
Abstract
Aldehyde dehydrogenase type 2 (ALDH2), a key enzyme in ethanol metabolism, processes toxic acetaldehyde to nontoxic acetate. ALDH2 deficiency affects 8% of the world population and 35–45% of East Asians. The ALDH2*2 allele common genetic variant has a glutamic acid-to-lysine substitution at position 487 (E487K) that reduces the oxidizing ability of the enzyme resulting in systemic accumulation of acetaldehyde with ethanol ingestion. With chronic ethanol ingestion, mutations in ALDH2 are associated with a variety of hematological, neurological, and dermatological abnormalities, and an increased risk for esophageal cancer and osteoporosis. Based on our prior studies demonstrating that a one-time administration of an adeno-associated virus (AAV) serotype rh.10 gene transfer vector expressing the human ALDH2 cDNA (AAVrh.10hALDH2) prevents the acute effects of ethanol administration (the “Asian flush syndrome”), we hypothesized that AAVrh.10hALDH2 would also prevent the chronic disorders associated with ALDH2 deficiency and chronic ethanol ingestion. To assess this hypothesis, AAVrh.10hALDH2 (1011 genome copies) was administered intravenously to two models of ALDH2 deficiency, Aldh2 knockout homozygous (Aldh2 −/−) and knockin homozygous (Aldh2 E487K+/+) mice (n = 10 per group). Four weeks after vector administration, mice were given drinking water with 10–15% ethanol for 12 weeks. Strikingly, compared with nonethanol drinking littermates, AAVrh.10hALDH2 administration prevented chronic ethanol-induced serum acetaldehyde accumulation and elevated liver malondialdehyde levels, loss of body weight, reduced hemoglobin levels, reduced performance in locomotor activity tests, accumulation of esophageal DNA damage and DNA adducts, and development of osteopenia. AAVrh.10hALDH2 should be considered as a preventative therapy for the increased risk of chronic disorders associated with ALDH2 deficiency and chronic alcohol exposure.
Introduction
One of the most common hereditary disorders, aldehyde dehydrogenase type 2 (ALDH2) deficiency, affects 560 million people, ∼8% of the world population. 1,2 Individuals of East Asian descent have the highest prevalence (35–45%). 2,3 ALDH2 belongs to a superfamily of enzymes that metabolize endogenous and exogenous aldehydes. 4 The autosomal gene codes for an enzyme localized to mitochondria. ALDH2 is ubiquitously expressed at low levels in all tissues, but is most abundant in the liver, the primary organ of ethanol metabolism. 5,6 ALDH2 is the second enzyme in the ethanol metabolism pathway and converts acetaldehyde, a toxic intermediate, to acetate (Supplementary Fig. S1). 7 Significant accumulation of serum acetaldehyde after ethanol ingestion results from mutations in ALDH2 that reduce the oxidizing ability of the enzyme. 3,8,9 The ALDH2 enzyme is a tetramer and the mutant protein functions as a dominant negative. 10,11 Heterozygotes have <50% ALDH2 enzymatic activity and homozygotes <4%. 12,13
The most common mutation is a glutamic acid-to-lysine substitution at position 487 (E487K), commonly referred to as the ALDH2*2 allele. 3,14 With acute ethanol ingestion, mutations in ALDH2 are responsible for the “Asian flush syndrome,” with erythema, nausea, headaches, and general systemic discomfort. 3,13,15 With chronic ethanol ingestion, ALDH2 mutations are associated with a number of serious medical conditions, including hematological, dermatological, and neurological abnormalities, a markedly increased risk for cancers of the oral cavity, pharynx, larynx, and esophagus, 23,16,17 and osteoporosis. 18 –24
There are two mouse models of ALDH2 deficiency that mimic many of the molecular and clinical phenotypes of humans carrying the ALDH2*2 allele. 12,25 In these mouse models, chronic administration of ethanol or acetaldehyde induces the chronic disorders associated with ALDH2 deficiency. 9,26,27 The Aldh2 knockout mouse (Aldh2 −/−) does not express detectable ALDH2 protein or enzymatic activity. 12 After ethanol administration, Aldh2 −/− mice have significantly higher levels of serum acetaldehyde than wild-type mice, and chronic exposure to ethanol results in weight loss and increased mortality. 28,29 The Aldh2 E487K knockin mouse (Aldh2 E487K+/+) has the lysine 487 mutation from the human ALDH2*2 allele. 25 These mice have reduced ALDH2 enzymatic activity and, when challenged with ethanol, have high serum acetaldehyde levels and exhibit increased ethanol-related behavioral abnormalities.
We have previously demonstrated that the acute effects of ethanol in these murine models of ALDH2 deficiency (the mouse equivalent of the “Asian flush syndrome”) can be corrected by systemic liver-directed serotype rh.10 adeno-associated virus (AAVrh.10hALDH2)-mediated delivery of the normal human ALDH2 coding sequence. 30 In this study, we have extended these observations in these two murine models of ALDH2 deficiency to assess whether this therapy will prevent the diseases associated with chronic ethanol ingestion. Strikingly, the therapy prevents chronic alcohol-mediated body weight loss, anemia, poor locomotion, skin pigmentation, elevated serum acetaldehyde, and liver malondialdehyde (MDA) levels. Importantly, AAVrh.10hALDH2 gene therapy also prevented esophageal DNA damage and adduct accumulation, precursors to the development of esophageal cancer, and the development of osteopenia, the murine equivalent of human osteoporosis.
Methods
Mouse models of ALDH2 deficiency
The ALDH2 knockout homozygous (Aldh2 −/−) mice, backcrossed with C57Bl/6 mice, were obtained from the Department of Environmental Health, University of Occupational and Environmental Health (Kitakyushu, Japan). 12 The Aldh2 E487K knockin homozygous mice (Aldh2 E487K+/+), a knockin mouse model of ALDH2 deficiency expressing mouse ALDH2 with the inactivating point mutation from the human ALDH2*2 gene, were obtained from the Department of Chemical and Systems Biology, Stanford University School of Medicine (Stanford, CA). 25 All mice were housed in microisolator cages and all food and water were autoclaved. Mice were bred as pairs (one female with one male) or trios (two females with one male). To generate homozygous mice, mice were bred as heterozygous pairs with genotyping of pups at 3 weeks of age by PCR (Transnetyx, Cordova, TN). The primers for Aldh2 −/− genotyping were forward–5′-GGACGTAGACAAGGCAGTGAAG, reverse–5′-CCCTACCCGGTAGAATTCGATATCA and those for Aldh2 E487K+/+ genotyping were forward–5′-GGAGCTGGGCGAGTATGG, reverse–5′-GAGTCTGAAGGCTGTGTACGTA 12,25 ). All experiments conformed to the relevant regulatory standards and were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Because alcohol consumption in the East Asian population is dominated by males 31 and because female mice respond to adeno-associated virus (AAV) vector with expression as much as 1 log less than males, 32 we utilized male mice for all studies.
AAV vectors
The AAVrh.10hALDH2 vector comprises the nonhuman primate-derived rh.10 capsid and an expression cassette including the 5′ and 3′ AAV2 inverted terminal repeats, the cytomegalovirus enhancer, chicken–β-actin promoter and intron, and rabbit β globin splice acceptor (CAG promoter), the human ALDH2 coding sequence with a hemagglutinin (HA) tag, and rabbit β-globin polyadenylation signal (Supplementary Fig. S2).
30
The HA tag was used for differentiating between human and mouse ALDH2 proteins because the mouse and human amino acid sequences are 96% homologous (NCBI Homologene:
The vector was produced using human embryonic kidney 293T cells as described previously. 32 In brief, the pAAV-CAG-hALDH2 expression plasmid (600 μg) and the AAVrh.10 packaging-Ad helper hybrid plasmid pPAKMArh.10 (1,200 μg) were cotransfected into 293T cells using polyethylenimine (PEI) transfection reagent (Polysciences, Warrington, PA). At 72 h post-transfection, cells were harvested and lysate was prepared using five cycles of freeze–thaw. The cell lysate containing the virus was clarified by centrifugation at 2675 × g for 15 min. The AAVrh.10hALDH2 vector was purified from the crude viral lysate by iodixanol gradient and HiTrap Q High Performance anion exchange chromatography (GE Healthcare, Piscataway, NJ), concentrated using a BioMax 100K membrane concentrator (Millipore, Billerica, MA) and stored in phosphate-buffered saline, pH 7.4 at −80°C.
