
Abstract
Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) is a rare disease caused by mutations to the UNC13D gene and the subsequent absence or decreased activity of the Munc13-4 protein. Munc13-4 is essential for the exocytosis of perforin and granzyme containing granules from cytotoxic cells. Without it, these cells are able to recognize an immunological insult but are unable to execute their cytotoxic functions. The result is a hyperinflammatory state that, if left untreated, is fatal. At present, the only curative treatment is hematopoietic stem cell transplantation (HSCT), but eligibility and response to this treatment are largely dependent on the ability to control inflammation before HSCT. In this study, we describe an optimized lentiviral vector that can restore Munc13-4 expression and degranulation capacity in both transduced FHL3 patient T cells and transduced hematopoietic stem cells from the FHL3 (Jinx) disease model.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is typified by high fever, splenomegaly, cytopenias, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, reduced NK cell activity, high ferritin levels, elevated levels of sIL2R, 1,2 and often hyponatremia. 3,4 In its inherited form, HLH is caused by mutations to the PRF1, UNC13D, STX11, STXBP2, and other genes that are associated with the exocytosis of lytic granules from cytotoxic cells. Consequently, infants with HLH are born seemingly healthy, but upon encountering an immunological insult such as a viral infection (e.g., Epstein-Barr virus [EBV]), their lymphocytes cannot execute cytotoxic degranulation, leading to a positive feedback loop of massive inflammation without clearing the infectious trigger. 5 –7 Left untreated, the disease is fatal. 8,9 To date, the only curative treatment available is an allogenic hematopoietic stem cell transplantation (HSCT). However, inflammation must be adequately controlled before HSCT, and despite recommendations for intensification of therapy in the HLH-2004 international treatment study, the reported pre-HSCT mortality was still high at 19%. 9 Survival rates vary depending on the conditioning protocol. 10 –19 Thus far, results from clinical trial NCT01998633 suggest that reduced intensity conditioning can improve overall 1-year survival of HLH patients to 82.4%, although larger and longer term studies will be needed to follow-up on these results. 20 For patients who do receive HSCT, the 5-year survival rate has improved from 54% to 61%. 16 –18,21 Thus, despite several advances made in the field of HLH disease treatment, the outcome is suboptimal and there is still substantial room for improvement.
HLH has been proposed as an excellent candidate for hematopoietic stem and progenitor cell (HSPC)-based gene therapy intervention for several reasons. First, modification of a patient's own HSPC would reconstitute immune system with functional cytotoxic cells, theoretically correcting the HLH phenotype for life. Second, this autologous HSPC-based gene therapy could eliminate the risk of graft versus host disease compared with an allogenic HSCT. Third, patients who receive an HLH diagnosis would not have to wait to find a matching HSCT donor. Finally, modification of a patient's own T cells could be used in autologous adoptive transfer to control infection and inflammation before HSCT. 22 –24
In this study, we focused exclusively on lentiviral (LV) gene therapy for familial HLH type 3 (FHL3), which is caused by mutations to the UNC13D gene and a subsequent absence or loss of function in the Munc13-4 protein. Studies conducted in Germany and North America found that FHL3 accounts for ∼25–30% of inherited HLH cases. 25,26 FHL3 is the most common form of FHL documented in white patients, 26 although multiple independent founder mutations leading to FHL3 have been described in populations from around the world. 26 –30 Missense and nonsense mutations, deletions, splicing error variants, and inversions have all been described in FHL3 patients, and no single exon of the 32 spliced exons seems to be favored as a site for mutation. 31,32
Munc13-4 is part of the UNC13 family of proteins, all of which are involved in processes relating to vesicle exocytosis. Munc13-4 has been implicated in the exocytosis of lytic granules from cytotoxic cells, 8,33 tertiary, and mucin-containing granules from neutrophils, 34,35 dense granules from platelets, 36 and secretory granules from mast cells during regulated exocytosis. 37 Although much of HLH research has focused on pathology arising from impairment of CD8 T cell and natural killer cell cytotoxic function, 8 it is worth noting that FHL3 patients also suffer from susceptibility to fungal infection, 38 neurological symptoms, 39 exacerbated response to lung infection, 26,39 and macrophage activation syndrome, 26 which could be related to impaired degranulation from other cell types. Although Munc13-4 is expressed in other tissues, 40 –42 the fact that HSCT results in cure suggests that its most critical role is in cells derived from the hematopoietic lineage, and therefore its role in nonhematopoietic tissues might be limited or complemented by other proteins in degranulation pathways.
