qPCR, amplification products were detected with Sybr Green and transcript levels were calculated by using the ΔΔCT method, 35 with normalization to RNU6B levels measured in the same samples.
To more confidently detect endogenous miR-33a-5p transcripts (based on size and restriction digests), cDNA was generated from RNA extracted from cells or EV, and this cDNA was used as a substrate for PCR by using the universal reverse primer and the miR-33a-5p-specific forward primer. The amplification reaction was divided into three equal volumes: One was not digested, one was digested with BsrDI (for which a restriction site is present in the gene encoding miR-33a-5p), and one was digested with NotI (for which the gene encoding miR-33a-5p lacks a restriction site). The products were then analyzed by tris-borate-EDTA-PAGE (4–20% Criterion™ TBE Polyacrylamide Gel; Bio-Rad, Hercules, CA).
To sequence the 3′ untranslated region of the bovine ABCA1 gene, total RNA was extracted from BASMC and BAEC, and 500 ng of RNA was used as a template for cDNA synthesis (GoScript™ Reverse Transcriptase; Promega, Madison, WI), using a gene-specific primer (see reverse primer below). The cDNA products were used as templates for PCR (Q5 High-Fidelity DNA Polymerase; forward primer: GTCCATATACGGTGGCTGGAAG; reverse primer: CTATATCACCGGGAAGCCAGTC). The amplicons were purified by agarose gel electrophoresis, excised, and sequenced (GenScript, Piscataway, NJ).
SDS-PAGE and immunoblotting
BAEC (freshly harvested) or BAEC-derived EV (either freshly isolated or thawed) were lysed (Roche cOmplete Lysis-M kit; Penzberg, Germany), and protein was quantified with the BCA assay (Thermo Scientific Pierce, Waltham, MA). To characterize protein expression in BAEC and EV, equal amounts of protein were separated on 10% Invitrogen NuPAGE Bis-Tris gels (Waltham, MA), followed by staining with Coomassie blue.
For immunoblotting, equal amounts of protein were separated on 4–15% Mini-PROTEAN® TGX gels (Bio-Rad), and they were transferred onto Immobilon-P membranes (Millipore, Billerica, MA); membranes were blocked with 5% (w/v) nonfat dry milk. Blots of BAEC and EV lysates were probed with: murine anti-CD9 (1:1,500 dilution, ab3923; Abcam, Cambridge, UK), goat anti-CD81 (1:300 dilution, sc-31234; Santa Cruz Biotechnology, Dallas, TX), rabbit anti-calnexin (1:300 dilution, sc-11397; Santa Cruz Biotechnology), or goat anti-calreticulin (1:300 dilution, sc-6468; Santa Cruz Biotechnology). Secondary antibodies included horseradish peroxidase-conjugated goat anti-mouse Immunoglobulin G (IgG) (1:7,500 dilution, 1706516; Bio-Rad), donkey anti-goat IgG (1:7,500 dilution, sc-2033; Santa Cruz Biotechnology), or goat anti-rabbit IgG (1:7,500 dilution, 1706515; Bio-Rad). Bound secondary antibody was detected with Amersham ECL Select (GE Life Sciences, Chicago, IL) and x-ray film.
BASMC and THP-1-derived macrophages were lysed and proteins were quantified, separated, and blotted as described earlier. Blots were probed with either murine anti-ABCA1 (1:500 dilution, sc-58219; Santa Cruz Biotechnology) or goat anti-GAPDH (1:1,500 dilution, sc-20357; Santa Cruz Biotechnology). Secondary antibodies included horseradish peroxidase-conjugated goat anti-mouse IgG (1:7,500 dilution, 1706516; Bio-Rad) or donkey anti-goat IgG (1:7,500 dilution, sc-2033; Santa Cruz Biotechnology). Bound secondary antibody was detected with Bio-Rad Clarity Western ECL substrate and imaged by using a charge-coupled device camera (Bio-Rad ChemiDoc, Hercules, CA). ABCA1 signal was normalized to GAPDH signal in the same lane, as a loading control. For all blots, signal was quantified by using NIH ImageJ software.
EV degradation assay
We used a modification of a published protocol 36 to assess whether EV-associated RNA was contained in lipid vesicles. Freshly isolated BAEC-derived EV were resuspended in ∼60 μL PBS and divided into three equal fractions. One fraction was treated with Tween-20 (1% v/v in PBS; Thermo Fisher Scientific) for 15 min at room temperature, followed by addition of pronase (final concentration 1 mg/mL; Roche) for 20 min at 37°C, inactivation of pronase (20 min at 85°C), addition of RNase A (final concentration 200 pg/mL; Thermo Fisher Scientific) for 30 min at 37°C, and addition of SUPERase• In™ RNase Inhibitor (final concentration 0.5 U/μL; Thermo Fisher Scientific) for 15 min at room temperature. The second fraction was treated with PBS without Tween-20; then, it received the same treatments as the first fraction.
The third fraction was handled and incubated in the same manner as the first two fractions, however without addition of Tween-20, pronase, or RNase. RNA was then extracted from the three fractions, as described earlier, and equal volumes of the RNA extracts were used to measure miR-33a-5p by reverse transcriptase-mediated quantitative PCR, as described earlier.
RNA, protein, and cholesterol-efflux studies in cells treated with BAEC-derived CM
For RNA and protein studies, BASMC and THP-1-derived macrophages were grown to 70–80% confluency in six-well plates. Growth medium was removed; cells were washed with PBS, then, they were fed with either BAEC-derived serum-containing CM (1 mL per well) or EV-depleted (via ultracentrifugation) BAEC-derived serum-containing CM (1 mL per well) as a control solution. As an alternative to treating BASMC and THP-1 macrophages with BAEC-derived EV-containing CM, we considered purifying EV from CM and treating the target cells with the purified EV.