Vector genome titers were determined by TaqMan qPCR using a CAG-specific primer–probe set (forward primer: 5′-GTCAATGGGTGGAGTATTTACGG, reverse primer: 5′-AGGTCATGTACTGGGCATAATGC) (Applied Biosystems, Foster City, CA). The purified AAVrh. 10hALDH2 vector was digested with proteinase K in the presence of 0.5% sodium dodecyl sulfate (SDS) plus 25 mM ethylenediaminetetraacetate (EDTA) at 70°C for 1 h followed by inactivation of the protease at 95°C for 15 min. The vector was then used as a template for TaqMan analysis using a pAAV-CAG-hALDH2 plasmid DNA standard of known copy number to generate a standard curve. The AAVrh.10control vector expresses an irrelevant transgene. 33
Overview of efficacy studies with AAVrh.10hALDH2
Male Aldh2 −/− and Aldh2 E487K+/+ mice (n = 10), age 6 to 10 weeks, were administered intravenously in 100 μL a one-time dose of AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were administered phosphate-buffered saline (PBS). Four weeks later, mice were given water or ethanol in water for 12 weeks (10% ethanol in water for the first 6 weeks and 15% ethanol in water for the second 6 weeks) ad libitum in the drinking water. Water or ethanol intake was assessed every week. Mice were evaluated for a variety of chronic disease phenotypes, including body weight, hemoglobin level, muscular coordination and strength (rotarod test), and skin pigmentation every 3 weeks. After 12 weeks ethanol ingestion, mice were sacrificed. Liver hALDH2 mRNA was evaluated by Taqman RT-qPCR and hALDH2 protein by Western analysis and immunohistochemistry. ALDH2 enzymatic activity and MDA levels in liver were quantified using commercially available assay kits. Serum acetaldehyde levels were measured by liquid chromatography/mass spectrometry (LC/MS) after derivatization. DNA damage on esophageal epithelium was evaluated by γH2AX immunohistochemistry. Esophagus DNA adducts were assessed by measuring N2-ethyl-2′-deoxyguanosine (N2-et-dG) by LC/MS. Osteopenia was assessed by microcomputed tomography (μCT) and histology.
Assessment of water or ethanol intake
To quantify water or ethanol intake, a test was performed with mice (n = 14) subgrouped into two sets of one, two, and four mice separated into different cages. Bottles (Techniplast, West Chester, PA) were filled with 500 mL water or ethanol in water (10% ethanol in water for the first 6 weeks and 15% ethanol in water for the second 6 weeks) and weighed before beginning the challenge. Each week, bottles were weighed, refilled to 500 mL, and placed back in the cage. Two control cages with the same type of bottle containing 500 mL water or ethanol were also set up as controls for evaporation and loss of liquid during cage movement. Water or ethanol intake for each cage of mice was calculated by the weight of the bottle in the cage minus weight of the water bottle from the control cage.
ALDH2 expression in liver
Human ALDH2 mRNA levels in the liver were assessed by TaqMan RT-qPCR using FAM dye labeled hALDH2-specific primer–probe sets (Assay ID; Hs01007998_ml) and murine 18S-specific primer–probe sets (Life Technologies, Waltham, MA). ALDH2 protein was quantified by Western analysis with an antibody against the HA tag (Sigma-Aldrich, St. Louis, MO). Paraformaldehyde-fixed livers were embedded in paraffin and cut into 5 μm sections. Vector-derived hALDH2 expression in the liver was detected by an anti-HA antibody (Histowiz, Brooklyn, NY). After sectioning, anti-HA immunohistochemical staining, and counterstaining with hematoxylin, digital images of liver cross-sections were acquired using a 20 × objective. The enzymatic activity of ALDH2 in liver was analyzed using the colorimetric mitochondrial aldehyde dehydrogenase (ALDH2) activity assay kit (ab115348; Abcam, Cambridge, MA) according to the manufacturer's protocol using 200 μg of total protein from liver homogenate. C57Bl/6 mice administered PBS were used as controls.
Acetaldehyde
Acetaldehyde measurements were performed at the Proteomics Resource Center, Rockefeller University 9,34 (n = 5 mice/group). Acetaldehyde levels were determined through derivatization with dinitrophenylhydrazine (DNPH) using butyraldehyde–DNPH (Accustandard, New Haven, CT) as an internal standard. Blood (120 μL) was deproteinized by addition of an equal volume of cold acetonitrile (ACN) and centrifuged for 30 min at 3,500 g at 4°C. Supernatant (48 μL) was mixed with 2 μL of 10 mM 13 C-acetaldehyde. Then, 15 μL of 16 mM DNPH in ACN and 5 μL of 1 M citric acid (pH 4.0) were added. After incubation at 25°C for 30 min, the reaction was quenched with 60 μL of 0.1% formic acid in water, and samples were kept in −80°C until analysis. Before LC/MS, 49 μL of sample was spiked with 1 μL of butyraldehyde–DNPH standard for a final concentration of 20 pm/μL. Acetaldehyde–DNPH (Accustandard, New Haven, CT) was used as an external calibrant to verify retention time. Samples were analyzed on a Dionex U-3000 HPLC system coupled to a TSQ Vantage triple-quad mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with a heated electrospray ionization source (HESI). Chromatographic separation was performed using a Thermo Acclaim 120 C8 (2.1 × 150 mm, 3 μm particle size) column at a flow rate of 200 μL/min, using 0.1% LC-grade formic acid (Pierce, Thermo Fisher Scientific) in water as buffer A and 100% methanol (Optima, Fisher Scientific, Hampton, NH) as buffer B. The gradient was the following: 5% buffer B (0–2 min), increased to 60% buffer B (2–10 min), increased to 80% buffer B (10–20 min), increased to 100% buffer B (20–21 min), held at 100% buffer B (21–26 min), returned to 50% buffer B (26–26.1 min) and held at 50% buffer B (26.1–30 min) for re-equilibration of the column. The mass spectrometer was operated with the following parameters; negative ion polarity; spray voltage, 3500 V; ion transfer capillary temperature, 350°C; source temperature, 37°C; sheath gas, 35 (arbitrary units); auxiliary gas, 10 (arbitrary units); collision gas, argon. The S-Lens voltage (S-lens) and collision energy (CE) were manually optimized for each multiple reaction monitoring (MRM) transition. The MRM transitions monitored were: Acetaldehyde-DNPH, m/z 223→122 (CE = 21, S-Lens = 65), 223→163 (CE = 13, S-Lens = 65), 223→181 (CE = 18, S-Lens = 65); 13 C-Acetaldehyde-DNPH, m/z 225→122 (CE = 21, S-Lens = 65), 225→163 (CE = 13, S-Lens = 65), 225→181 (CE = 18, S-Lens = 65); Butyraldehyde-DNPH, m/z 251→122 (CE = 27, S-Lens = 70), 251→152 (CE = 19, S-Lens = 70), 251→163 (CE = 13, S-Lens = 70). Quantitation was performed using a known amount of 13 C-acetaldehyde standard in the sample and an external calibration curve of acetaldehyde–DNPH prepared in parallel. Rate constants of acetaldehyde consumption were determined by fitting time–concentration values to a first-order decay model. Butyaldehyde–DNPH was used to evaluate instrument performance.
Malondialdehyde
MDA in liver homogenate was assayed by TBARS Assay Kit (Cayman, MI) according to a modified manufacturer's protocol. In brief, the mixture of 25 μL liver homogenate, 25 μL SDS, and 1 mL color reagent was boiled at 99°C for 1 h. After cooling down on ice for 10 min, the sample was centrifuged at 1,600 g for 10 min and the absorbance was read at 540 nm by a spectrophotometer. MDA concentration was normalized by the amount of total protein determined by Pierce™ BCA Protein Assay Kit (Thermo Scientific).
Assessment of body weight, hemoglobin, locomotion, and skin pigmentation
To assess the effects of chronic ethanol challenge on total body mass, mice were weighed at pre-ethanol, 3, 6, 9, and 12 weeks time points.
Hemoglobin levels were determined in blood collected in EDTA tubes from the tail vein at pre-ethanol, 3, 6, 9, and 12 weeks time points and were automatically counted using the ADVIA 120 Hematology System (Siemens Healthineers, Erlangen, Germany).
The rotarod behavior test was used to evaluate mouse strength and coordination every 3 weeks during ethanol challenge. An automated four-lane rotarod unit (AccuScan Instruments, Columbus, OH) was used to evaluate locomotor activity. The apparatus has 7 cm diameter drums with grooves to improve grip. Drums were rotated at a fixed speed of 2 rotations/min (rpm) for the first 20 s, accelerated up to 30 rpm in the next 100 s, and then up to 60 rpm in the last 60 s. The time and rpm when mouse fell from or failed to walk on a drum were recorded. Failure to walk was defined as a mouse that did not fall from the drum, but clung in one position and went around twice. Tests were performed twice at each time point and the average rpm was calculated.
