
Abstract
Recent advances in genome sequencing have greatly improved our ability to understand and identify the causes of genetic diseases. However, there remains an urgent need for innovative, safe, and effective treatments for these diseases. CRISPR-based genome editing systems have become important and powerful tools in the laboratory, and efforts are underway to translate these into patient therapies. Therapeutic base editing is one form of genome engineering that has gained much interest because of its simplicity, specificity, and effectiveness. Base editors are a fusion of a partially deactivated Cas9 enzyme with nickase function, together with a base-modifying enzyme. They are capable of precisely targeting and repairing a pathogenic mutation to restore the normal function of a gene, ideally without disturbing the rest of the genome. In the past year, research has identified new safety concerns of base editors and sparked new innovations to improve their safety. In this review, we provide an overview of the recent advances in the safety and effectiveness of therapeutic base editors and prime editing.
Gene Therapy Background
There are currently more than 6,250 known monogenic diseases, which affect roughly one in every one hundred Canadians and account for 40% of the costs in pediatric hospitals. 1,2 Worldwide, ∼76 million people are affected by monogenic diseases alone and this does not account for cancers and complex diseases that affect hundreds of millions more. Despite our rapidly growing understanding of genetic etiologies, less than 5% of genetic diseases have clinically approved treatments and these often have low therapeutic benefit.
Many of the current treatment options are focused on symptom management of the ongoing disease, are often very expensive, and require long-term, continuous treatment plans. For example, enzyme replacement therapy for lysosomal storage diseases costs >$200,000 USD per year, and it requires lifelong intravenous injections; however, many of the disease symptoms persist. 3 As an alternative, gene therapy is a strategy to treat diseases by either adding new functional copies of a gene into the patient's cells to compensate for the defective gene (gene augmentation) or directly repairing in situ the disease-causing mutation (gene repair). Because these treatments target the root cause of the disease, they limit the likelihood of adverse drug reactions and have the potential for life-long treatment from a single administration of a gene therapy drug.
In the past 10 years, from 2008 to 2018, a total of 41 new treatments have been approved by the FDA for the treatment of genetic diseases. 4 This is an average of 4.1 newly approved treatments per year (range of 1–6 per year). At this rate of implementation, it would take more than 1,500 years to develop treatments for all monogenic diseases. Since the goal of gene therapy is to treat diseases at the DNA level, once a gene therapy strategy has been developed for one disease that is effective, safe, and cost-effective, this strategy could be applied to the treatment of many different diseases with only minor modifications. Recent advances in CRISPR/Cas9 and other gene editors have provided new tools for gene repair that have the potential to directly repair disease-causing mutations in patients.
CRISPR/CAS9 Gene Editing Technology
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) along with CRISPR Associated Protein 9 (Cas9) has quickly become a powerful gene-editing tool in the laboratory. 5 Compared with previous technologies such as TALENs and ZFNs, CRISPR/Cas9 gene editing is much more cost efficient and simpler in design due to the fact that it uses an RNA molecule as the targeting system, known as a guide RNA (gRNA), as opposed to a string of DNA-binding protein motifs. 6 Precision targeting to a specific target sequence relies on homologous DNA–RNA binding and can be achieved by using only a target-specific gRNA and Cas9 protein (Fig. 1a). 5