For three reasons, we decided to use EV-containing CM rather than purified EV, with EV-depleted CM as the negative control. First, we were concerned that use of purified EV could yield a falsely positive result, due to exposure of the target cells to supraphysiological concentrations of EV. Second, we judged that EC-derived CM would be a more physiological medium than any medium typically used to resuspend purified EV. Third, we decided that we could confirm an active role for EV in BAEC-derived CM by removing the EV from CM by ultracentrifugation and testing the EV-depleted CM for activity. Ultracentrifugation removes EV from CM but does not deplete CM of biomolecules that could be a source of false positivity. All aliquots of CM used to treat cells were frozen and thawed only once before use. Cells were harvested either 24 h after addition of CM (for RNA analyses) or 48 h after addition of CM (for protein analyses).
To measure apoAI-mediated cholesterol efflux from THP-1-derived macrophages, cells were grown to 70–80% confluency in 48-well plates. Growth medium was removed; cells were washed with PBS, and then they were treated either with BAEC-derived serum-free CM (200 μL per well, containing fatty acid-free albumin (FAFA); 2 mg/mL; Sigma-Aldrich) or with EV-depleted serum-free CM. Twenty-four hours later, CM was removed; cells were washed with PBS and loaded with cholesterol by addition of serum-free RPMI 1640 (200 μL per well) containing FAFA (2 mg/mL) and [ 3 H] cholesterol (2 μCi/mL; PerkinElmer Life Sciences, Waltham, MA). After 24 h, cholesterol-loading medium was removed; cells were washed with PBS, and then they were fed with serum-free RPMI-1640 (200 μL per well, containing 2 mg/mL FAFA), either with or without purified human apoAI (50 μg/mL). Twenty-four hours later, medium and cells were collected for measurement of [ 3 H] cholesterol. 21
Compared with macrophages, cultured SMC are relatively resistant to cholesterol loading. 37 Therefore, to measure apoAI-mediated cholesterol efflux from BASMC, we varied the protocol described earlier. BASMC were grown to 70–80% confluency in 48-well plates. Growth medium was removed; cells were washed with PBS, and then they were loaded with cholesterol by addition of serum-free DMEM (200 μL per well) containing FAFA (2 mg/mL) and [ 3 H] cholesterol (2 μCi/mL). Seventy-two hours later, cholesterol-loading medium was removed; cells were washed with PBS, and they were treated with BAEC-derived serum-free CM (200 μL per well; with 2 mg/mL FAFA) either with or without purified human apoAI (100 μg/mL; Academy Bio-Medical Company, Houston, TX) or with EV-depleted serum-free CM. Seventy-two hours later, medium and cells were collected for measurement of [ 3 H] cholesterol, as described. 21
Statistics
Data were analyzed with SigmaPlot (Systat Software, Inc., San Jose, CA). We tested equal variance and normality assumptions with Brown-Forsythe and Shapiro-Wilk tests, respectively. When these assumptions were met, we performed an unpaired two-tailed t-test. When these assumptions were violated, we used a nonparametric Mann–Whitney rank-sum test. When technical controls were included in individual experiments (e.g., EV-depleted CM in an experiment designed to compare EV-containing CM harvested from EC transduced with different viral vectors), results with these controls are presented, but not included in the statistical analyses. Therefore, with one exception, statistical analyses compare only two groups. The exception is the three-group EV degradation experiment, for which we used Kruskal–Wallis analysis of variance (ANOVA), with Dunn's method for post hoc correction for multiple comparisons.
For our analyses, we considered technical replicates to be repeated measurements of the same sample; for example, duplicate PCRs performed on the same cell extracts. 38 We considered biological replicates to be experiments performed on different wells of cells and with different aliquots of test reagents. For example, three wells of cells plated separately and experimented on at a later time point were considered to be n = 3 biological replicates. In all cases, the consistency of data generated from biological-replicate wells of cells was further tested by repeating the experiments on separate days, using cells derived from separate vials of cryopreserved cells. We refer to these and other non-contemporaneous replicate experiments, performed with newly thawed cells or with separately cultured cells as “independent experiments.”
Results
Cultured BAEC release exosomes
To test whether BAEC are a suitable model system for developing exosome-mediated gene therapy, we first determined whether exosomes are released from cultured BAEC. Analysis of global protein expression (on Coomassie-stained gels) revealed differences between BAEC lysates and lysates of material isolated by ultracentrifugation of BAEC-CM, suggesting that BAEC release EV 29 into CM (Fig. 1A). Immunoblotting of these samples provided further evidence that exosomes are present in BAEC-CM: The exosome markers CD9 and CD81 39 were enriched among CM-derived proteins, whereas calnexin and calreticulin (both of which are absent in exosomes) 29 were depleted among CM-derived proteins (Fig. 1B).