To assess skin pigmentation from chronic ethanol ingestion, pictures were taken of exposed ear, genitals, tail, and sole skin of each mouse. For analysis, darkness of the paw sole was quantified with ImageJ. All sole areas were traced and intensity of every dot in the traced area was measured as mean red–green–blue (RGB) intensity. Positive pigmentation was quantified as an intensity with mean RGB intensity of 90 or less after normalization to the mean RGB intensity of white background.
DNA damage and adducts
DNA damage was assessed by immunohistochemistry for γH2AX. γH2AX was stained with an antibody to phosphohistone H2A.X (Ser 139) (clone: 20E3, 1:100, Cell Signaling Technology, MA), and the number of γH2AX-positive cells and total epithelial cells on the basal membrane of the esophagus were counted using a 40 × objective. Measurement of the DNA adduct N2-et-dG in esophagus DNA was performed at the Proteomics Resource Center, Rockefeller University for n = 5 mice per group. DNA was extracted from a whole mouse esophagus using the QIAGEN (Germantown, MD) DNAeasy Blood & Tissue Kit, according to the manufacturer's protocol except with the addition of sodium cyanoborohydride (NaBH3CN) to each reagent of the kit (150 mM to cell lysis buffer and 100 mM to other reagents). Extracted DNA was dissolved in 60 μL of 10 mM Tris–HCl/5 mM EDTA buffer (pH 7.5), and the amount of DNA was measured by Nanodrop. Extracted DNA was mixed with 1.2 μL of 1 M citrate buffer (pH 6.0), 0.8 μL of 750 mM CaCl2, 45 U micrococcal nuclease (Worthington Biochemical Corporation, Lakewood, NJ), 0.15 U spleen phosphodiesterase (Worthington Biochemical Corporation), 4.8 μL of 8 M NaBH3CN, and 1 μL of 5 pm/μL internal standard. The mixture was incubated at 37°C for 3 h, and then 2.5 μL of 2 M Tris-HCl (pH 8.5), 2 μL of 45 mM ZnSO4, 6 U alkaline phosphatase (Sigma-Aldrich), 1 μL of 8 M NaBH3CN, and 1.5 μL distilled water were added. After incubation at 37°C for 3 h, nucleosides were extracted twice with 600 μL of chilled methanol and the combined supernatant was evaporated to dryness. The dried samples were resuspended in 100 μL LC/MS grade water (Optima™; Fisher Scientific), sonicated for 10 s, centrifuged at 4°C for 10 min, and supernatant was subjected to liquid chromatography tandem mass spectrometry (LC-MS/MS) analyses.
LC-MS/MS analyses were performed using a Vantage TSQ triple-stage quadrupole mass spectrometer (Thermo Fisher Scientific) equipped with a heated electrospray ionization source. The mass spectrometer was operated with the following parameters: positive ion polarity; spray voltage, 3,500 V; ion transfer capillary temperature, 300°C; source temperature, 250°C; sheath gas, 30 (arbitrary units); auxiliary gas, 10 (arbitrary units); collision gas, argon; and dwell time, 200 ms. The S-Lens voltage (S-lens) and collision energy (CE) were manually optimized for each MRM transition. The MRM transitions monitored were deoxyguanosine (dG), m/z 268 → 152 (CE = 12, S-lens = 50); [ 15 N5]-N2-et-dG, m/z 301 → 185 (CE = 14, S-lens = 57); and N2-et-dG, m/z 296 → 180 (CE = 12, S-lens = 60). Chromatographic separation was performed on a Dionex Ultimate 3000 HPLC equipped with a Thermo Hypersil Gold aQ column (2.1 mm × 150 mm × 3 μm particle size). Column temperature was maintained at 36°C and the autosampler was set to 4°C. Mobile phase A consisted of 0.1% LC-grade formic acid (Pierce; Thermo Scientific) in water and mobile phase B consisted of 0.1% LC-grade formic acid in ACN. Separation was achieved using the following gradient (flow rate set at 0.4 mL/min): 0% B (0–6 min), 1% B (6–7.65 min), 6% B (7.65–9.35 min), held at 6% B (9.35–10 min), increased to 50% B (10–12 min), 75% B (12–14 min), held at 75% B (14–17 min), returning to 0% B (17–17.5 min), and re-equilibrating (17.5–30 min). The injection volume was 1–3 μL with ≥2 technical replicates per biological replicate. The amount of N2-et-dG in each DNA sample was determined from the ratio of the peak area of N2-et-dG relative to the internal standard [ 15 N5] N2-et-dG and by using a calibration curve (0.6–2,500 fmol/μL, r 2 ≥ 0.98). Owing to the high concentrations of dG in each DNA sample, the original resuspension stock was further diluted 100-fold followed by injection of 0.5–1 μL of this mixture onto the column. The amount of dG was then determined using a calibration curve (20–1,250 fmol/μL, r 2 ≥ 0.97).
μCT and histological analysis of femurs
Femurs fixed in 4% paraformaldehyde then stored in 70% ethanol were scanned using a high-resolution Scanco μCT 35 (Scanco Medical AG, Bruttisellen, Switzerland). Specimens were scanned with an isotropic voxel size of 7 μm. For analysis of femoral bone mass, a region of trabecular bone 2.1 mm wide was contoured, starting 280 μm from the proximal end of the distal femoral growth plate. Femoral trabecular bone was thresholded at 211 permille and femoral cortical bone was thresholded at 350 permille. A Gaussian noise filter optimized for murine bone was applied to reduce noise in the thresholded two-dimensional (2D) image. Three-dimensional reconstructions were created by stacking the thresholded 2D images from the contoured regions.
For histological analysis, femurs were decalcified with 0.5 M EDTA (pH 8.0) and embedded in paraffin. Sections were deparaffinized and stained with hematoxylin and eosin. Metaphysis was defined as the area within 2 mm from the bottom of growth plate. Areas of metaphysis for each trabecular bone were traced and quantified by ImageJ. The ratio of trabecular bone in metaphysis was calculated by dividing the sum area of each trabecular bone in metaphysis by total area of metaphysis.
Statistical analysis
All data are presented as means ± standard error of the mean (SEM) unless otherwise stated; the “n” value for each group is stated in the figure or figure legend. Differences between groups were analyzed using an unpaired two-tailed Student's t-test. The behavior score and body temperature after ethanol exposure were also correlated with acetaldehyde levels. These correlations were evaluated using the correlation test for paired samples. r 2 values >0.8 indicate a strong relationship between the test groups. p-Values <0.05 were considered significant for all comparisons.
Results
AAVrh.10hALDH2-mediated expression of hALDH2
Aldh2 −/− and Aldh2 E487K+/+ mice were treated with AAVrh.10hALDH2 (Supplementary Fig. S2) or AAVrh.10control (1011 gc) by intravenous administration. Sixteen weeks later, hALDH2 mRNA, protein levels, and ALDH2 enzymatic activity in liver were analyzed. AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice had significantly higher hALDH2 mRNA expression in liver than AAVrh.10control-treated mice (Aldh2 −/−, p < 10−4; Aldh2 E487K+/+, p < 10−5; Fig. 1A). Western analysis revealed liver hALDH2 protein expression in AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice (Aldh2 −/−, p < 10−3; Aldh2 E487K+/+, p < 10−2; Fig. 1B). Liver immunohistochemical staining of AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice was consistent with the Western analysis. Liver hALDH2 positive cells were found mainly around hepatic and portal veins in AAVrh.10hALDH2-treated mice, whereas no positive cells were observed in AAVrh.10control-treated mice (Fig. 1C). In addition, AAVrh.10control-treated mice had undetectable levels of ALDH2 liver enzyme activity, whereas AAVrh.10hALDH2-treated mice showed high levels of enzymatic activity (Aldh2 −/−, p < 10−4; Aldh2 E487K+/+, p < 10−5) comparable with the wild-type C57Bl/6 mice (Aldh2 −/−, p > 0.3; Aldh2 E487K+/+, p > 0.4; Fig. 1D).

In vivo liver expression of human ALDH2 12 weeks after a single intravenous administration of AAVrh.10hALDH2 or AAVrh.10control (1011 gc) to Aldh2
−/− and Aldh2
E487K+/+ mice. C57Bl/6 mice were intravenously administered PBS.
Water versus ethanol intake
Four weeks after vector administration, mice were supplied water or ethanol in their daily drinking bottles for 12 weeks (10% ethanol in water for the first 6 weeks and 15% ethanol in water for the second 6 weeks) ad libitum. Water or ethanol intake was measured every week. In all genotypes, water or ethanol intake increased proportionally to the number of mice in the cage (all r 2 > 0.96, p < 10−9, Supplementary Fig. S3A). Ethanol intake was found to be significantly less than water intake for wild-type C57Bl/6, Aldh2 −/− and Aldh2 E487K+/+ mice (C57Bl/6, p < 10−4; Aldh2 −/−, p < 10−8; Aldh2 E487K+/+, p < 10−7; Supplementary Fig. S3A–C). In contrast, AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice drank significantly more ethanol than AAVrh.10control-treated mice (Aldh2 −/−, p < 10−5; Aldh2 E487K+/+, p < 10−4; Supplementary Fig. S3B, C).