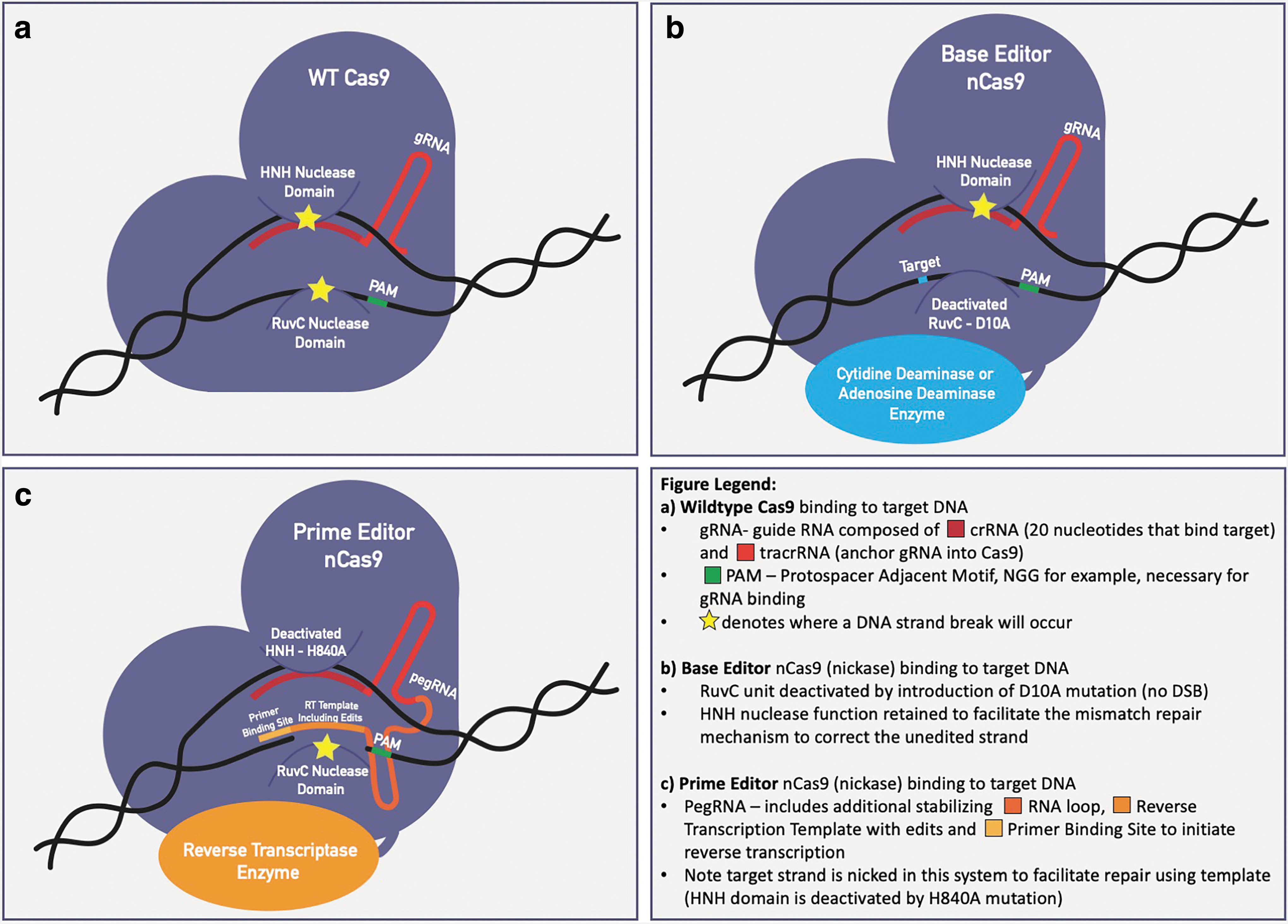
Currently, however, gene therapy applications have been limited due to the low efficiency of homology directed repair (HDR) mechanisms in cells to repair the double stranded break (DSB) generated, without causing insertions or deletions. 5,7 –9 More specifically, the current gene editing technologies have relied on nucleases, such as Cas9, to generate DSBs, which are repaired by either Non-Homologous End Joining (NHEJ), which induces random insertions, and/or deletions at break site (in most cases is not suitable for gene repair) or HDR, which uses a homologous donor template to repair the break, has an extremely low efficiency (often <1%), and is limited to actively dividing cell types. 7,10 –14 The NHEJ primarily results in small insertions or deletions of up to 20 bp (indels); however, recent studies have also observed the formation of large deletions (1.5 kb or more) and complex rearrangements at the target locus after wildtype (WT) Cas9 introduction. 15
Lastly, a major concern for utilizing nucleases to generate DSBs at target locations within the genome is with the possibility of breaks occurring at additional, undesired locations (i.e., off target mutagenesis) that will oftent create indels, which can cause frameshifts or premature stop codons when coding genes are affected.
One novel approach to overcoming the low efficiency of CRISPR/Cas9 gene repair by HDR has been to design a Cas9 with only single-stranded nuclease function (nCas9) fused with a nucleotide-modifying enzyme, such as a deaminase, that is capable of generating a single nucleotide substitution without creating DSBs in the DNA (Fig. 1b). The first generation of base editors used cytidine deaminase rat APOBEC1, which mediates the direct conversion of cytosine to uracil, referred to as the cytosine base editors (CBEs). 16 This approach, first designed by David Liu's Lab at MIT, combines the simplicity and efficiency of CRISPR gene targeting with the DNA editing capabilities of cytidine deaminase. 9,16 –19
More recently, a second formulation of adenine base editor (ABE), capable of substituting adenine for inosine (read as guanine), was developed. 18 Several advantages of ABE are its high efficiency of base editing (>50% has been reported), high product purity, and low rates of indels. This may be because DNA-targeting adenine deaminase is not a naturally occurring enzyme. ABE has the potential to correct almost 50% of all known pathogenic single nucleotide variants (SNVs) and when the CBEs are also considered, these have the potential to target more than 62% of all human pathogenic SNVs by using variants of these two base editors. 18,20 There are currently ∼47,000 known pathogenic single nucleotide polymorphisms reported in Clinvar and as the range of base editors and other technologies expands, this will provide a novel opportunity to directly repair many pathogenic disease mutations. 1,18
Prime editing is the newest approach that has the potential to correct all types of pathogenic single base changes as well as short insertions and deletions (roughly less than 40 bp insertions or 80 bp deletions). 21 This new technology greatly expands our potential to correct pathogenic variants. It is important to note that when prime editing is directly compared with base editing at an optimal site for base editing, it outperforms prime editing in terms of correction efficiency. Therefore, prime editing should be considered as an additional gene editing tool and not as a replacement for base editing. Prime editing, coined “search and replace,” works by fusing a reverse transcriptase enzyme “replace” with the cas9 enzyme “search” and uses an extended length gRNA sequence (pegRNA) as the replacement sequence template (Fig. 1c).