The first gene therapy study for FHL3 was recently published and described by transducing activated peripheral blood mononuclear cells (PBMCs) from three FHL3 patients using either a vesicular stomatitis virus G protein (VSV-G) or a measles H/F pseudotyped LV vector that encoded for the complementary DNA (cDNA) product from the human UNC13D gene. When NSG mice were transplanted with EBV-induced tumors and then infused with transduced FHL3 patient T cells, reduction in tumor size was observed. 24 The same group then showed transduction of hematopoietic stem cells from Munc13-4 null (Jinx) mice—the mouse model for FHL3—showed improvements to in vitro metrics and viral clearance as a result of gene therapy. 23 Finally, a second group transduced FHL3 patient cells with a gamma retrovirus-based gene therapy construct demonstrated that transduced cells had improved degranulation capacity and cytotoxic abilities. 22 Both groups used an Ef1α promoter and codon-optimized transgene. 22,23 Here, we describe several new LV vectors, establish the minimum chimeric threshold needed for restoring adequate degranulation, and demonstrate that our described vector can be used to transduce both patients T cells and HSPC from the FHL3 mouse model.
Materials and Methods
Cloning
All cloning was carried out into the LTG 1337 backbone, supplied by Expression Therapeutics. In humans, UNC13D comprises 32 exons, 8 therefore in constructing our UNC13D LV vector, we used the sequence from the Homo sapiens unc-13 homolog D (UNC13D) messenger RNA (mRNA; NCBI reference Sequence: NM_199242.2). When translated, this nucleotide sequence yields isoform 1 of the Munc13-4 protein (protein Sequence NP_954712.1, UniProt identifier Q70J99-1). Codon optimization was performed by GenScript according to the algorithm previously described in our laboratory. 43 All clones were screened using restriction digest and sequenced by Genewiz.
LV vector packaging and titering
The third generation, self-inactivating, VSV-G pseudotyped LV vectors were constructed using a standard HEK 293T transfection protocol.
44
Specifically, HEK 293 T/17 cells were transfected with a four-plasmid system. After transfection, the following day the media was replaced with Dulbecco's modified Eagle's medium (DMEM), 1 × with 4.5 g/L glucose,
Transduction
All transductions were carried out in the presence of polybrene-supplemented media (8 μg/mL; Sigma). Cell lines used in in vitro experiments were transduced once with a single overnight application of LV vector using the specified multiplicity of infections (MOIs). HSCs that were used for transplant studies were transduced twice in 50% viral media (mean MOI of 42), with the first hit of the virus being incubated on the cells overnight and the second hit of virus lasting for 6 h.
Transplants
Whole bone marrow was flushed from the tibias and femurs of donor Jinx (Unc13dJinx, UNC13D; Jackson Laboratories) or CD45.1+ mice. Sca-1+ cells were isolated according to manufacturer protocols using Sca-1 antibody (553334; BD Biosciences), biotin-labeled magnetic beads (130-090-485; MACS; Miltenyi Biotec), and MACS magnetic separation unit (Miltenyi Biotec magnet and stand; 130-042-303 and 130-042-109, respectively). These isolated Sca-1 cells were then cultured in stimulation media consisting of StemPro nutrient-supplemented media (10640-019; Gibco), recombinant mouse interleukin 3 (IL-3) (20 ng/mL; 403-ML; R&D), recombinant human IL-11 (100 ng/mL; 218-IL; R&D), recombinant human Flt3/Fc (100 ng/mL; 368-ST; R&D), mouse mSCF (100 ng/mL; 455 MC; R&D),
VCN analysis
The same RRE primers, SYBR Green Power PCR Master Mix, and PCR protocol that were used for the titering of new LV vectors were also used in the qPCR-based copy number analysis of transduced cells.
Infection
Jinx mice were infected by intraperitoneal injection with 2 × 105 plaque-forming units (PFU) of lymphocytic choriomeningitis virus (LCMV) Armstrong (generously provided by the Ahmed Lab).
Colony-forming unit assays
One thousand cells per milliliter of Methocult Culture media (03434; StemCell Technologies) were plated and incubated for 7–10 days. Colonies were subsequently counted, assessed using a viability stain, and the number of successfully transduced colony-forming units was determined either by copy number assay or flow cytometry on the pooled cells.