Systemic acetaldehyde and MDA levels after chronic ethanol exposure
To assess whether augmentation of ALDH2 enzyme levels in the liver by gene therapy could prevent the effects of chronic ethanol ingestion, Aldh2 −/− and Aldh2 E487K+/+ mice were administered AAVrh.10hALDH2 or AAVrh.10control intravenously 4 weeks before the addition of ethanol in the daily water supply. Serum acetaldehyde levels of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice given ethanol were significantly higher than those of mice given water for 12 weeks (Aldh2 −/−, p < 10−2; Aldh2 E487K+/+, p < 0.02; Fig. 2A). However, serum acetaldehyde levels in AAVrh.10hALDH2 vector-treated Aldh2 −/− and Aldh2 E487K+/+ mice given ethanol for 12 weeks were reduced to levels similar to the Aldh2 −/− or Aldh2 E487K+/+ mice given water and the wild-type C57Bl/6 mice given ethanol (all p > 0.7, Fig. 2A). In addition to metabolizing acetaldehyde, ALDH2 also processes reactive aldehydes derived from oxidative stress such as MDA, which is implicated in DNA adduct formation and mutagenesis. 35,36 The level of MDA in the livers of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice given ethanol for 12 weeks was significantly higher than that of mice given water (Aldh2 −/−, p < 10−3; Aldh2 E487K+/+, p < 10−2; Fig. 2B). In contrast, the MDA level in AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice was not significantly different from that of the ALDH2-deficient mice given water or the wild-type C57Bl/6 mice (all p > 0.1, Fig. 2B).

Effect of AAVrh.10hALDH2 therapy on serum acetaldehyde and liver MDA levels with chronic ethanol exposure. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks.
Body weight, hemoglobin, locomotion, and dermatological abnormalities
Prior studies have demonstrated that with chronic ethanol ingestion, Aldh2 −/− and Aldh2 E487K+/+ mice have decreased body weight and blood cell counts. 9,37 To assess whether AAVrh.10hALDH2 therapy could prevent these effects of chronic ethanol consumption, mice were evaluated every 3 weeks for body weight, hemoglobin levels, and rotarod locomotion. Body weight and blood hemoglobin levels of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice decreased significantly for 12 weeks (Fig. 3A and Supplementary Fig. S4A, body weight, p < 10−3; Fig. 3B and Supplementary Fig. S4B, hemoglobin, p < 10−5). In parallel, these mice performed poorly on the rotarod test of locomotion (p < 10−2, Fig. 3C and Supplementary Fig. S4C). However, the body weight, hemoglobin levels, and rotarod performance for ALDH2-deficient mice treated with AAVrh.10hALDH2 were similar to those of the wild-type C57Bl/6 mice given ethanol for 12 weeks (Fig. 3 and Supplementary Fig. S4).
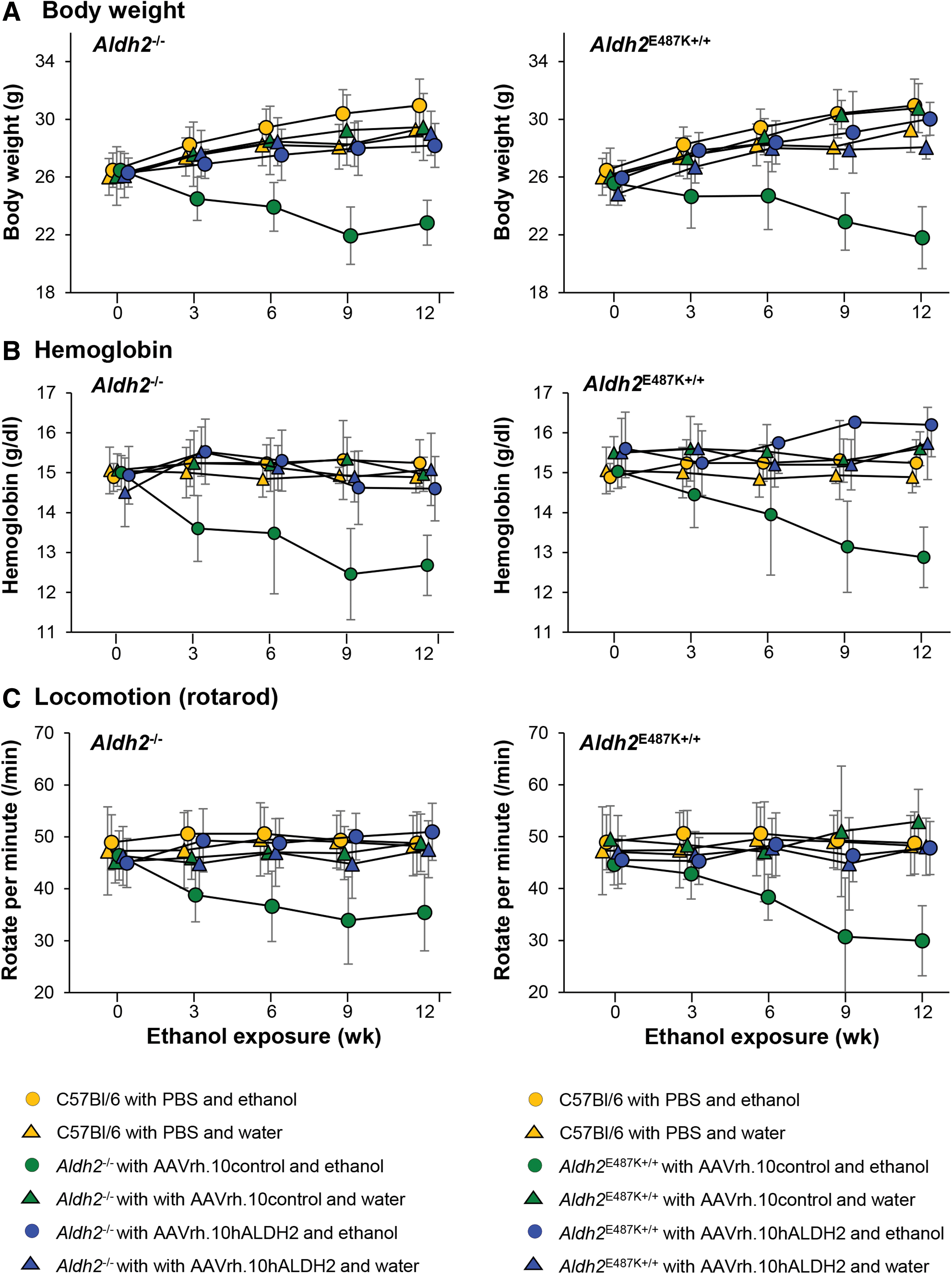
Effect of AAVrh.10hALDH2 therapy on body weight, hemoglobin levels, and locomotion (rotarod) during chronic ethanol exposure. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks. Tests were performed pre-exposure and 3, 6, 9, and 12 weeks during ethanol exposure.
ALDH2*2 individuals as well as ALDH2-deficient mice develop skin hyperpigmentation with chronic ethanol consumption. 37,38 AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice had increased pigmentation on the skin of the ears, genitals, tail, and soles with chronic ethanol ingestion, compared with littermates given water (Fig. 4A). Quantification of sole pigmentation showed that AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol ingestion had significantly higher levels of sole pigmentation than the ALDH2-deficient mice with water or the wild-type C57Bl/6 mice (all p < 10−7, Fig. 4B and Supplementary 5A, B). In comparison, sole pigmentation of AAVrh.10hALDH2-treated mice with ethanol exposure was significantly less than that of AAVrh.10control-treated mice (Aldh2 −/−, p < 10−2; Aldh2 E487K+/+, p < 10−4; Fig. 4B).

Effect of AAVrh.10hALDH2 therapy on skin hyperpigmentation after 12 weeks chronic ethanol exposure. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks.