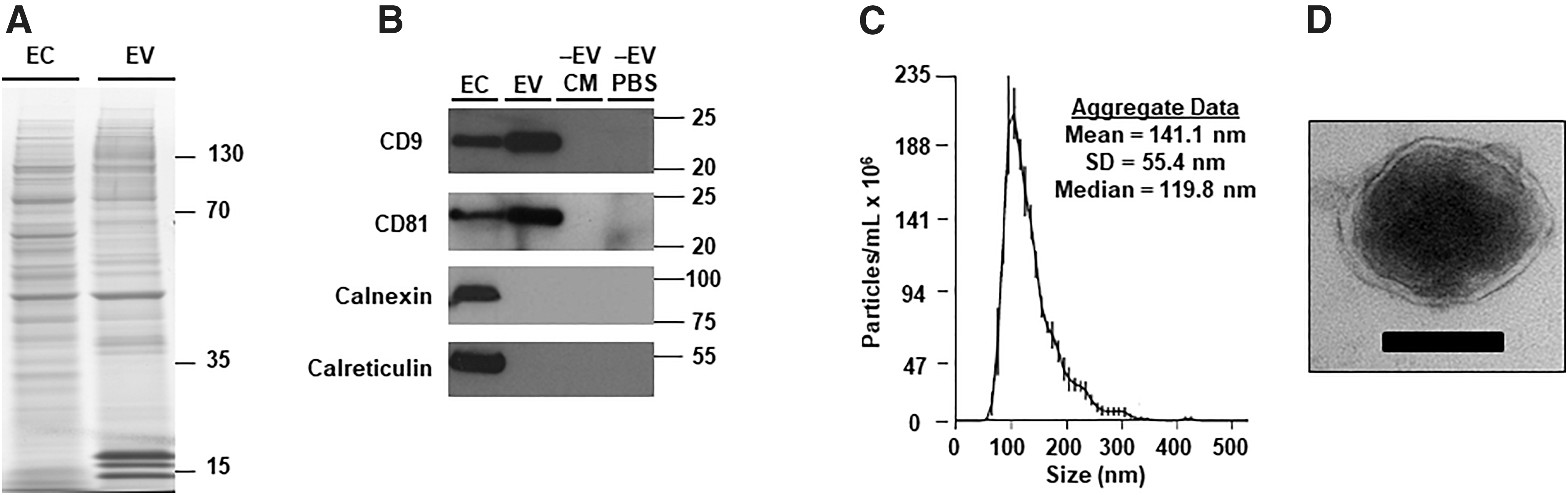
BAEC release EV that are predominately exosomes.
We next characterized the physical properties of particles in the ultracentrifuged BAEC-CM. Nanoparticle tracking analysis showed that the majority of these particles were in the size range of exosomes (30–150 nm 40 ; Fig. 1C) rather than microvesicles (diameter up to 1,000 nm). 41 As expected, this analysis also showed that EV-depleted BAEC-CM and the EV-depleted PBS wash (Fig. 1B) had >99% fewer particles compared with the resuspended ultracentrifugation pellet (data not shown), confirming that the ultracentrifugation protocol efficiently isolates EV. Imaging of the resuspended pellet with transmission electron microscopy revealed exosome-sized particles (∼100 nm diameter) with an apparent lipid bilayer and an electron-dense core, characteristic of exosomes. 36,42 (Fig. 1D and Supplementary Fig. S2) Therefore, cultured BAEC release EV that are predominantly exosomes.
EV from EC are enriched in small RNA and contain miR-33a-5p
A main function of EV, particularly exosomes, is to transfer small RNA to recipient cells. 41 We, therefore, characterized EV-associated RNA by measuring the size distribution of EC-associated and EV-associated RNA and by testing whether the EV-associated RNA was contained in lipid-bound vesicles. As expected, EC-associated RNA consisted primarily of larger RNA, especially rRNA (Fig. 2A), whereas EV-associated RNA was predominantly less than 500 nucleotides (Fig. 2B). To determine whether EV-associated RNA was contained in lipid vesicles, we first tested whether we could detect miR-33a-5p (an important regulator of lipid metabolism 43 and the target of our gene-therapy strategy) in RNA extracted from either EC or EV.
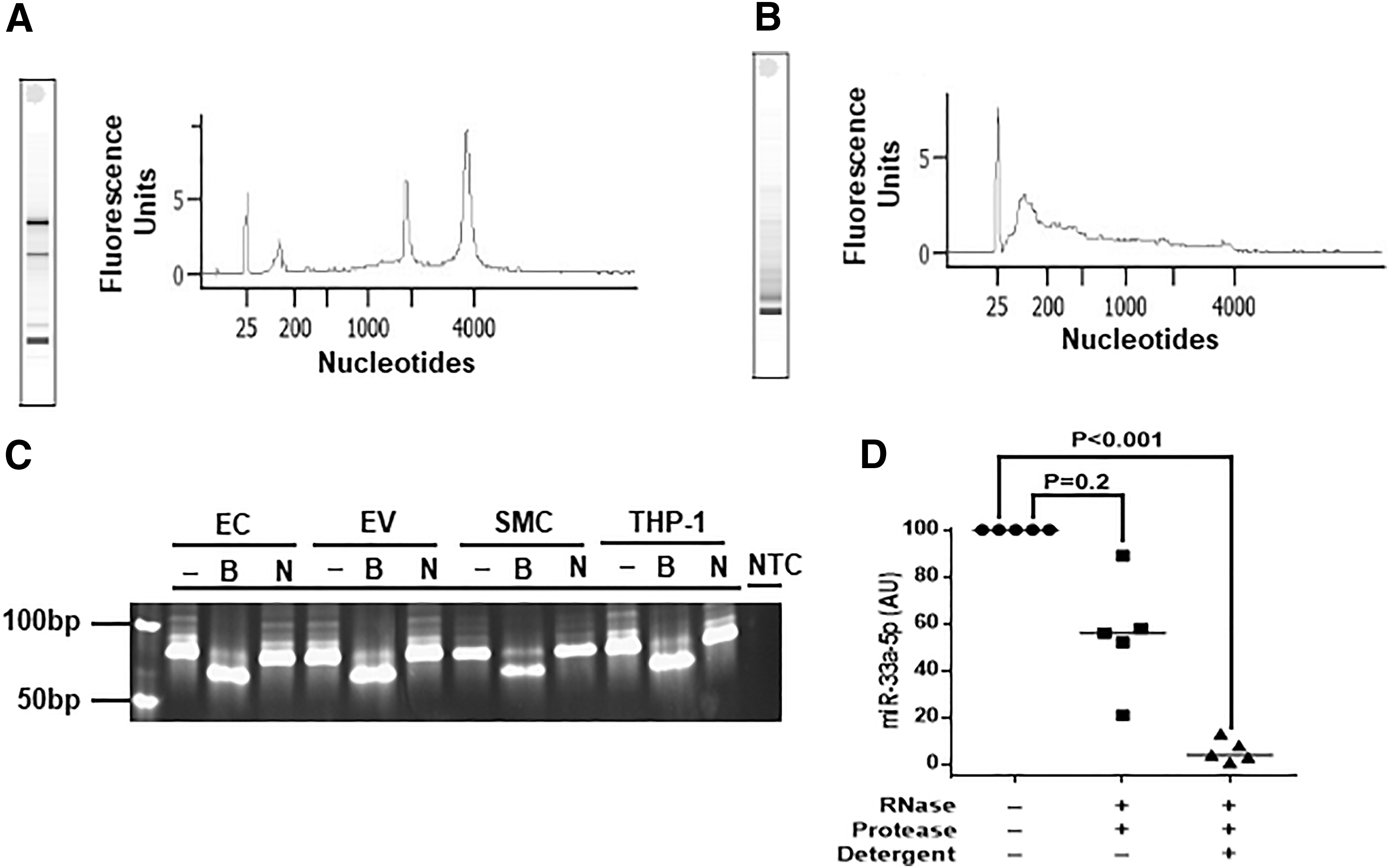
BAEC-derived EV contain small RNA, including miR-33a-5p. Size distribution of total RNA from EC
We are not aware of any prior reports of miR-33a-5p expression from primary EC; however, production of miR-33a-5p by EC and its release in EV could negatively impact our strategy to use EC and EC-derived EV to deliver anti-miR-33a-5p to neighboring cells. Indeed, reverse transcriptase-mediated PCR (using primers designed to amplify miR-33a-5p) generated an amplicon of the predicted size (∼85 bp; personal communication; Qiagen) from both total EC RNA and EV-associated RNA (Fig. 2C). We confirmed the identity of the amplicon with a BsrDI restriction digest, which yielded the predicted fragments of ∼70 and ∼15 bp (Fig. 2C and data not shown). A negative control NotI digest (no NotI sites are present in the gene encoding miR-33a-5p) left the amplicon intact, as predicted (Fig. 2C, Supplementary Fig. S3).