Western blot
A standard Western blot protocol 45 was performed by blotting onto a PVDF membrane (162-0261; BioRad) and staining for Munc13-4 (Clone ERP4914, ab109113; Abcam) and beta-actin (Clone 2D1D10, A00702; GenScript). Secondary antibodies (IRDye 800 CW and IRDye 680; Licor; catalog numbers 926-32213 and 026-68072, respectively) were used to clearly distinguish between the Munc13-4 and beta-actin bands, and the blots were imaged using a LI-COR Odyssey CLx imaging device using Image Studio Version 4.0 software.
Degranulation assay
Homogenized splenic tissue was passed through a 70 μm filter, washed with phosphate-buffered saline containing 5% FBS, and plated at 300,000 cells/FACS tube in RPMI containing 10% FBS and 1% penicillin and streptavidin and supplemented with mouse IL-2 (10 ng/mL, 575402; BioLegend). Cells were then incubated for 30 minutes in either the presence or absence of 3 μg/mL of CD3 (Clone 145-2C11, 100301; BioLegend) and 1 μg/mL of CD28 antibody (Clone 37.51, 102101; BioLegend) to produce “stimulated” and “unstimulated” sample groups, respectively. Subsequently, monensin (420701; BioLegend) and CD107a antibody (BV421, 121617; BD Biosciences) were added to all the cultures, and the cells were incubated for an additional 8 h. Staining for analysis consisted of a live dead cell stain, CD8 (PE,108739; BioLegend), CD44 (UV379, 740215; BD Biosciences), and CD69 (FITC, 557392; BD Biosciences). The chimeric transplant studies also incorporated the use of CD45.1 and CD45.2 antibodies (PerCP-Cy5.5, 560580, BD Biosciences; and BUV737, 564880, BD Biosciences, respectively) to distinguish between cells derived from Jinx and CD45.1 engrafted Sca-1 cells.
Cytotoxicity assay
Frozen PBMCs from an FHL3 patient were thawed and stimulated using CD3 and CD28 (7 and 0.5 μg/mL, respectively) for 24 h. These cells were subsequently double transduced with an MOI of 10 for both TPO-CAR and UNC13D LV vectors according to the aforementioned transduction protocol. Five days after transduction, these cells were coincubated with M-07e cells. Cell killing was assessed by flow cytometry by looking for cell death markers Annexin V and propidium iodide (PI).
Primary cell and cell lines
Healthy donor T cells were isolated by negative selection from donor PBMC sourced from leukoreduction filters, as previously described.
46,47
The following cell lines were used in this study: NIH 3T3 cells (ATCC number CRL-1658), Jurkat clone E6-1 cells (ATCC number TIB-152), M-07e cells (ACC 104). Munc13-4-deficient T cells were isolated from FHL3 patients treated in the Children's Healthcare of Atlanta (CHOA), Emory University School of Medicine (Atlanta, GA). The patient had biallelic genetic defects: a c.551G>A (
Cell culture
The following reagents were used for cell culture: DMEM (10-017-CV; Corning), RPMI (10-040-CV; Corning), StemPro (10640-019; Gibco), FBS (S11150H; Atlanta Biologicals), and penicillin/streptomycin (09-757F; Lonza).
Flow cytometry
The following reagents were used throughout the various flow cytometry-based experiments: eFluor 780 viability staining (65-0865-14; ThermoFisher), PE mCD8 (108739; BioLegend), UV379 mCD44 (740215; BD Biosciences), FITC mCD69 antibody (557392; BD Biosciences), BV421 mCD107a (121617; BD Biosciences), PerCP-Cy5.5 CD45.1 (560580; BD Biosciences), BUV737 CD45.2 (564880; BD Biosciences), Annexin V, and PI.
Statistical analysis
All statistical tests were performed using GraphPad Prism version 8.
Results
Stable expression of Munc13-4 in healthy donor cells
To design an optimal LV-based expression system, we set out to assess the dynamic change in the expression of degranulation proteins at baseline and immune activation. Western blot was performed on PBMCs from healthy donors to assess the expression of perforin, Munc13-4, Rab27a, and STXBP2 before and 120 h after stimulation with CD3/CD28 beads (Fig. 1A–C). The results indicate that perforin was upregulated ∼20-fold from its baseline levels in response to stimulation and activation, but that Munc13-4, Rab27a, and STXBP2 protein levels were either not significantly different from baseline (Munc13-4 and STXBP2) or only twofold increased from baseline (Rab27a). This suggests that a constitutive promotor driven LV expression system would be appropriate for correcting FHL3-related degranulation defects.