Esophageal damage after chronic ethanol challenge
Ethanol and acetaldehyde treatment of ALDH2-deficient mouse models leads to accumulation of molecular and genetic changes including DNA damage and adducts that are the precursors of the genetic mutations leading to esophageal cancer development in humans. 27,39 Acetaldehyde predominantly induces double-strand breaks, and phosphorylated H2A histone family member X (γH2AX) is a well-established marker for detecting these lesions. 27,40 Immunohistochemistry using a specific γH2AX antibody was performed on longitudinal esophageal sections of mice chronically treated with ethanol or water, and the number of positive cells in the epithelial layer was counted. AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice with ethanol exposure had significantly more γH2AX-positive cells in the esophageal epithelium than mice given water (Aldh2 −/−, p < 10−5; Aldh2 E487K+/+, p < 10−4; Fig. 5A, B) and the wild-type C57Bl/6 mice (Aldh2 −/−, p < 10−4; Aldh2 E487K+/+, p < 10−4; Fig. 5A, B). In contrast, the number of γH2AX-positive cells in the esophageal epithelium of AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice with ethanol exposure was significantly reduced compared with AAVrh.10control-treated mice (Aldh2 −/−, p < 0.02; Aldh2 E487K+/+, p < 10−3; Fig. 5B). However, the number of γH2AX-positive cells in AAVrh.10hALDH2-treated mice with ethanol was still higher than that in the Aldh2 −/− and Aldh2 E487K+/+ mice given water or the wild-type C57Bl/6 mice (both p < 10−2, Fig. 5B). The extent of DNA damage was correlated with ALDH2 enzymatic activity (Supplementary Fig. S6).

Effect of AAVrh.10hALDH2 therapy on γH2AX-positive cells in the esophageal epithelium after 12 weeks chronic ethanol exposure. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks.
Acetaldehyde also causes mutations leading to esophageal cancer by forming DNA adducts. N2-et-dG is the primary acetaldehyde-derived DNA adduct found in humans and is considered the most sensitive marker for acetaldehyde exposure-related DNA damage in mice. 26 DNA was extracted from the whole esophagus of mice chronically exposed to ethanol or water, and N2-et-dG and dG nucleosides were quantified by LC/MS. AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice had significantly elevated levels of N2-et-dG after 12 weeks of chronic ethanol exposure compared with mice that received water and C57Bl/6 wild-type mice (all p < 10−3, Fig. 6). AAVrh.10hALDH2 treatment significantly reduced the number of N2-et-dG adducts in the esophageal DNA compared with AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice (both p < 10−2, Fig. 6).
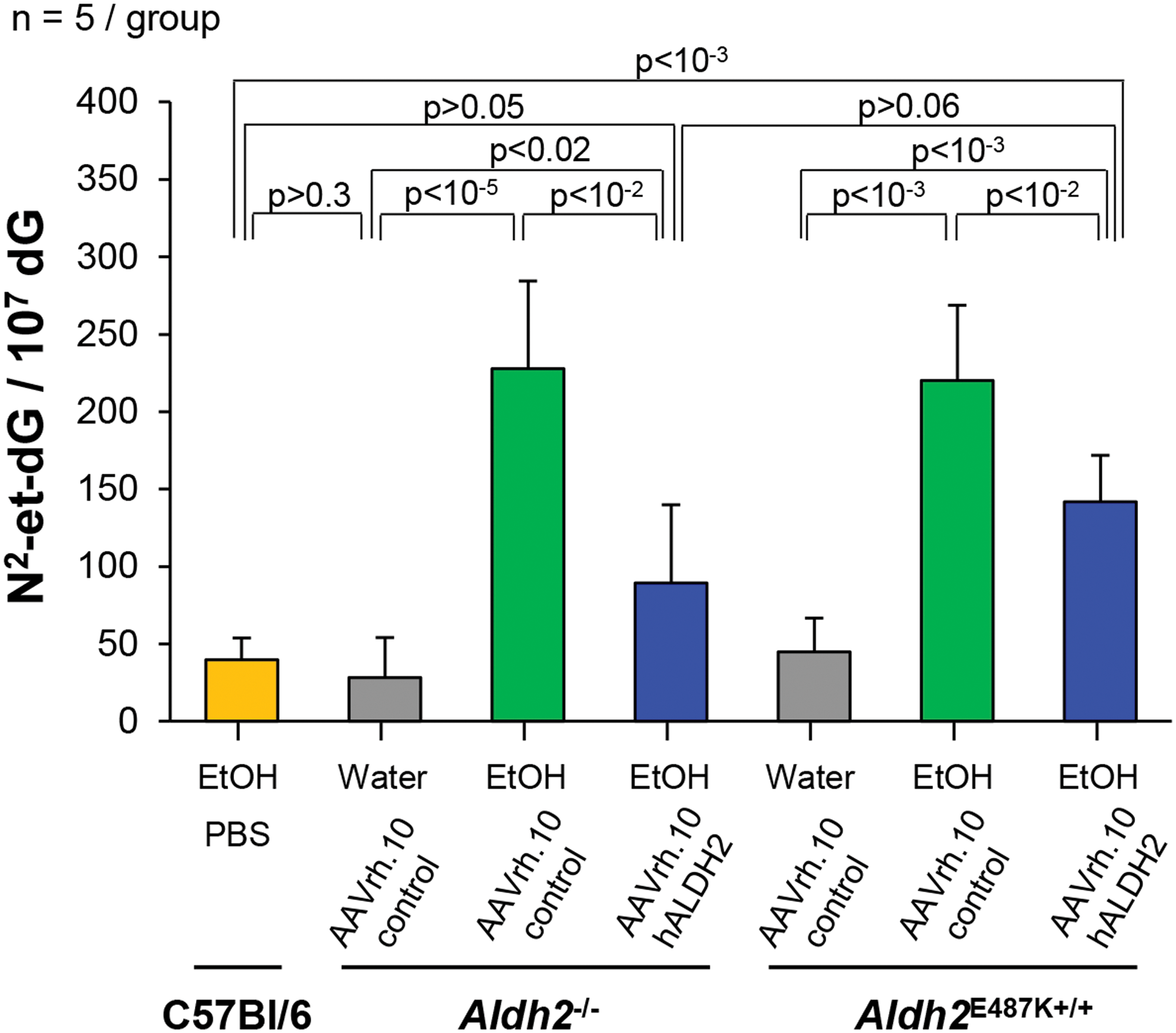
Effect of AAVrh.10hALDH2 therapy on DNA adducts in the esophagus after chronic ethanol exposure. Aldh2 −/− and Aldh2 E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks. DNA was extracted from whole esophagus and N2-et-dG adducts were quantified by liquid chromatography tandem mass spectrometry. Values are presented as means ± SEM. N2-et-dG, N2-ethyl-2′-deoxyguanosine. Color images are available online.
Skeletal phenotypes of AAVrh.10hALDH2-treated ALDH2-deficient mice
Chronic ethanol ingestion by Aldh2 knockout mice results in decreased bone mass compared with wild-type mice, 41 and transgenic mice overexpressing the ALDH2*2 allele have reduced bone mass compared with wild-type controls. 20 After 12 weeks of ad libitum chronic ethanol consumption, femurs from all mice were analyzed by μCT scans of the trabecular bone and midshaft cortex. 42,43 C57Bl/6 mice given water or ethanol had similar trabecular bone mass (Fig. 7A, B). Aldh2 −/− and Aldh2 E487K+/+ mice administered AAVrh.10control or AAVrh.10hALDH2 and given water had trabecular bone mass similar to C57Bl/6 mice (Fig. 7C, E, G, I). In striking contrast, Aldh2 −/− and Aldh2 E487K+/+ mice administered AAVrh.10control with chronic ethanol ingestion had severe loss of trabecular bone mass compared with mice given water or C57Bl/6 wild-type mice given either water or ethanol (Fig. 1D, H). Importantly, the Aldh2 −/− and Aldh2 E487K+/+ mice treated with AAVrh.10hALDH2 gene therapy with ethanol consumption had significantly increased trabecular bone mass compared with AAVrh.10control-treated mice with no visual difference from wild-type mice (Fig. 7F, J).

Three-dimensional μCT reconstructions of femur trabecular bone for AAVrh.10hALDH2 therapy after chronic ethanol ingestion. Aldh2
−/−and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks. Fixed femurs were scanned by μCT. Shown are representative three-dimensional reconstruction of trabecular bone in metaphysis from one representative animal of each group.