We considered that EV-associated miR-33a-5p might co-purify with EV as an extravesicular RNA or RNA/protein complex, rather than being contained within EV. 41 To differentiate these two possibilities, we performed an EV degradation assay, 44 in which purified EV are treated with RNase and protease with or without pre-treatment with detergent (Supplementary Fig. S4), followed by measurement of miR-33a-5p by reverse transcriptase-mediated quantitative PCR.
Treatment of EV with RNAse and protease alone caused a variable decrease in miR-33a-5p that did not achieve statistical significance (p = 0.2; Fig. 2D). However, if EV were treated with detergent before exposure to RNAse and protease, miR-33a-5p was decreased by >95% (p < 0.001; Fig. 2D). Taken together, our results do not exclude the possibility that some of the miR-33a-5p in the EV-containing pellet is extravesicular; however, they do show that a substantial amount of EV-associated miR-33a-5p is contained in lipid-bound vesicles, which are predominantly exosomes.
In parallel with our measurements of miR-33a-5p in EC and EC-derived EV, we measured miR-33a-5p in bovine SMC and human THP-1 cells. Detection of miR-33a-5p in cultured SMC and THP-1 cells was a prerequisite to use of these cells in experiments that test efficacy of anti-miR-33a-5p in reversing miR-33a-5p-mediated suppression of cholesterol efflux. Consistent with others' results, 22,45 miR-33a-5p was present in both cell types (Fig. 2C).
Addition of the X-motif increases EV uptake of anti-miR-33a-5p transcripts and enhances transfer of anti-miR-33a-5p to target cells
EC-derived miR-33a-5p (Fig. 2C) could be transferred to macrophages and SMC via exosomes, inhibiting cholesterol efflux and driving atherosclerosis. If abundant, endogenous EC-derived exosome-contained miR-33a-5p could interfere with our gene-therapy strategy by binding and inactivating HDAd-expressed anti-miR-33a-5p. Therefore, we pursued two strategies aimed at using EC-derived exosomes to deliver a miR-33a-5p-targeted antagomiR to adjacent vascular cells.
First, we constructed an HDAd vector that expresses an miR-33a-5p-targeted antagomiR (HDAdAntimiR33a5p; Supplementary Fig. S1), transduced EC with this HDAd, and measured anti-miR-33a-5p in both EC and EC-derived exosomes. In parallel, to address our concern that endogenous EC-derived exosome-contained miR-33a-5p could block anti-miR-33a-5p activity, we constructed a second anti-miR-33a-5p-expressing HDAd (HDAdXMoAntimiR33a5p; Supplementary Fig. S1), in which we added a short nucleotide sequence (the “X-motif”) that increases uploading of small RNA into exosomes (Supplementary Fig. S1). 32,46 Incorporation of the X-motif decreased EC-associated anti-miR-33a-5p by ∼98% (p = 0.008; Fig. 3A) and increased exosome-associated anti-miR-33a-5p by ∼80-fold (p = 0.008; Fig. 3B).