Transduction of FHL3 patient T cells
A LV construct that expresses the cDNA product from the human UNC13D gene (GenBank accession number AJ578444.1) under the control of the Ef1α promoter was designed (Fig. 2A). Stimulated PBMCs from an FHL3 patient were transduced using a standard polybrene transduction protocol, and it was observed that these transduced cells expressed Munc13-4 (Fig. 1D). Stimulation of the transduced cells with CD3 and CD28 induced increased expression of the exocytosis marker CD107a (LAMP-1) at the plasma membrane compared with nontransduced patient cells, indicating functionality of the transgenic Munc13-4 (Fig. 1E and Supplementary Fig. S1). The patient's T cells used in this study were naive to EBV; hence, we could not assess cytotoxicity against EBV-transformed lymphoblastoid cell lines. To assess the cytotoxic abilities of the transduced cells, we double transduced FHL3 patient PBMCs with both the UNC13D construct and a human thrombopoietin (TPO) chimeric antigen receptor (CAR) construct. This subsequently rendered the double-transduced cells cytotoxically competent and able to recognize cells expressing TPO receptor (MPL). A cytotoxicity assay performed by coculturing these cells with MPL expressing target cells (M-07e cells) demonstrated an increase in cytotoxic function in transduced patient cells compared with nontransduced patient cells (Fig. 1F and Supplementary Fig. S2).

Optimization of the LV construct
To determine the optimal expression system for Munc13-4, several alternative LV vectors were designed (Fig. 2A). Starting with the previously described construct, we added the sequence for green fluorescent protein to act as a marker for transduced cells. Second, we replaced the Ef1α promoter with the synthetic MND promoter that was adapted from Moloney murine leukemia virus and myeloproliferative sarcoma virus elements. 48 Third, we included a Kozak sequence in some of our constructs to enhance translation of transgene mRNA. 49 Finally, we created a codon optimized version of the canonical UNC13D transgene using the optimization strategy we previously described. 50 All cloned plasmids were checked for their accuracy using restriction digests and DNA sequencing.
The cloned plasmids were integrated into a four-plasmid transfection system to generate VSV-G pseudotyped LV vectors. Previous FHL3 gene therapy studies found that H/F pseudotyped vectors produced efficient transduction of T cells from FHL3 patients. 22,24 We chose to pseudotype our LV vectors with VSV-G because VSV-G is pantropic, 51 and clinical trials using VSV-G pseudotyped LV vectors are underway (NCT02234934, NCT01560182, NCT03315078, NCT02333760, and NCT03818763 52,53 ). Also, we were concerned about the findings from some studies that have shown that H/F pseudotyped LV vectors can induce syncytia formation in transfected cells, leading to cell death and subsequent low titers. 54,55 In addition, it has been shown that H/F pseudotyped LV vectors have difficulty targeting murine cells, 56 and also that native-form H protein induces a humoral immune response in measles-vaccinated individuals, which can lead to neutralization and clearance of H/F pseudotyped LV vectors. 57
We were reliably able to produce these self-inactivating LV vectors at similar titers (Fig. 2B) using our previously described HEK 293T transfection protocol. 58 We demonstrated that these vectors could be used to transduce NIH 3T3 cells (a mouse embryonic fibroblast cell line that does not express Munc13-4) and robustly express Munc13-4 protein. We achieved higher levels of protein expression from cells that were transduced with the Ef1α_Kozak_Codon-Optimized-UNC13D construct (Fig. 2C), and therefore we chose to confine our subsequent transduction experiments to use this particular LV construct.
Identifying potentially therapeutic Munc13-4 expression levels
Jinx mice were irradiated twice with 550 rads of gamma radiation and transplanted with different mixtures of Jinx whole bone marrow (CD45.2+) and wild-type (WT) bone marrow (CD45.1+). Flow cytometry analysis showed that 6 weeks after transplantation both WT and Jinx bone marrow engrafted in the recipient mice in proportions consistent with the original transplant. Furthermore, the ratios of CD45.2 to CD45.1 positive cell remained consistent across specific CD8+, CD4+, and NK1.1+ cell subpopulations (Fig. 3). At baseline, we established that following LCMV infection, there was a robust increase in CD44+ CD8 T cells, and this memory response was comparable between WT and Jinx mice (Fig. 4A–E). Twenty-two weeks post-transplantation, the mice were killed and their splenocytes were stimulated with CD3 and CD28 free antibody and analyzed for the expression of plasma membrane-bound CD107a (Lamp-1) and CD69. CD8+, and CD44+ cells from the mice transplanted with WT CD45.1+ bone marrow degranulated similarly to those from nontransplanted WT mice, indicating that HSCT did not affect degranulation capacity (Fig. 4E). Consistent with previous research, splenocytes from WT, Jinx, and chimeric mice all increased CD69 expression after stimulation (Fig. 4G), corroborating the evidence that the presence or absence of Munc13-4 expression does not affect the mechanism of T cell activation. 59 Mice with 15% of CD45.1+ mouse bone marrow showed CD107a upregulation comparable with that of a WT mouse (Fig. 4F). Together, these data show that similar to studies carried out in the FHL2 gene therapy mouse model, 60 a 15% correction might be sufficient to recapitulate the WT degranulation phenotype in FHL3 disease model mice.