Data from several parameters gathered from the μCT were assessed including bone volume density (bone volume/total volume) of trabecular bone, thickness of bone cortex at the midshaft, trabecular number, trabecular thickness, and trabecular space (Fig. 8). The bone volume density of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol ingestion was significantly lower than counterparts given water and C57Bl/6 wild-type mice with chronic ethanol ingestion (all p < 10−2, Fig. 8A). These decreases were corrected with AAVrh.10hALDH2 gene therapy in both Aldh2 −/− and Aldh2 E487K+/+ mice (both p < 10−2, Fig. 8A). Importantly, the bone volume density of AAVrh.10hALDH2-treated Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol ingestion was equivalent to that of C57Bl/6 wild-type mice with ethanol (both p > 0.4, Fig. 8A). The cortical thickness of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol ingestion was significantly lower than the mice given water (both p < 10−2, Fig. 8B) and C57Bl/6 wild-type mice with chronic ethanol ingestion (Aldh2 E487K+/+, p < 0.04; Aldh2 −/−, p > 0.1; Fig. 8B). The cortical thickness was increased by AAVrh.10hALDH2 treatment equivalent to that of C57Bl/6 mice in both Aldh2 −/− and Aldh2 E487K+/+ mice (Aldh2 −/−, p > 0.3; Aldh2 E487K+/+, p > 0.5; Fig. 8B). The trabecular number and trabecular thickness for AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol ingestion were both lower (all p < 0.04, Fig. 8C, D), whereas the trabecular space parameter was significantly greater than that of either ALDH2-deficient mice with water or C57Bl/6 mice with chronic ethanol ingestion (p < 10−2; Fig. 8E). All of these parameters were significantly corrected by AAVrh.10hALDH2 therapy in both Aldh2 −/− and Aldh2 E487K+/+ mice to measurements equivalent to C57Bl/6 mice with chronic ethanol ingestion (all p > 0.1, Fig. 8C–E).
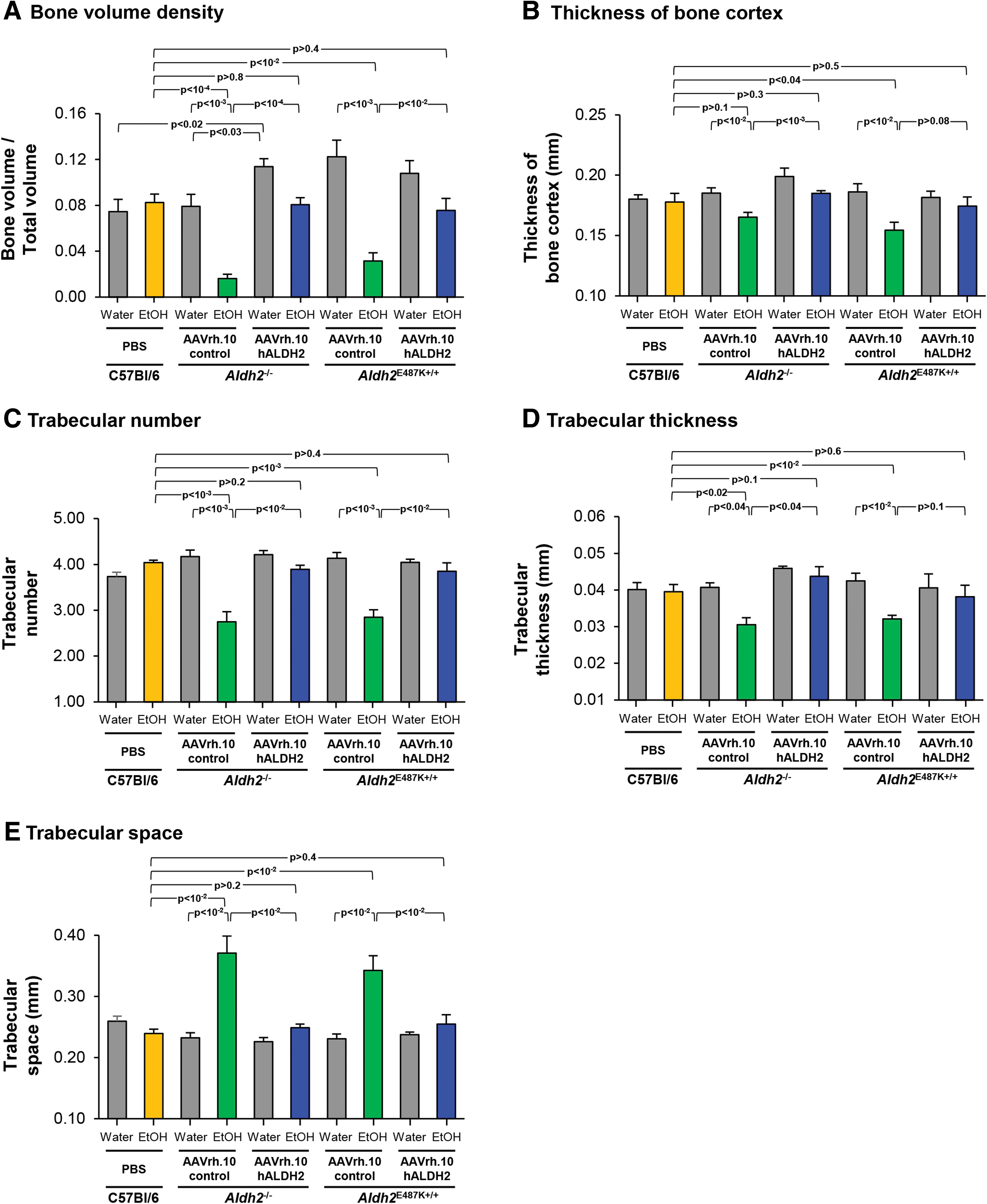
Quantitative μCT assessment of AAVrh.10hALDH2 therapy on bone structure of femurs of ALDH2-deficient mice after chronic ethanol ingestion. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks. Fixed femurs were scanned by μCT at the trabecular bone and midshaft cortex.
Longitudinal sections of the femurs were stained for histological analysis with hematoxylin and eosin (Fig. 9). The histological findings support the results of the μCT scans. Femur histology of C57Bl/6 wild-type mice given either water or ethanol had no visible differences (Fig. 9B). Histological assessment of Aldh2 −/− and Aldh2 E487K+/+ mice administered AAVrh.10control and given water showed no notable difference from that of wild-type mice (Fig. 9C, E, G, I). However, the presence of trabecular bone in the metaphysis of AAVrh.10control-treated Aldh2 −/− and Aldh2 E487K+/+ mice after chronic ethanol ingestion was significantly reduced (both p < 10−2, Fig. 9D, H and Supplementary Fig. S7). Interestingly, AAVrh.10hALDH2 vector-treated Aldh2 −/− and Aldh2 E487K+/+ mice with ethanol had significantly more trabecular bone than AAVrh.10control vector-treated Aldh2 −/− and Aldh2 E487K+/+ mice given ethanol (both p < 10−3, Fig. 9F, J and Supplementary Fig. S7). Indeed, the trabecular bone levels with AAVrh.10hALDH2 treatment were similar to the levels of trabecular bone in mice given water (Aldh2 −/−, p < 0.02; Aldh2 E487K+/+, p > 0.8; Supplementary Fig. S7) or wild-type mice given ethanol (Aldh2 −/−, p > 0.07; Aldh2 E487K+/+, p > 0.3; Supplementary Fig. S7).