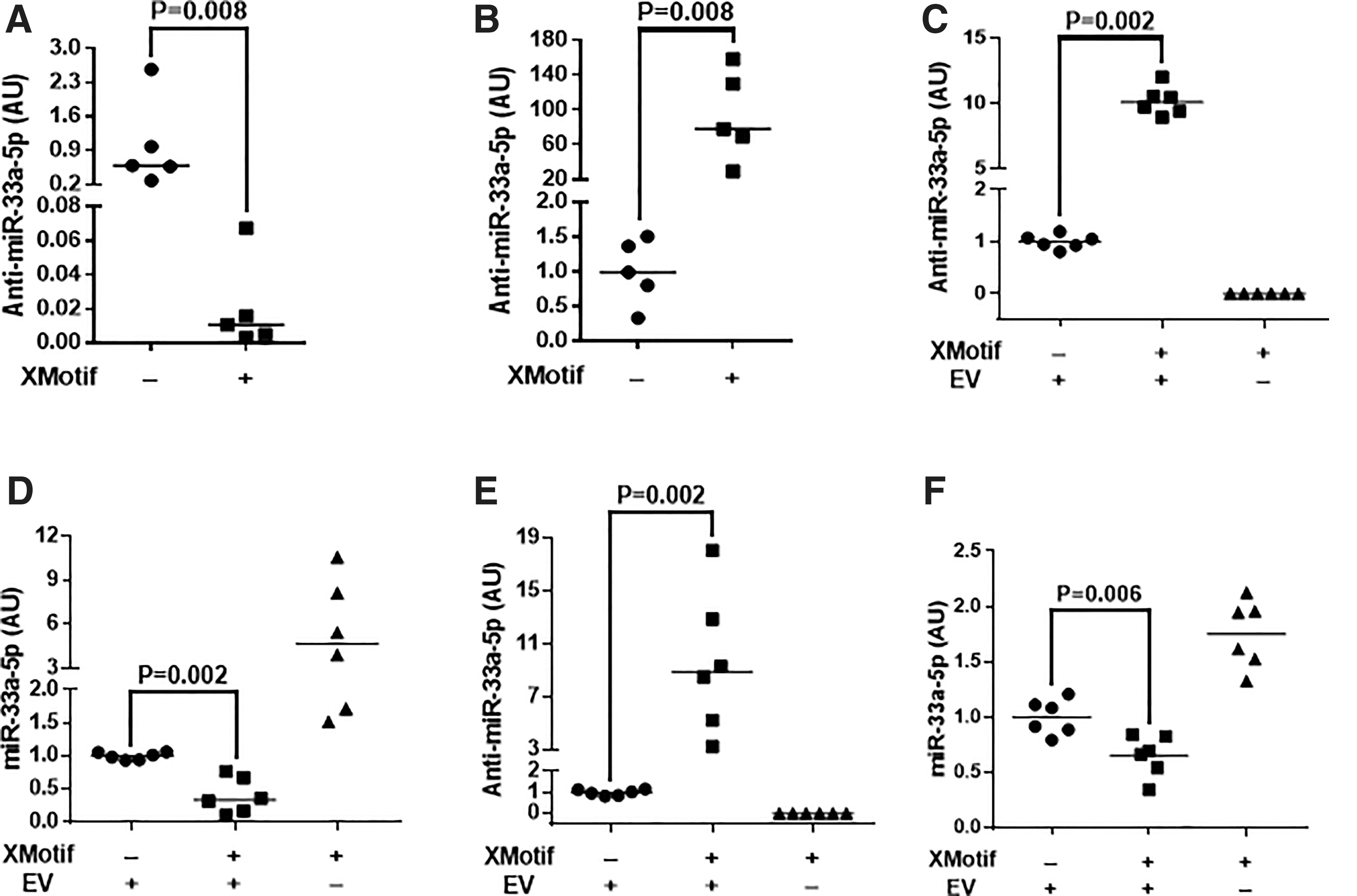
EV-mediated transfer of anti-miR-33a-5p to macrophages and SMC decreases miR-33a-5p expression: effect of X-motif.
We next harvested CM from EC transduced with either HDAdAntimiR33a5p or HDAdXMoAntimiR33a5p, and we incubated macrophages and SMC with this CM. Anti-miR-33a-5p was detected in macrophages exposed to CM from either HDAdAntimiR33a5p- or HDAdXMoAntimiR33a5p-transduced EC (Fig. 3C). In contrast, anti-miR-33a-5p was undetectable in macrophages exposed to EV-depleted CM. Addition of the X-motif to the anti-miR-33a-5p transcript increased macrophage anti-miR-33a-5p by ∼10-fold (p = 0.002).
When compared with control macrophages (treated with EV-depleted CM), exosome-mediated transfer of anti-miR-33a-5p effectively decreased macrophage miR-33a-5p levels. Treatment of macrophages with CM from HDAdAntimiR33a5p-transduced EC decreased miR-33a-5p by ∼80% (Fig. 3D). Macrophage miR-33a-5p levels were decreased by an additional ∼60% by treatment with CM from HDAdXMoAntimiR33a5p-transduced EC (p = 0.002).
Similar results were obtained with SMC. Anti-miR-33a-5p was detected in SMC exposed to CM from HDAdAntimiR33a5p-transduced EC. SMC treated with CM from HDAdXMoAntimiR33a5p-transduced EC contained ∼9-fold higher levels of anti-miR-33a-5p (p = 0.002; Fig. 3E). In contrast, anti-miR-33a-5p was undetectable in SMC exposed to EV-depleted CM.
Exosome-mediated transfer of anti-miR-33a-5p decreased SMC miR-33a-5p levels. Compared with control SMC, exposure of SMC to CM from HDAdAntimiR33a5p-transduced EC reduced levels of miR-33a-5p by ∼70%, and exposure to CM from HDAdXMoAntimiR33a5p-transduced EC decreased miR-33a-5p levels by an additional ∼35% (p = 0.006; Fig. 3F). Therefore, EC-derived exosomes can deliver anti-miR-33a-5p to macrophages and SMC and suppress miR-33a-5p levels. Addition of the X-motif to the antagomiR-expression cassette significantly increases both antagomiR delivery and target miRNA suppression.