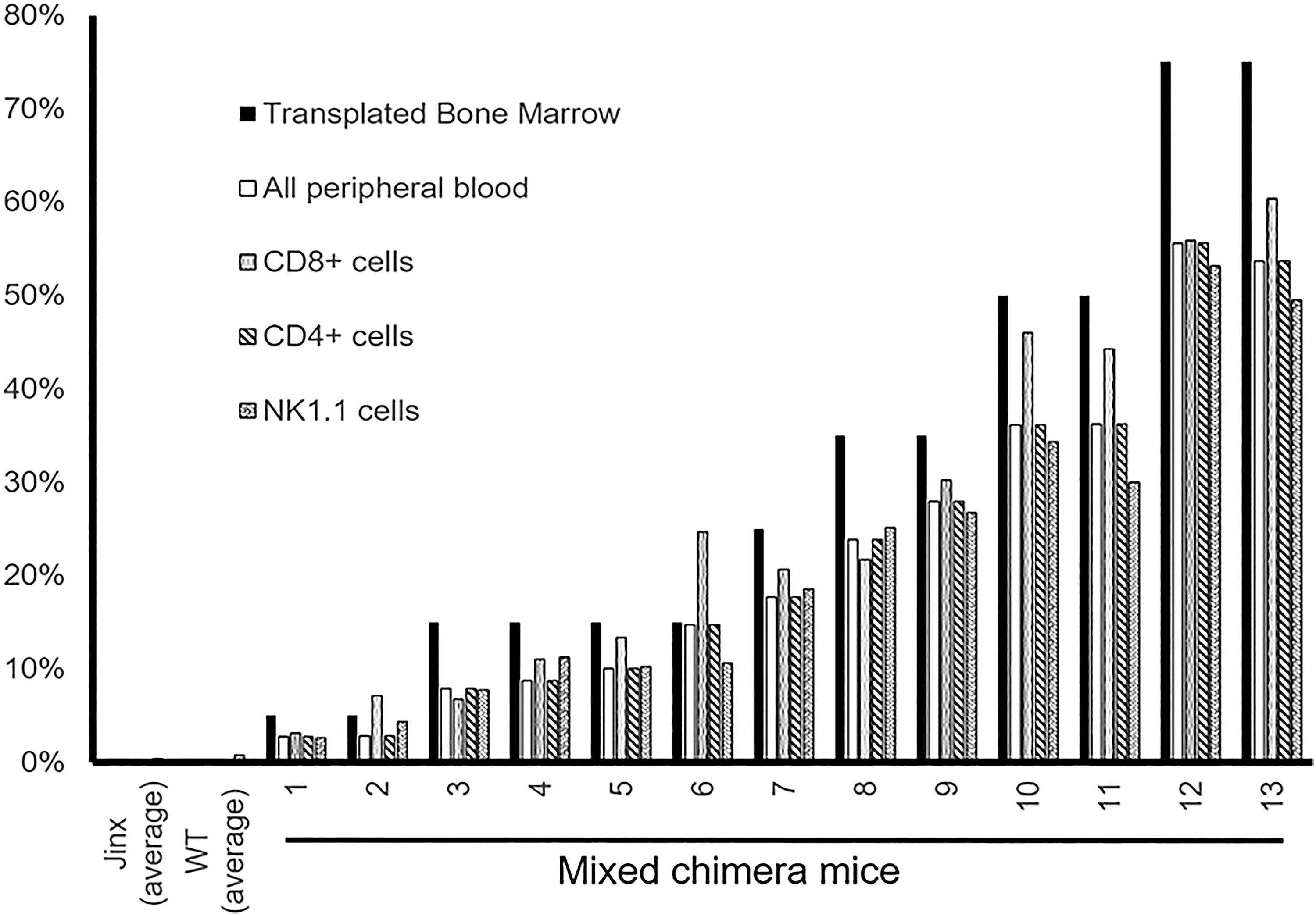
Six weeks after transplant, flow cytometry analysis indicated that the chimerically transplanted mice maintained their chimerism within the bulk peripheral blood and also across the CD8+, CD4+, and NK1.1+ cell populations.