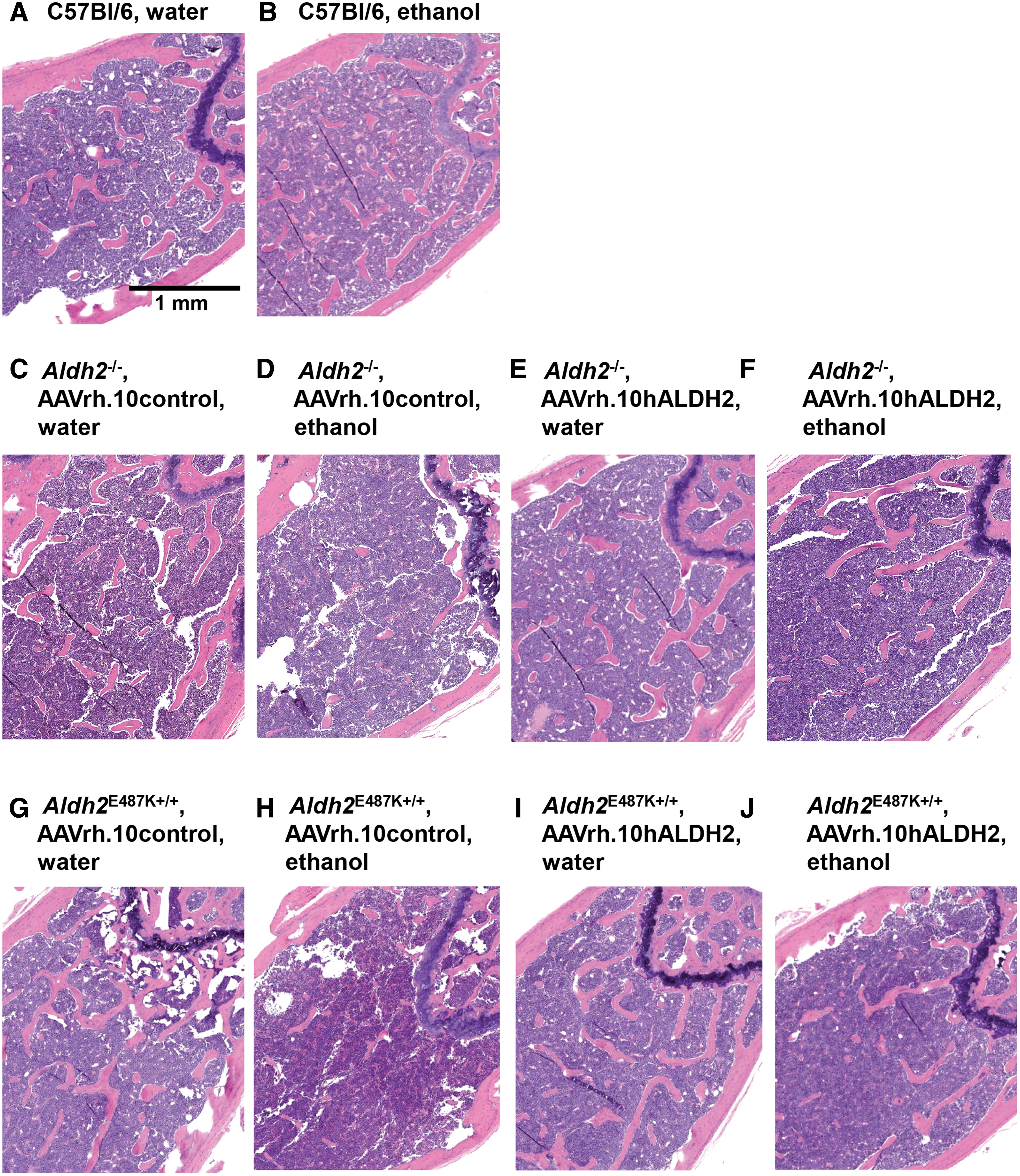
Histological assessment of AAVrh.10hALDH2 therapy on femurs of ALDH2-deficient mice after chronic ethanol ingestion. Aldh2
−/− and Aldh2
E487K+/+ mice were intravenously administered AAVrh.10hALDH2 (1011 gc) or AAVrh.10control (1011 gc). C57Bl/6 mice were intravenously administered PBS. Four weeks after vector administration, mice were challenged with water or ethanol for 12 weeks. Fixed decalcified femurs were embedded in paraffin and longitudinal sections were stained with hematoxylin and eosin. Examples from animals in each group are shown.
Discussion
ALDH2 deficiency is one of the most common hereditary disorders affecting 560 million people, ∼8% of the world population, including 35–45% of East Asian descendants. 1 –3 ALDH2 is a key enzyme in the ethanol metabolism pathway and works primarily in the liver converting acetaldehyde to acetate. 5,7 The most common genetic variant causing this deficiency is a glutamic acid-to-lysine substitution at position 487 (E487K) designated the ALDH2*2 allele. 3,14 Heterozygote and homozygote individuals for the ALDH2*2 allele have reduced enzymatic activity and ability to process acetaldehyde. 12,13 The ALDH2 enzyme is a tetramer and the mutant protein functions as a dominant negative 10,11 ; heterozygotes have <50% ALDH2 enzymatic activity and homozygotes <4%. 12,13 The major consequence of the mutation comes from accumulation of acetaldehyde primarily after ethanol consumption. The acute response to ethanol of individuals with ALDH2 deficiency is the famous “Asian flush syndrome,” appearing as alcohol-induced facial flushing, tachycardia, nausea, and headaches. 3,13 ALDH2 deficiency is also linked with a number of serious chronic medical disorders, including neurological, endocrine, cardiovascular and dermatological disorders, and aberrant drug metabolism. 2,3 Importantly, the ALDH2*2 allele is associated with a marked 7–12-fold increase in the risk of upper aerodigestive tract cancer of the oral cavity, pharynx, larynx, and esophagus with chronic ethanol ingestion. 3,17 Individuals carrying the ALDH2*2 allele who also smoke cigarettes and consume alcohol have a very high cancer risk (odds ratio 50:1) with a 25-year earlier onset of esophageal carcinoma. 1,44,45 In addition to the high risk for aerodigestive cancer, chronic ethanol consumption with ALDH2 deficiency is associated with a high risk for osteoporosis. 18 –24 The data demonstrate that pretreatment with AAVrh.10hALDH2, an AAV serotype rh.10 nonhuman primate vector coding for human ALDH2, prevents many of these chronic disorders in two mouse models of ALDH2 deficiency with chronic ethanol consumption.
Body weight, hemoglobin, locomotion, and dermatological abnormalities
Aldh2 −/− and Aldh2 E487K+/+ mice chronically consuming ethanol had increased serum acetaldehyde levels, higher MDA levels, lower body weight, lower hemoglobin counts, worse performance on a locomotion test, and increased skin hyperpigmentation than wild-type mice or ALDH2-deficient mice given water. All of these abnormal parameters were prevented by pretreatment with AAVrh.10hALDH2 therapy.
Loss of body weight in ALDH2-deficient mice with ethanol consumption has been observed in several previous studies. 9,33,46 Even without alcohol consumption, the body mass index is lower in individuals with the ALDH2*2 allele. 47,48 Long-term exposure to acetaldehyde impeded growth of Wistar rats in a dose-dependent manner, 49 suggesting the increased systemic acetaldehyde after ethanol consumption in ALDH2-deficient individuals may contribute to the decreases in body weight.
Hematological abnormalities such as anemia and leukocytopenia are a frequent complication of alcoholism even in normal individuals, 50 and individuals heterozygous for the ALDH2*2 allele who are alcoholics have increased susceptibility to these conditions. 51 In a study of Japanese men, heterozygous ALDH2*2 individuals who were moderate to heavy drinkers had lower red cell counts and hemoglobin levels than never or rare drinkers. 52 In addition, acetaldehyde causes DNA damage in hematopoietic stem cells, impairs blood production, and promotes bone marrow failure. 53,54 The ALDH2*2 allele is associated with increased risk of aplastic anemia and accelerated progression of disease in patients with Fanconi anemia. 55,56
The ALDH2*2 allele is associated with neurodegenerative disorders including Alzheimer's disease and Parkinson's disease. 2,57 –60 Aged Aldh2 −/− × Aldh1a1 −/− mice exhibit deficits in motor performance. 61 ALDH2 also detoxifies two endogenous aldehydes, 4-hydroxynonenal (4-HNE), and 3,4-dihydroxyphenylacetaldehyde (DOPAL), which have been implicated in neurotoxicity and the development of Parkinson's disease. 2,61 Chinese individuals homozygous for the ALDH2*2 allele have a 4.87-fold increase in the risk for Parkinson's disease. 59
Chronic alcohol use can precipitate a number of skin disorders including hyperpigmentation. 62 The hyperpigmentation of exposed skin has been observed in ALDH2-deficient mice at levels similar to those of the Aldh2 −/− and Aldh2 E487K+/+ mice with chronic ethanol consumption in this study. 9,38 The mechanism of melanin deposition is not well understood but is dependent on the dose of alcohol. 38 In humans, esophageal melanosis occurs at higher incidence in individuals with the ALDH2*2 allele. Alcoholic Japanese men have a 2.66-fold increased risk for these abnormalities, 63 suggesting a link to high acetaldehyde exposure as a consequence of the inactive ALDH2 enzyme.
