
Abstract
Although the sequence of the AAV inverted terminal repeat has been known for 40 years, there are still unanswered questions about functions attributable to the terminal 125 nucleotides.
Barrie and I met at a Gordon Conference on Animal Cells and Viruses in the early 1970s; he had essentially followed me in working with Jim Rose in the Laboratory of Biology of Viruses, NIAID. We both had an interest in the molecular biology of adeno-associated virus (AAV) and subsequently both of our laboratories did a lot of work in parallel on physically mapping the AAV genome using restriction enzymes and developing a transcription map. We even coauthored an article entitled “Genome localization of adeno-associated virus RNA.” 1 We were both studying AAV2 and not a lot of attention had yet been given to the other known serotypes.
Of particular note, we both were interested in the reported ability of AAV to inhibit both adenovirus and herpesvirus cell transformation and oncogenesis. Jeff Ostrove, Donna Duckworth, and I reported that AAV infection reduced the oncogenicity of adenovirus in golden Syrian hamsters; this was correlated with a decrease in the expression of the adenovirus E2B oncogene. 2 At the same time, Barrie and Luis de la Maza published an article indicating that AAV defective interfering (DI) particles that contained just the ends of the AAV genome could inhibit adenovirus oncogenicity. 3
AAV has become the favorite viral vector for gene therapy. Our laboratory has had a longstanding interest in the AAV itr. This has proven to be fortuitous, because the itr is the only part of the AAV genome that is required for the prototypical AAV vector. We have long had detailed knowledge of the sequence, structure, and biological function. Our original data indicated that it was a natural terminal repeat, 4 whereas Jim Rose and Frank Koczot had published data showing that the ends of AAV were an inverted terminal repeat. 5 To reconcile the two conclusions, Hugh Gerry and I suggested that the terminal sequence was a palindrome. This model was subsequently proven to be correct by sequence analysis, although more complex than a simple palindrome.
The terminal 125 nucleotides were palindromic, but the overall palindrome was interrupted by two smaller palindromic sequences, one on either side of nucleotide 63 (the axis of symmetry). When the overall 125 nucleotide sequence was folded on itself, a T-shaped structure formed. Only seven nucleotides were not base paired, three T's at the tip of one of the cross arms and three A's at the tip of the other cross arm, plus either an A or T separating the two internal palindromes. Of note, there were two sequences determined for the terminal 125 nucleotides. 6 This was the consequence of the inversion of the itr during DNA replication. The model for replication invoked the formation of the hairpin structure at the 3′ itr to serve as the primer for DNA replication. (Initiation of DNA replication requires a primer, which must eventually be resolved to restore the original 5′ terminus of the genome, lest there be sequential shortening of the genome with each round of replication. 7 )
Resolution of the now covalent hairpin linking both the primer and progeny strands involves nicking at a point opposite the original 3′ terminus, with subsequent transfer of the original 3′ terminal sequence from the primer strand to become the 5′ end of the progeny strand. The gap left at the 3′ end of the parental strand can be filled by repair synthesis primed by the shortened 3′ parental strand.) (Note: were the terminal sequence a simple palindrome, inversion would yield a sequence identical to the original one; but because of the two smaller internal palindromes, the inversion results in a different sequence. The two sequence orientations are referred to as “Flip” and “Flop.,” as given in Fig. 1).