Gene transfer into the FHL3 disease mouse model
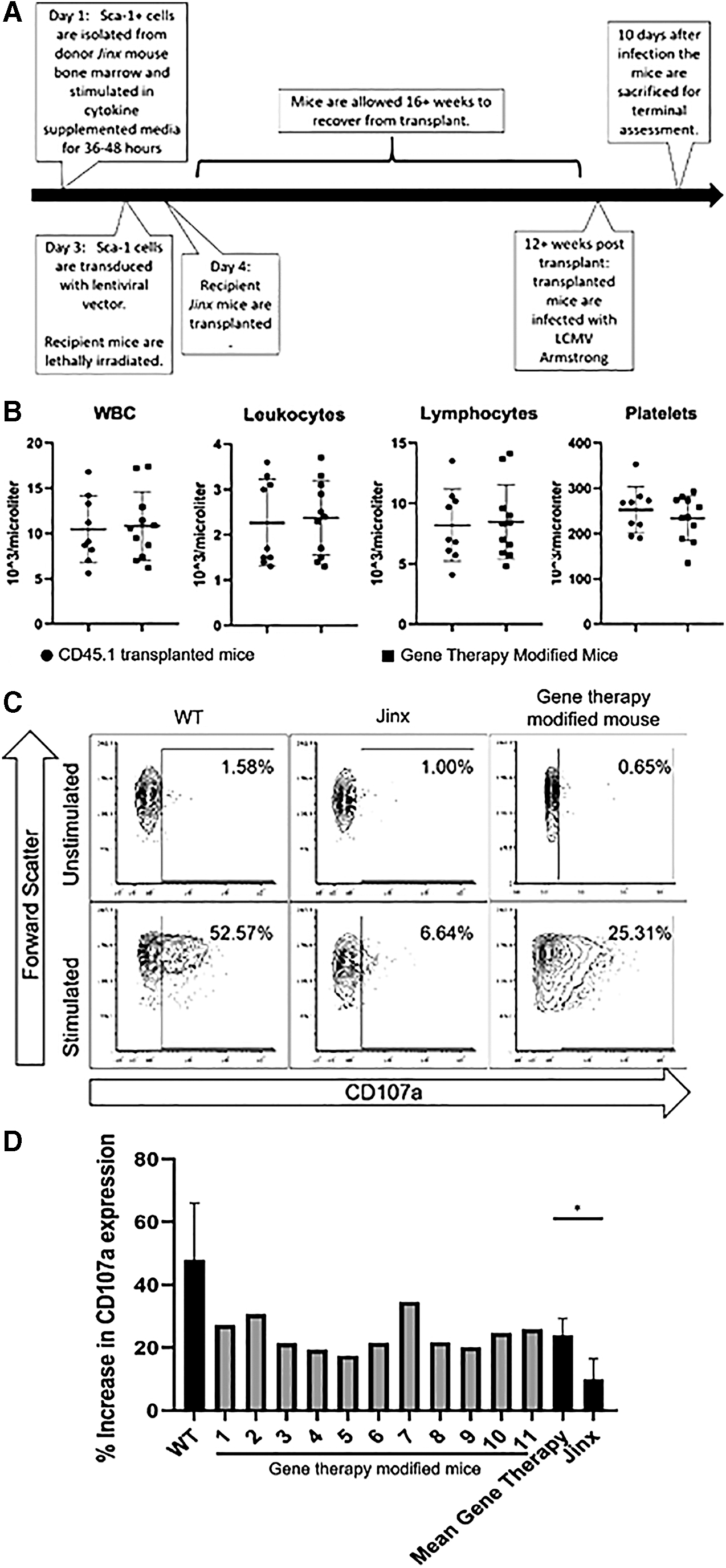
To determine the in vivo efficacy of our gene therapy approach within the FHL3 mouse model, Sca-1 cells were isolated from donor Jinx mice and transduced with the Ef1α_Kozak_CodonOptimizedUNC13D LV vector. Subsequently, one million of these cells were transplanted into Jinx mouse recipients according to our previously published protocols (Fig. 5A). 61 Within 12 weeks, the mice had normal complete blood counts (CBCs) (Fig. 5B) and an average copy number of 0.15 in the peripheral blood. Twenty weeks after transplantation, the mice were infected with 2 × 105 PFU LCMV Armstrong to induce the FHL3 disease phenotype. 59 Ten days after infection, the mice were killed, and degranulation assay results ascertained a statistically significant increase in CD107a expression in stimulated cells from gene therapy-modified mice compared with nontransplanted Jinx mice (Fig. 5C, D). The mean viral copy number (VCN) in the bulk splenocyte population was 0.30 copies per cell. Despite LCMV challenge, we did not observe an obvious HLH phenotype in our Jinx mice.