Exosome-mediated transfer of anti-miR-33a-5p upregulates ABCA1 expression and enhances apoAI-mediated cholesterol efflux
To provide a more optimal control vector for testing whether exosome-mediated delivery of anti-miR-33a-5p increases expression of the miR-33a-5p target ABCA1 and increases cellular cholesterol efflux, we generated a control HDAd that expresses a scrambled antagomiR 22 and includes the X-motif (HDAdXMoScr; Supplementary Fig. S1). After transduction of EC with HDAdXMoScr, the scrambled antagomiR was incorporated into EV and accumulated in EC-derived CM at similar levels as XMoAntimiR33a5p, establishing the suitability of HDAdXMoScr to serve as a control for HDAdXMoAntimiR33a5p (Fig. 4B).

MiR-33a-5p transcripts in macrophages and SMC are decreased by treatment with EC-derived EV containing anti-miR-33a-5p. BAEC were transduced with HDAd vectors expressing either:
We then repeated the experiments described earlier (Fig. 3C–F), only this time we compared CM from HDAdXMoAntimiR33a5p-transduced EC with CM from HDAdXMoScr-transduced EC. As expected, compared with cells treated with CM from HDAdXMoScr-transduced EC, exposure to CM from HDAdXMoAntimiR33a5p-transduced EC decreased miR-33a-5p expression in both macrophages and SMC (70–80%; p < 0.001; Fig. 4C, D). EV-depleted CM from HDAdXMoAntimiR33a5p-transduced EC had no effect on miR-33a-5p levels in either cell type.
Before testing whether EV-mediated delivery of anti-miR-33a-5p altered ABCA1 expression in human THP-1-derived macrophages and bovine SMC, we sequenced the 3′ UTR of the Bos taurus ABCA1 gene to determine whether consensus miR-33a-5p binding sites (CAATGCA; present in both the Mus musculus and Homo sapiens ABCA1 3′ UTRs) 22 are conserved in Bos taurus. Three consensus miR-33a-5p binding sites were identified (Supplementary Fig. S5). We then treated THP-1-derived macrophages and bovine SMC with CM either from EC transduced with HDAdXMoAntimiR33a5p or from EC transduced with HDAdXMoScr. CM from HDAdXMoAntimiR33a5p-transduced EC increased ABCA1 protein by ∼2.2-fold in macrophages (p < 0.001; Fig. 5A, B) and ∼1.6-fold in SMC (p = 0.01; Fig. 5C, D). In both cell types, these effects were eliminated when exosomes were removed from the CM samples.
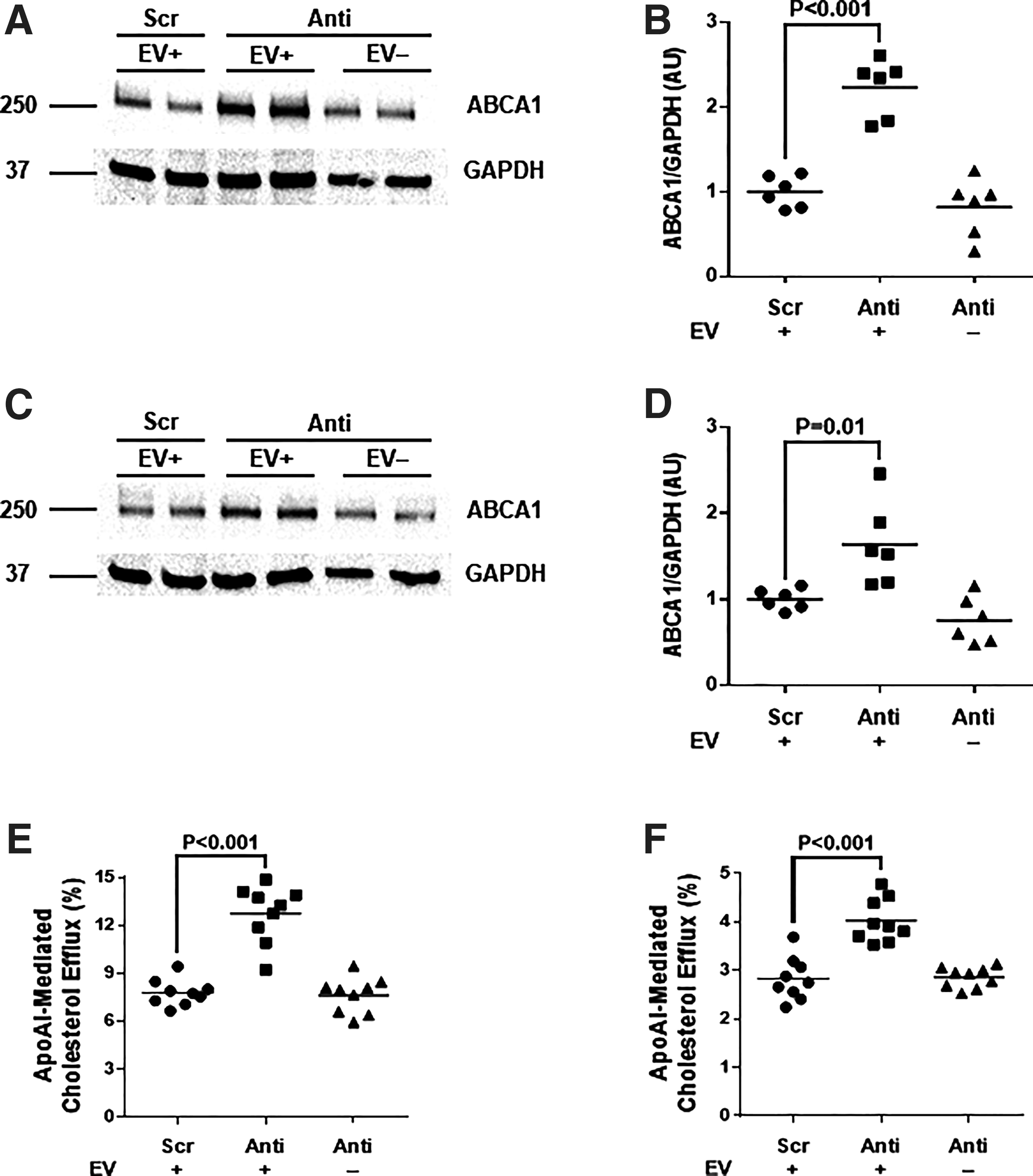
ABCA1 protein and apoAI-mediated cholesterol efflux are increased by treatment with EC-derived EV containing anti-miR-33a-5p. THP-1-derived macrophages
Finally, we loaded both THP-1-derived macrophages and bovine SMC with cholesterol and tested whether exposure to CM from HDAdXMoAntimiR33a5-transduced EC increased cholesterol efflux to exogenous apoAI. Compared with CM that contains exosomes with scrambled antagomiRs, CM containing exosome-enclosed anti-miR-33a-5p increased apoAI-mediated cholesterol efflux by 40–60% (p < 0.001 for both cell types; Fig. 5E, F). These effects were absent when EV were removed from the CM by ultracentrifugation.