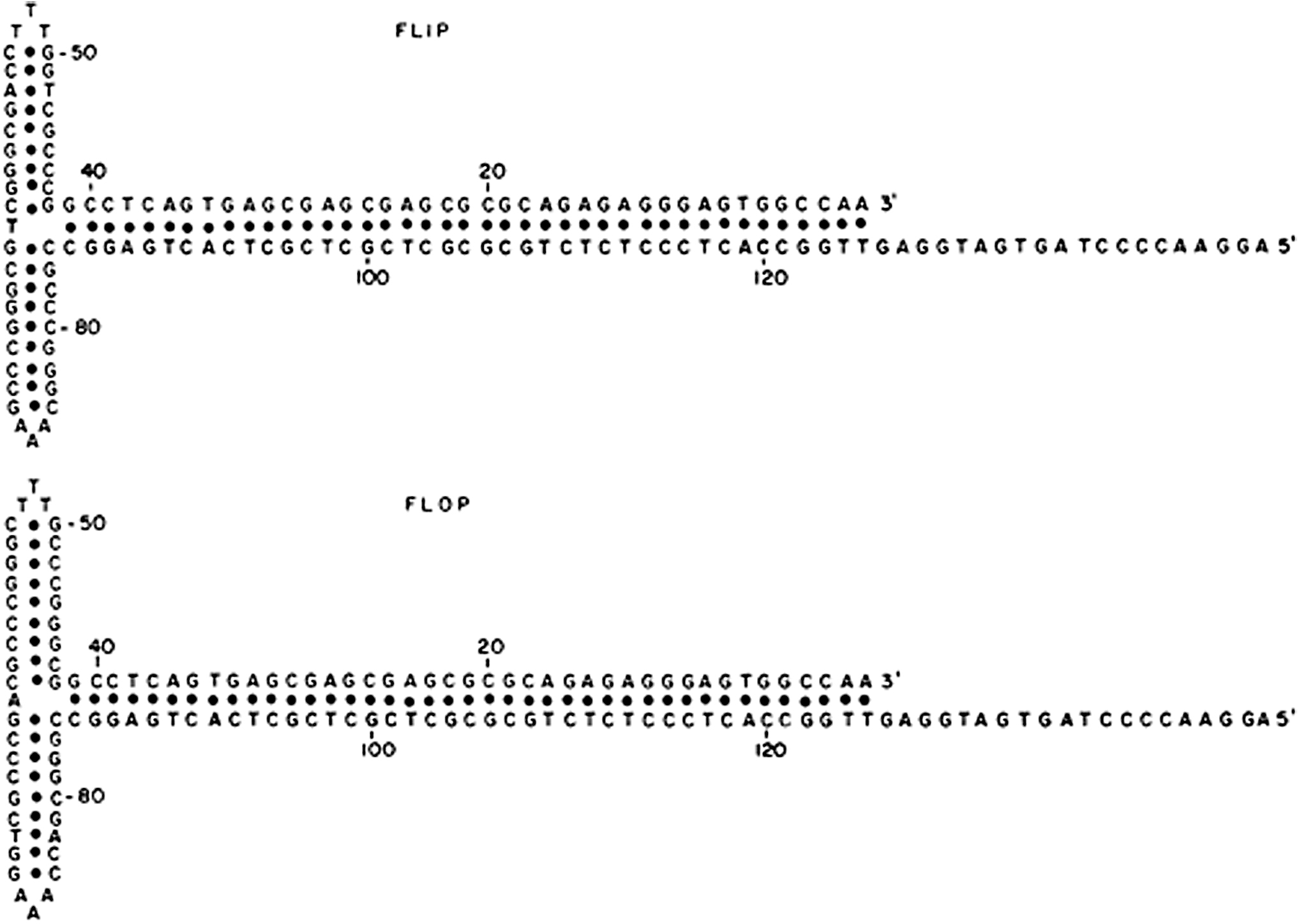
Nucleotide sequences of the inverted terminal repetition in AAV2 DNA. The second sequence (flop) represents an inversion of the first 125 nucleotides. The sequences are represented in the form that contains the maximum amount of self-base pairing. 6 AAV, adeno-associated virus.
The overall itr is 145 nucleotides long. Not only does the itr have interesting physical properties, but the itr and nearby unique internal sequences have numerous biological and regulatory properties, not all of which are even now fully understood. Among these are binding sites for several transcriptional transactivators. 8 –11 The itr has been implicated as being important in both the regulation and priming of AAV DNA replication. 12,13 Conversely, in a nonpermissive milieu the itr functions in cis as a negative regulator of both replication and transcription.