Discussion
Current treatment for HLH entails an allogeneic HSCT, but the success of this procedure is greatly influenced by the ability to control disease-related inflammation before transplantation and the availability of HLA-matched HSCT donors. Inadequate inflammation control increases the risk of a patient experiencing transplant-related mortality, usually caused by infections, veno-occlusive disease, pneumonitis, graft failure, and/or graft-versus-host disease. 62 Only 53% of HLH patients achieve full resolution of their inflammation before HSCT, and of these patients, 13% experience disease reactivation. 9 Furthermore, the inability to control inflammation before HSCT accounts for 22% of diagnosed HLH patients dying before HSCT. 9 In light of suboptimal outcomes, we and others have proposed a gene therapy treatment by which a patient's T cells would be isolated, transduced with a corrective construct, and transfused back into the patient such that the transduced cells could resolve the cause of inflammation and if necessary, allow for additional HSPC-based autologous gene therapy or allogeneic HSCT. 22,24
LV gene therapy utilizing a constitutive promoter is better suited for genetic defects where the protein is consistently expressed without much dynamic change upon activation. Our results showed limited to no significant change in expression levels of degranulation proteins Munc13-4, STXBP2, and Rab27a after TCR stimulation. Thus, these genetic defects are ideal candidates for constitutively expressing LV-based gene therapy strategies. In contrast, perforin seems to be dynamically regulated, and therefore gene therapy for perforin deficiency will likely necessitate the use of a perforin-specific promoter as described in previous studies 3,63 or maybe a better candidate for gene editing rather than gene addition strategies.
We were able to successfully augment Munc13-4 expression in activated patient T cells, and restores in vitro degranulation. We note that our results corroborate those from other studies that have shown that T cells from FHL3 patients, despite having been subjected to a hyperinflammatory environment and massive in vivo expansion, are permissive to LV transduction. 22,24,64 Furthermore, using a dual transduction protocol we were able to restore cytotoxic capacity in FHL3 T cells, demonstrating the effectiveness of our LV vector and the potential of a dual UNC13D and CAR-T transduction protocol as a therapeutic option to reduce viral-induced inflammation before a patient received HSCT.
Recent reports showed that human T cells and mouse hematopoietic stem cells can be transduced with LV vectors that express a codon-optimized UNC13D cDNA under the control of the Ef1a or EFS promoters. 22,23 These cells achieved VCNs of 224 and 5 copies per cell after transduction using MOIs of 100. 22 –24 Scaling LV production to be able to produce such large quantities of LVs for treating a patient remains a major challenge for the field of gene therapy, and so we created multiple LV constructs with the intent of producing a superior and scalable vector. Each construct was reliably and efficiently produced, but transduction into cell lines and subsequent western blot analysis showed that the codon-optimized version produced the highest expression of Munc13-4, which was comparable with constitutive expression levels in human PBMCs, even at relatively low MOIs.