Discussion
We tested the concept that transducing EC with a vector that expresses an exosome-targeted therapeutic antagomiR could achieve delivery of the antagomiR into neighboring vascular cells, thereby enhancing cholesterol efflux. Our major findings are: (1) Primary cultures of EC express miR-33a-5p and release it into exosomes. (2) Anti-miR-33a-5p, expressed in EC, enters exosomes that can mediate transfer of anti-miR-33a-5p to other cells. (3) Addition of the X-motif to the anti-miR-33a-5p transcript increases levels of anti-miR-33a-5p within exosomes and enhances anti-miR-33a-5p delivery to target cells. (4) Exposure of macrophages and SMC to CM from EC expressing anti-miR-33a-5p transfers anti-miR-33a-5p to these cells, reduces miR-33a-5p, and increases ABCA1 protein as well as apoAI-mediated cholesterol efflux (Supplementary Fig. S6).
MiR-33a-5p is highly expressed in macrophages and hepatocytes, in which it plays a major role in regulation of cholesterol efflux and HDL biogenesis as well as fatty acid synthesis and oxidation. 22,47 In macrophages, miR-33a-5p also plays an important role in regulating metabolism, immune cell polarization, and autophagy. 48,49 Roles for miR-33a-5p in SMC are less well understood. We found only one study in which miR-33 expression is directly measured in SMC. 45 Another study infers SMC expression of miR-33 based on indirect evidence. 50 Neither study documents a role for SMC miR-33 in regulating cholesterol efflux.
Roles for miR-33a-5p in EC have never been described. MiR-33 is expressed in immortalized lines of cultured EC, 22,51 but it is essentially undetectable in cultured primary human umbilical vein EC. 22 We are aware of no other reports that EC express miR-33 either in vitro or in vivo, and a recent review of EC miRNA and atherosclerosis does not mention miR-33. 52 Our observation that BAEC express miR-33a-5p and release it in exosomes raises the possibility that EC miR-33 could contribute to atherogenesis both by promoting lipid accumulation in EC and—via exosome-mediated transfer—by augmenting endogenous miR-33a-5p in suppressing cholesterol efflux from neighboring intimal macrophages and SMC. The concept that EC miRNAs can be atherogenic and that antagomiRs directed at these miRNA could be therapeutic is well established in relation to other miRNA (e.g., miR-92a and miR-712). 52 –54 Our data extend this concept to miR-33a-5p.
The strategy developed here exploits EC transduction with anti-miR-33a-5p to achieve an ultimate goal of transferring this antagomiR to sub-endothelial intimal cells. Expression of anti-miR-33a-5p that is limited to EC could be an effective means of treating atherosclerosis, because lipid accumulation in EC contributes to atherogenesis. 55 However, lipid accumulation in intimal macrophages and SMC is also an important driver of atherosclerosis, 56 and numerous studies in transgenic mice show that manipulation of gene expression in intimal macrophages and SMC can have profound atheroprotective effects. 57 –62 Our strategy exploits gene transfer to EC, because EC are accessible to HDAd vectors and can be stably transduced in vivo. 13
Exosomes can cross complex biological barriers such as the intestinal lining and blood
Other strategies for delivering therapeutic proteins and RNA to the arterial intima include systemic infusions of proteins, 67 ex vivo produced exosomes (see below), 63 other types of viral vectors, 11,68 nanoparticles, 69 and chemically modified oligonucleotides. 70 Compared with these approaches, our strategy of stable EC expression of an exosome-targeted antagomiR has theoretical advantages of site specificity, efficiency, cost, durability, lack of immunogenicity, and low toxicity. These advantages will be tested in future in vivo studies.