Furthermore, the itr facilitates recombination of the viral genome with the cellular genome. This happens at many sites in the cellular genome in both nondividing and dividing cells and is not dependent on the presence of the large Rep regulatory protein. In dividing human cells in culture, the AAV genome can integrate preferentially at a specific site on chromosome 19q13.4. 14 –18 Preferential integration requires the large Rep protein and the itr and the entire genome and tandem repeats of the genome may be integrated. This occurs in a nonpermissive milieu (no helper virus coinfection). The integrated state has been reported to persist for >100 passages in culture. 19 Rescue of the integrated genome and production of wild-type AAV is induced by adenovirus super-infection of the latently infected cell or by exposure of the cell to other stressful conditions.
In a latently infected transformed cell line in culture, an antibiotic resistance gene carried by the integrated AAV genome continues to be expressed; there is no indication of suppression of gene expression. A similar phenomenon is observed in cells transduced by AAV vectors that persist as extrachromosomal circles. Thus, unlike most other viral vectors for gene therapy, transgenes in AAV vectors continue to be expressed for much longer periods of time. What is it that differentiates AAV vectors from other viral vectors? We do not know with certainty, but a good possibility is that the AAV itr by virtue of its very stable potential hairpin structure is able to assume structural conformation(s) that block protein DNA complex formation(s) that could lead to suppression of gene expression.
Because the AAV itr is able to form a hairpin to serve as the primer for DNA replication, it would seem likely, and indeed has been shown, that replication could initiate from either the right or left end of the duplex form of AAV DNA. If both the left and the right itrs were in the flip orientation, progeny genomes had itrs in both orientations and the orientation at one end of the progeny genome did not seem to determine the orientation of the itr at the other end. 6 One question is whether there is any biological difference between the two itrs. A series of experiments has indicated that the left itr of the duplex form of AAV DNA is dominant. 20 Several experiments have been carried out to see whether the absolute sequence of the itr is required for replication. The cloned AAV genome can be transfected into a permissive cell cotransfected by a helper adenovirus; the cloned AAV genome is rescued from the integrated state and replicated.
If one of the itrs in the cloned genome had a deletion mutation, the mutation was rescued and repaired during replication. 21,22 In other experiments, several nucleotides at the tip of one of the cross arms of the T were replaced by a second sequence that maintained the palindromic nature of the cross arm, the ability of the AAV genome to be rescued and repaired was retained. 23 However, if a nonpalindromic sequence were inserted, the genome could not be rescued and replicated. Thus, conformation of the itr is an important consideration. However, as might be expected, when both wild-type and a viable mutant were simultaneously cotransfected, wild type was predominant.
A more complex result was seen when one itr in the cloned genome had a wild-type sequence and the other had a mutant sequence. 21,22 If the left itr was wild type, all progeny genomes had wild-type itrs at both ends. However, if the left itr was the mutant and the right was wild type, the resulting progeny genomes had a mixture of wild type and mutant itrs. We do not understand the molecular basis for polar difference in the results. Clearly the mechanism of DNA replication and/or rescue is more complex than current models can account for.
Forty years ago, experiments were conducted to characterize nonhomologous recombination between AAV and SV40 viruses. 24 On the one hand, this was a model system to evaluate potential recombination between SV40 and host cell DNA using a receptor genome that could be characterized by the technology available at that time (both the SV40 and AAV genomes had just been sequenced 25,26 ). African Green Monkey cells were co-infected with SV40 and AAV2. After two passages, infectious centers were assayed for cell colonies that contained AAV sequences. Because AAV cannot independently replicate in monkey cells, any colonies that contained detectable AAV sequences must have contained hybrid recombinant SV40/AAV viruses that contained the SV40 origin of DNA replication.
The hybrid SV40/AAV were further characterized to determine which AAV sequences were present. This was of some interest because the assay was not designed to be selective for a particular region of the AAV genome. Contrary to expectations (always a more interesting result), all the AAV/SV40 hybrids were positive for sequences from the right end of the AAV genome. Characterization of the recombinants revealed that about half of them were short tandem repeats of the SV40 origin of replication and ∼200–400 nucleotides from the right end of AAV; about half of the AAV sequence was just inboard from the boundary of the right itr and the rest of the sequence extended about half was through the itr, terminating frequently in the region of the small internal palindromes. 27 Why the left itr was never detected in the recombinants suggested a polar difference most likely associated with the short unique sequence just inboard from the right itr. Further consideration will be given to this issue in the next section.