Studies of HLH patients that received HSCT reported that between 20% and 30% bone marrow chimerism is protective against disease reactivation, although the correlation between donor chimerism and patient outcome is by no means perfect. 65 Similarly, gene therapy studies in the perforin-deficient FHL2 mouse model showed that as little as 10–20% engraftment of WT cells within the bone marrow of transplanted mice was sufficient to restore immune function. 60 We sought to determine what percent correction in a Munc13-4 null mouse model (Jinx mouse model) would be sufficient to correct the Munc13-4-deficient degranulation defect. To this end, varying donor chimerism was established in Jinx mice using WT (CD45.1) and Jinx (CD45.2) bone marrow. As expected, positive correlation was noted between the proportion of engrafted CD45.1 cells and degranulation capacity. No correlation was observed between the proportion of engrafted CD45.1 cells and cell activation as assessed by cell surface CD69 expression. This is because Munc13-4 deficiency does not interfere with immune activation and T cell from Jinx mice are capable of immune activation. Although the overall trends of this experiment are not surprising, what is important is the proportion of degranulation in 15% WT mixed chimera mice is comparable with that of WT mice. This result is consistent with clinical data, 65 and the finding that engraftment of 10–20% WT bone marrow in perforin−/− mice was enough to reduce serum levels of interferon γ (IFN-γ) after LCMV infection, 60 and that a perforin−/− mouse with as little as 8–15% of gene therapy-modified CD8 T cells or >30% gene therapy-modified HSCs is protective against LCMV. 63,66 Although it would be an overreach to definitely say at this point that FHL3 gene therapy requires a minimum of 15% degranulation competent T cell compartment, the existing body of evidence suggests that even a smaller subset of immunocompetent cells can compensate and adequately regulate the immune system. Further in vivo studies are needed to definitely determine the threshold needed for disease phenotype correction.
There were some concerns if the constitutional expression of Munc13-4 in HSPC could impair engraftment. We noted robust engraftment of UNC13D gene-transduced HSPC. By 12 weeks post-transplantation, these mice had normal complete blood counts compared with control Jinx mice that had received one million CD45.1 Sca-1 cells. This indicates that UNC13D gene therapy modification does not negatively affect engraftment potential.
Twenty weeks after transplantation, mice were injected with LCMV Armstrong to induce the FHL3 disease phenotype. 59 For our degranulation assays, we specifically observed changes in CD107a expression within the memory CD8 T cell population (CD44+ and CD8+ cells). Upon stimulation, these cells showed significantly higher expression of CD107a compared with nonmodified LCMV-infected Jinx mice. Although an improvement, CD107a upregulation was less than that observed in WT mice, perhaps as a result of transgene integration into transcriptionally inactive genome sites and low copy number achieved. CD107a expression on cytotoxic cells is an indicator of the restoration of cytolytic ability. 67 Therefore, the restoration of CD107a expression in our degranulation assays indicates successful gene therapy modification and potential for FHL3 disease correction.
It is worth noting that a more clinically relevant gene therapy model would involve first inducing illness in Jinx mice through LCMV infection and then correcting it with HSPC-based gene therapy. Furthermore, comparing the work presented here with previous FHL3 gene therapy mouse studies may be difficult owing to how we elected to infect our mice with LCMV Armstrong rather than the more virulent WE strain of LCMV, which was used in previous FHL3 gene therapy studies and produces a more clinically robust disease model. 23 The LCMV Armstrong strain was used because of the well-described CD8 T cell response in Jinx mice. 59,68 Indeed, we were able to recapitulate previous findings showing a robust CD8 T cell memory response upon LCMV Armstrong infection, and gene therapy improved degranulation of these cells. However, despite the Jinx mice having a fixed degranulation defect, we could not adequately model human FHL3 disease utilizing less virulent LCMV Armstrong strain as an infection trigger. Future studies with either a higher dose of LCMV Armstrong or the use of more virulent WE strain of LCMV or use of a different infectious trigger could better recapitulate features of human FHL3 such as cytopenia, elevated liver enzymes, elevated IFN-γ, sIL2R, and ferritin. Once this is achieved, it will help in the assessment of in vivo disease activity and laboratory parameters correction after HSC-based gene therapy. Also, additional studies such as lineage-specific VCN analysis, deeper immunophenotyping, viral clearance, integration site analysis, and secondary transplant studies could give valuable preclinical data critical for initiating gene therapy studies for FHL3 in humans.
Overall, an UNC13D LV vector was optimized to effectively modify FHL3 patient T cells and hematopoietic stem cells from the FHL3 (Jinx) mice. Furthermore, we showed that having 15% degranulation-competent cells is still sufficient to produce degranulation response comparable with WT. Therefore, a significant body of preclinical data supports the pursuit of autologous HSCT or T cell LV vector gene therapy as a transformative approach to FHL3 disease management or cure.
Footnotes
Acknowledgments
Stocks of LCMV Armstrong were generously supplied by Dr. Rafi Ahmed Lab.
Author Disclosure
The authors declare no conflict of interest.
Funding Information
NHLBI 1K08HL141635-01A1, Atlanta Pediatric Scholars Program K12 Scholar supported by grant K12HD072245 and U54AI082973.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.