We developed a strategy of engineering EC in situ to produce therapeutic exosomes based—in part—on a large body of work involving an injection of in vitro generated exosomes. This work includes injection of unmodified exosomes, of exosomes loaded in vitro with synthetic RNA (mRNA, siRNA, or shRNA), and of exosomes harvested from cultured cells that are engineered to express therapeutic RNA. 63 In all cases, these exosomes are injected in vivo with the expectation that they will deliver therapeutic RNA to specific target cells. There are many practical hurdles associated with this approach, including scalability, inefficiency, non-specificity, immunogenicity, a potential need for repeated re-administration, and off-target effects. 63,71 Nevertheless, there are reports of successful application of this approach in preclinical animal models of cardiovascular disease. For example, direct myocardial injection of EV produced by cultured hypoxic EC improved systolic function and decreased fibrosis after experimental myocardial infarction in rats. 72 Similar results in mice were reported by using a direct myocardial injection of in vitro mesenchymal stem cell-generated exosomes, including a demonstration that exosome-mediated therapeutic effects were mediated by miR-125b. 73 Others used an engineered-exosome approach to show that exosomes purified from EC engineered to overexpress KLF-2 and injected intravenously into atherosclerosis-prone mice reduced aortic lipid accumulation in an miR-143/145-dependent manner. 24 Compared with these precedents, our approach of in vivo gene transfer leading to local release of exosomes avoids the need for sterile large-scale in vitro exosome production and invasive exosome injection. Our approach would also provide a particularly high concentration of therapeutic exosomes in close proximity to the targeted cells.
We selected miR-33a-5p as a target, because suppression of miR-33a-5p increases ABCA1 expression in vascular wall cells, and increases cholesterol efflux, which should be atheroprotective. Moreover, miR-33 regulation of ABCA1 expression and cholesterol efflux is highly conserved, including in nonhuman primates, 22,47 enhancing translational potential. Importantly, as concerns our approach, increased ABCA1 levels are consistently atheroprotective in the endothelium, 74 –76 and increased ABCA1 expression in EC does not cause cellular toxicity. 21
Most studies that examined ABCA1 expression in macrophages of atherosclerosis-prone mice also find that increased macrophage ABCA1 expression is associated with less atherosclerosis 77 –79 (the single exception is a model of systemic myeloid-specific ABCA1 deletion, not an overexpression model). 80 The effects of altered ABCA1 expression in SMC on atherosclerosis are less well studied in vivo, although data in this study as well as others' in vitro work suggest that upregulation of SMC ABCA1 would be atheroprotective via enhanced cholesterol efflux. 81,82
Although ABCA1 is likely the most biologically important target of miR-33a, 83 numerous other genes are regulated either directly or indirectly by miR-33a. 43,84,85 Anti-miR-33a de-repression of some of these genes, including the cholesterol transporter ABCG1, IRS2 (a positive regulator of insulin signaling), and efferocytosis-promoting genes, 22,47,49 could also have atheroprotective effects. The effects of de-repressing other miR-33a targets—including genes associated with fatty acid metabolism 47,85 —in EC and neighboring vascular cells are unknown and will require further investigation.
It is likely that anti-miR-33a would have salutary effect in vascular cells; however, the consequences of miR-33a antagonism and ABCA1 upregulation in hepatocytes are less predictable. Atheroprotective effects of hepatic miR-33a inhibition were evident in early short-term murine studies, 22 but longer-term treatments did not reduce atherosclerosis 86 and were associated with hypertriglyceridemia and hepatic steatosis. 85 Increased hepatic ABCA1 expression is associated with both increased and reduced atherosclerosis, depending on the animal model. 87,88
Although our approach is designed to deliver antagomiRs directly to the vessel wall, it is likely that at least some of the antagomiR-containing exosomes released from transduced EC would enter the circulation. Because a significant number of intravenously injected exosomes traffic to the liver, 89 anti-miR-33a-5p-containing exosomes released from transduced EC might also enter hepatocytes. We expect that effects on the liver would be minor, due to a low number of exosomes released systemically from focally transduced EC, but it will be important in future in vivo studies to examine hepatocyte expression of ABCA1, hepatocyte miR-33a-5p and anti-miR-33a-5p levels, plasma lipoprotein levels, and liver histology. The potential for toxic effects of anti-miR-33a on peripheral blood and bone marrow will also require evaluation; however, an injection of high levels of synthetic anti-miR-33a into nonhuman primates was not associated with hematologic toxicity. 47
Incorporation of an X-motif into the anti-miR-33a-5p transcript increased EV-anti-miR-33a-5p transcripts by ∼80-fold and increased anti-miR-33a-5p transfer to target cells by ∼10-fold. In other studies, these values have ranged from ∼2-fold to >1,000-fold 32,46,90 and are likely transcript- and cell-type dependent. Increased loading of anti-miR-33a-5p into exosomes is advantageous for targeting neighboring cells but decreases the amount of anti-miR-33a-5p in EC, potentially limiting its atheroprotective activity.
Strategies for overcoming this limitation include co-expression of an anti-miR-33a-5p transcript that lacks an X-motif and co-expression of an ABCA1 transcript. We and others have shown that overexpression of ABCA1 in EC effectively increases ABCA1 protein and cholesterol efflux and is non-toxic. 21,75 The large cloning capacity of HDAd (>30 kB) 91 would enable inclusion of all three expression cassettes in a single HDAd. Future expression cassettes might achieve even higher levels of anti-miR-33a-5p by substitution of the U6 promoter for the H1 promoter used in this study. 92
In summary, our in vitro data support feasibility of a gene-therapy strategy that generates antagomiR-containing EC-derived exosomes that enter neighboring intimal cells, increase cholesterol efflux, and prevent or reverse atherosclerosis. Future studies will examine in vivo efficacy and probe for off-target effects both in vascular cells and in the liver. If successful, we speculate that this strategy might be applied to treat other diseases of the sub-endothelium and vascular media such as aneurysms and medial calcification, in which both miRNA and exosomes play significant roles. 70,93
Footnotes
Acknowledgment
The authors thank AdVec, Inc. for permission to use the HDAd reagents.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported in part by NIH grant R01HL114541, the John L. Locke Jr. Charitable Trust, a gift from Sam and Lynn Stevens, and the Cellular Imaging Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704). Dr. Stamatikos was supported by NIH grant T32HL007828 and by a postdoctoral fellowship from the American Heart Association 17POST33650120.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.