In 1996, I was asked to speak with a National Institutes of Health committee that had been established on an ad hoc basis to consider whether knowledge about AAV was sufficient to consider using it in clinical trials as a vector for gene therapy. At that moment there was great enthusiasm for starting human trials, but I was hesitant because I thought a lot of fundamental information was still lacking. The information we did have was all positive; first, AAV was not known to be the agent of any human disease; second, we knew the genetic maps and complete DNA sequences, we knew that under some conditions AAV2 could integrate in a site-specific manner into the human genome, but that this required Rep gene expression, and we knew that transgenes carried by AAV vectors continued to be expressed for long periods of time (the life time of a mouse).
We also had a good idea of the requirements for AAV replication, so that nonreplicating vectors could be constructed. However, very little was known about the biology of AAV after infection, either at the cellular level or at the level of an intact host, especially a human. We did not know critical factors about the ability of the vector to gain entry into the nucleus, interaction of the vector with the host immune system, or tissue specificity. Numerous human serotypes had been identified and differences among the serotypes with regard to the issues listed previously had not been defined.
Nevertheless, clinical trials with AAV vectors were begun. Results were uniformly negative. Although the AAV vectors seemed quite safe, expression of the transgenes carried by the vectors occurred at disappointingly low levels. What expression was observed was not long lived. Despite the general notion that AAV is not highly immunogenic, it was found that an immune response was elicited. Despite early indications that AAV2 the prototype could infect multiple tissues, more detailed studies revealed that different serotypes had different tissue distributions after infection and that the route of infection was an important consideration.
Viruses are pathogens, so an important consideration is that the vector does not contain sequences that contribute to pathogenicity. In addition, for safety considerations, generally, it is considered desirable that vectors were not be able to replicate. Over time additional studies on the fate of AAV and AAV vectors after infection enabled modifications in the construction of vectors to the point that AAV vectors have been developed that have enabled apparent cures for several monogenic human diseases, including a form of Leber's disease (congenital blindness), 28 –31 hemophilia (A and B), 32 –36 lipoprotein lipase deficiency, 37 and spinal muscle atrophy. 38
A major reason that AAV has found favor as a vector for human gene therapy is that until recently there has been no known association with human disease. In recent years this conclusion has been called into question. Any viral DNA genome that enters the nucleus has the potential to integrate into the host genome. Indeed, in model systems it has been estimated that AAV vectors recombine at a frequency of 2 × 10−7. 39 However, no adverse consequences have ever been noted in gene therapy trials in humans or nonhuman primates.
There have been several reports that tumors were observed in certain strains of newborn mice infected with AAV or AAV vectors. 40,41 The initial observations have been reproduced and so additional evidence that wild-type AAV or AAV vectors might be oncogenic has been sought. Several years ago, a French group reported that AAV sequences were detectable in liver tissue from some patients who had primary hepatic cell carcinoma that was not otherwise attributable to cirrhosis or chronic infection with hepatitis virus B or C. 42 The original report was confusing because the tumors were not clonal and, in some cases, the AAV sequences were detected in cells apart from the tumor.
Although the original report generated a heated response from several laboratories, 43,44 who were leaders in gene therapy and who disputed whether the data reported supported the notion that AAV vectors might be oncogenic, subsequent studies have supported the original report. Analysis of the AAV sequences present in the liver tissue has indicated the presence of a short sequence (∼250–400 nucleotides) from the right end of the AAV genome. About half the sequence was derived from the itr with a boundary in the itr in the region of the smaller internal palindromes. The other half of the sequence extended from the boundary between the right itr and internal unique sequences for ∼200–300 nucleotides.
In additional studies it was reported that this sequence had a predilection to integrate into regulatory regions of genes characteristically expressed in cells of primary hepatic cell carcinoma. Insertion of the sequence into the regulatory regions induced expression of these genes. 45 Thus, it does seem likely that under the specialized conditions described that AAV might indeed be oncogenic. It should be noted that the identity of the inserted sequences recapitulates the data we reported over 30 years; there is something curious about the right terminus of the AAV genome that predisposes to nonhomologous recombination, or, in other words, what goes around comes around.