Acetaldehyde and cancer
Prior studies have shown that esophageal cancer per se cannot be induced with long-term ethanol ingestion in mouse models of ALDH2 deficiency. 64,65 However, mouse models of ALDH2 deficiency do accumulate more DNA damage and adducts in their esophagus with chronic ethanol ingestion. 27,66 The carcinogenicity of acetaldehyde has been shown experimentally using long-term acetaldehyde exposure in the water supply of Sprague–Dawley rats. Increased development of malignant tumors was found in various organs dependent on the acetaldehyde levels. 67 Acetaldehyde is categorized as a group I human carcinogen by the International Agency for Research on Cancer. 68,69 Acetaldehyde can form DNA–DNA and DNA–protein cross-links, induce DNA adduct formation, and interfere with DNA replication and the DNA repair machinery. 2,70 –72 Several reports have suggested that the accumulation of acetaldehyde-induced DNA adducts in cells leads to the development of cancer. 26,73,74 The most abundant of the adducts is N2-et-dG; accumulation of this adduct in the esophagus has been observed in ALDH2-deficient human alcoholics and mice exposed to ethanol and is often used as a biomarker for acetaldehyde-related DNA damage as a precursor of esophageal cancer. 27,75 Other acetaldehyde-induced adducts, α-methyl-γ-OH-propano-deoxyguanosine and N2-(2,6-dimethyl-1,3-dioxan-4-yl)-deoxyguanosine, are detected in the blood of ALDH2-deficient alcoholics. 75 Mammalian cells and rodents treated with ethanol or acetaldehyde develop DNA interstrand crosslinks and double-stranded breaks leading to chromosome rearrangements and genomic instability. 54,66,70
Osteoporosis
Osteoporosis caused by chronic alcohol consumption is detrimental to quality of life and a significant financial burden on both individuals and society. 76,77 Worldwide, 67 million individuals >50 years old with ALDH2 deficiency are at increased risk for osteoporosis with chronic alcohol intake. 2,3,78 –80 Alcoholism is a risk factor for osteoporosis even in individuals without ALDH2 deficiency. 18,21,22 Acetaldehyde, the intermediate product of alcohol metabolism, interferes with bone metabolism. 19,20,81,82 The increased accumulation of serum acetaldehyde resulting from ALDH2 deficiency further increases the risk of developing osteoporosis. 2,3,19,20
Gene therapy
We hypothesized that genetic modification of the ALDH2-deficient liver to express the normal human ALDH2 coding sequence would restore ALDH2 enzymatic function, reduce persistent elevated levels of circulating acetaldehyde, and prevent the ALDH2-deficient phenotypes associated with chronic ethanol consumption. The data demonstrated that administration of AAVrh.10hALDH2 reconstituted ALDH2 protein expression and enzymatic activity in two murine models of ALDH2 deficiency at 16 weeks after administration. In Aldh2 −/− and Aldh2 E487K+/+ mice, AAVrh.10hALDH2 treatment corrected chronic ethanol-induced elevated serum acetaldehyde levels and liver MDA levels as well as a variety of abnormal phenotypes including body weight loss, lower hemoglobin levels, poor locomotion, skin pigmentation, esophageal DNA damage and adduct accumulation, and osteopenia.
The results for AAVrh.10hALDH2 gene therapy prevention of chronic ethanol-induced esophageal DNA damage and adducts in the ALDH2-deficient mice are consistent with reports suggesting that Alda-1, a small molecule activator of ALDH2, can reduce the accumulation of DNA damage and adducts in the esophagus of mice with ALDH2 deficiency. 64 Although AAVrh.10hALDH2 treatment significantly reduced both the number of γH2AX-positive cells and the level of N2-et-dG adducts in the esophagus, these markers of DNA damage remained higher than in ethanol-exposed wild-type mice or ALDH2-deficient mice given only water. This may be due to acetaldehyde produced locally in the esophagus contributing to the induction of DNA damage and ultimately esophageal cancer. 64,66,83 The data show that AAVrh.10hALDH2 treatment prevents the increased level of circulating acetaldehyde in the blood in ALDH2-deficient mice given alcohol. However, mucosal cells, salivary glands, or microbes in the oral cavity also are able to process ethanol to acetaldehyde after alcohol exposure, which may contribute to acetaldehyde-induced damage in the esophagus. 84 –86 Because AAVrh.10hALDH2 mainly targets the liver and esophageal epithelial cells have a high turnover rate, long-term AAV gene therapy for the esophagus is difficult. However, a liver-directed gene therapy approach may be unable to completely reduce the elevated local levels of acetaldehyde in the oral cavity and esophagus. Even so, our data indicate that reducing the burden of systemic acetaldehyde by restoring ALDH2 expression in the liver substantially lowers the accumulation of DNA damage and DNA adducts that precede esophageal carcinogenesis in ALDH2-deficient mice that chronically consumed ethanol.
μCT quantification and histological assessment of femurs from the ALDH2-deficient mouse models demonstrated that chronic ethanol ingestion for 12 weeks induced osteopenia. Consistent with previous reports, the bone density loss was especially striking in trabecular bone and was also significant in the midshaft cortex. 20,41 AAVrh.10hALDH2 therapy prevented the development of osteopenia in ALDH2-deficient mice with chronic ethanol ingestion and maintained bone density similar to C57Bl/6 mice with chronic ethanol exposure or ALDH2-deficient mice with water. This is consistent with a previous study showing that the ALDH2 activator Alda-1 could reverse trabecular bone loss in ovariectomized osteopenic rats. 87
Implications for human therapy
Regardless of their sensitivity to alcohol, there has been a rise in alcohol dependence and abuse among the populations of ALDH2*2 carriers in East Asian countries and their descendants. 88,89 In Japan, 26% of heavy drinkers are ALDH2*2 heterozygotes, 90 and 58% of Asian American college students with an inactive ALDH2*2 allele are regular drinkers. 91 This presents a large population with a highly increased risk of chronic disorders such as upper aerodigestive tract cancer and, without intervention, an emerging public health crisis. 2,3,92 Abstaining from alcohol is currently the only treatment for reducing the high risk of upper aerodigestive tract cancer and osteoporosis associated with ALDH2 deficiency. Our data support the concept that AAV-mediated gene therapy presents a possible effective preventative therapy for the chronic disorders associated with chronic ethanol consumption in individuals with the ALDH2 deficiency state.
Although AAVrh.10hALDH2 may prevent the deleterious physiological effects of chronic alcohol consumption for individuals with the ALDH2*2 allele, the therapy may be limited in addressing the underlying reward–addiction cycle relevant for alcoholism. The ALDH2*2 allele is generally associated with a lower risk of alcoholism because of the immediate deleterious physiological effects of alcohol consumption. 93 However, ALDH2*2 alcoholics become tolerant of these physiological effects with continued alcohol consumption. 94 Studies of Chinese and Korean alcoholics found that heterozygous ALDH2*2 individuals scored significantly higher on the positive alcohol expectancies scale and lower on the negative alcohol expectancies scale than individuals with a functional ALDH2. 95,96 In animal studies, acetaldehyde demonstrates several properties that are similar to addictive drugs, including activation of dopamine receptors, locomotor stimulation, and self-administration. 97 –103 Interestingly, administration of a lentiviral vector encoding ALDH2 into the brain ventral tegmental area of rats reduced ethanol consumption and binge drinking in rats bred for alcohol preference. 104 In this context, AAVrh.10hALDH2 reduces systemic acetaldehyde levels with drinking alcohol, and thus may help reduce addictive behaviors in addition to protecting against the serious physiological effects of chronic alcohol consumption in ALDH2*2 individuals.
There is also epidemiological evidence that ALDH2 deficiency is implicated in several other diseases including neurological disorders, diabetes, cardiac ischemia, stroke, hepatocellular carcinoma, chronic obstructive pulmonary disease, severity of Fanconi anemia, and dermatitis. 2,3 ALDH2 not only converts acetaldehyde to acetate in the ethanol metabolism pathway, but also metabolizes numerous other aldehydes such as 4-HNE and MDA, by products of oxidative stress, and acrolein, an environmental aldehyde found in tobacco smoke, car exhaust, and other pollutants. 2,105 In this study, significantly more MDA was observed in the liver of Aldh2 −/− and Aldh2 E487K+/+ mice with ethanol exposure, which was prevented by AAVrh.10hALDH2 therapy. These aldehydes induce cytotoxicity because of their ability to spread through cell membranes and produce adducts with DNA, proteins, and lipids that disrupt their function. 106 –108 Without a functioning ALDH2 enzyme, the clearance mechanisms available to protect cells and tissue from aldehyde-mediated damage are limited.
In summary, the data show that AAVrh.10hALDH2 gene therapy prevents chronic ethanol consumption-induced disease-related phenotypes in the two mouse models of ALDH2 deficiency. The overall effects of ALDH2 deficiency on human health, specifically the marked increase risk for cancer and osteoporosis, support the concept of developing AAVrh.10hALDH2 as a preventative therapy in high-risk ALDH2-deficient individuals.
Footnotes
Acknowledgments
We thank S. Cronin, A. Camilleri, S. Nag, and F. Hart for assistance with these studies; J. Fernandez and H. Molina at the Proteomics Resource Center at Rockefeller University for assisting with the LC/MS acetaldehyde and DNA adducts assays; S. Rafii for the use of the ADVIA hematology system; and N. Mohamed for editorial assistance.
Author Disclosure
R.G.C. has equity in LEXEO and R.G.C. and O.E.P. are participants in a patent disclosure regarding gene therapy for ALDH2 deficiency.
Funding Information
These studies were supported, in part, by National Institute of Health R41 AA027730 01 to LEXEO Therapeutics and, in part, by the Department of Genetic Medicine, Y.M. was supported, in part, by a scholarship from the Uehara Memorial Foundation (Japan), and OEP was supported, in part, by a Parker B. Francis Fellowship. M.B.G. holds award DP5OD021351 from the Office of the Director of the NIH, a Career Award for Medical Scientists from the Burroughs Welcome Foundation, and a Pershing Square Sohn Prize for Young Investigators in Cancer Research. Research reported in this publication was supported by the NIAA/NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.