At least two questions follow from the above results. The first is a practical consideration; do the results raise serious considerations about the safety of AAV vectors, which up until now have appeared extremely safe? The prototypical AAV vector contains only the AAV itr at either end. Thus, the oncogenic concerns raised by the findings with hepatic cell carcinoma would not seem to be applicable to current AAV vectors. (It may well be pertinent to vectors that contain sequences from the right end.) Of course, the extent to which this conclusion may be overly simplistic clearly needs to be investigated. In cell culture, wild-type AAV infection leads to site-specific integration in transformed cells on chromosome 19q13.4. 17 Colonies with integrated AAV DNA can be detected by expression of a selectable transgene or ability to rescue viable AAV after super-infection with adenovirus.
Characterization of the integrated DNA has shown that the AAV DNA is present as either a head-to-tail or head-to-head tandem repeat. It has been hypothesized that, at the least, slightly more than a full length genome is required for rescue and replication. 19 A functional Rep gene is required for site-specific integration and the target sequence has been identified. 46,47 Site-specific integration occurs by nonhomologous end joining. 48 Of note is the fact that the palindromic part of the itr resembles a Holliday structure invoked as an intermediate in recombination. 49 Junctions between viral and cellular sequences have been mapped within the itr.
A very different picture was seen if recombination between AAV and cellular DNA was assayed by screening for AAV sequences. Integrated sequences were readily detected, most integration did not occur at 19q13.4, but rather at a variety of different sites on the genome, although, there were some more favored sites. 50,51 In some reports, recombination into 19q13.4 had occurred, but represented only a small fraction of the sites detected. Significantly, integrated sequences represented only a small part of the genome, although sequences from the itr were usually present at the junctions with cell DNA. Screening the fate of AAV vectors in animal models of gene therapy revealed that integration of vector sequences was very rare (10−7) and that most vectors were maintained in an extrachromosomal state. 52
Of interest, there have been isolated reports that suggest that AAV vectors used to transduce hepatic cells in vivo seem to persist by integration into the cellular genome. 53,54 This may reflect that persistent expression of the transgene requires integration because of the relatively rapid turnover of hepatocytes.
More recently, investigators have reported that dogs that were injected with AAV vectors to treat hemophilia A did express therapeutic levels of factor 8 for >9 years. 55 When autopsies were performed, evidence for integration of the vector into hepatic cell genomes was found. It is hypothesized that integration of the transgene vector was necessary to maintain expression in the dividing hepatic cells over such a prolonged time. Presumably, extrachromosomal vectors would have been diluted out with time. There was no evidence that the vector was oncogenic, but the evidence of integration does raise the possibility that the vector might integrate into an oncogene or its regulatory region with deleterious effects (Personal communication: D. Sabatino in talk given at ASH meeting 2019). 55 Thus, it was suggested that patients receiving AAV-based gene therapy be followed for an extended period of time beyond the currently recommended 5 years. Candidly, it seems to this author to be a good recommendation for anybody treated with any form of gene therapy or modification.
Are the reports of integration of fragments of AAV different or more frequent than seen with other nuclear DNA viruses. My personal bias is that any piece of DNA, linear, circular, single, or double stranded has a small, but finite chance of recombining with the cellular genome. The palindromic structure of the AAV itr may well confer special biological properties rendering the AAV genome particularly recombinogenic. This seems particularly probable given the results originally seen in the study of heterologous recombination between AAV and SV40.
When asked in 1995 whether knowledge of AAV had progressed to a point where human gene therapy trials were advisable, I had some hesitancy. We have now achieved effective clinical results using AAV vectors; much of this success is directly attributable to fundamental studies on the biology of AAV. What is paradoxical is that the only component of the AAV genome required in the vector particle is the itr, the sequence of which we have known since 1980. Yet we still do not understand the details of its higher order structure or the full range of its biological properties. The beauty of science is that each advance opens up new questions to be resolved.
Footnotes
Acknowledgments
I thank Arun Srivastava for his critical review of, and helpful assistance with, this article.
Author Disclosure
K.I.B. declares no conflict of interests in the publication of this article.
Funding Information
No funding was received for this article.