One of the most exciting aspects of prime editing is that it requires precise sequence complementarity in three steps of the editing process, adding additional safety measures to the process. 21 For the Cas9 domain to initially bind the target site and nick the protospacer adjacent motif (PAM) strand, there must be complementarity between the spacer and target sequences. Next, the target site must have complementarity to the primer binding site to initiate pegRNA-templated reverse transcription. Lastly, there must be complementarity between the target site and the product of reserve transcription to resolve the flap created by the cas9 nickase. In each of these steps, if there is a lack of complementarity, successful editing will theoretically not occur.
Another important consideration with prime editing is that it does not require the PAM sequence to sit in as precise of a position as with base editing. This adds considerably more flexibility to the guide design; however, it is important to note that it is still not as flexible as WT Cas9 gene knockouts or HDR guide designs. The PAM still needs to sit within a window near the target mutation, and the position does have an effect on the efficiency. This means that the development of more flexible PAM-tolerating cas9s has a great deal to offer for both base and prime editing technologies, which are discussed next.
One noted safety concern for prime editing has been the observation of the low-frequency insertion of the pegRNA scaffold sequence at the target site, which could occur as a result of reverse transcription extending beyond the intended length. Anzalone et al. observed this event at an average of 1.7% ± 1.5% of total insertion events across 66 experiments in four loci. Similarly, Lin et al. observed on target editing byproducts (mainly scaffold insertions) at 6 of the 21 target sites they tested in rice and wheat, with average byproduct observations ranging from 0.5% to 4.9% at a given site. 22 Although the relative frequency of these events is low, it represents a current safety limitation of the system for therapeutic applications and presents an opportunity to further fine tune this system to be less error-prone.
Lastly, due to the addition of the multiple added components of the prime editing gRNA (i.e., primer binding site length, reverse transcription template length and composition, and flexibility in guide position choice), the design and optimization of prime editing becomes far more complex than that of base editing, where generally one or only a few guides can be tested for a single target. The development of robust, high-throughput, quantitative techniques to test and compare the effects of varying individual aspects of prime editing will greatly improve our understanding of the technologies' editing mechanism and streamline the time-consuming and challenging optimization of this new technology. It will be interesting to see with more studies whether prime editing will hold up to the rigorous scrutiny that base editing has been subjected to in terms of safety and efficiency.
How Do We Define Safety in the Context of Gene Editing Technologies?
Safety and efficacy are on the forefront of nearly every discussion revolving around therapeutic gene editing. Ideally, there should be a global consensus on the thresholds of both safety and efficacy that need to be achieved before CRISPR/Cas9 base editing therapeutics are used in preclinical and clinical trials. By clearly highlighting safety concerns, defining the methods to measure them, and encouraging the open publication of all related safety data, this will help facilitate the development of safe and effective base editing strategies. Discussions of safety and efficacy require the opinions of experts and non-experts from a wide-ranging audience to be weighed and considered. Government officials, health care providers, patients, researchers, and the general public all need to have equal voices in this evolving discussion. A definition of “what is safe” will allow us to better define concrete goals and thresholds to evaluate current and future technologies.
The scientific community is currently divided on what level of safety is appropriate for any technology that has the potential to mutagenize the human genome. In a webinar hosted by The Scientist in May 2019, Dr. David Segal expressed his opinion that even the safest base editor we can possibly imagine, for example, 99% accurate, is still too unsafe to ever be used in therapeutics. 23 He argued that when you consider the context of editing hundreds of thousands of cells in the human body, it is highly likely that some off-target mutation will occur somewhere in a cell in your body. If this is extrapolated to a population-level analysis, the risk of an unfortunate outcome is even greater.
On the other hand, this may fail to take the baseline somatic mutation rate into consideration. Recent estimates using whole exome sequencing suggest that the somatic mutation rate across all cell types averages roughly 50 × the germline mutation rate of 1.1–1.7 × 10–8 per nucleotide site per generation (for base substitutions alone). 24 –29 This broad estimation varies greatly between cell types (likely based on differences in cell division rates), between individuals, and over time. 24,30 –32 Risk factors such as cessation and exposure to asbestos, for example, greatly increase the somatic mutation rate, but in truth likely thousands of environmental factors all contribute varying amounts to fluctuations in this average, estimated baseline rate.
As Dr. Michael Lynch concisely puts it, “few other species willingly expose themselves to environmental mutagens to the extent that humans do.” 24 In fact, the human body is very good at dealing with these mutations. Studies have shown that there is a safeguarding effect to ongoing transcription, which allows cells to maintain low mutation frequencies in actively transcribed regions in response to mutagenesis. 33 –39 We suggest that a goal of reducing the CRISPR/Cas9 mutagenesis rate to levels at or below the estimated somatic mutation rate could be one logical means to define the threshold of safety for a potential gene editing therapeutic.
Another key factor in determining a threshold of safety that needs to be considered is balancing the ethical considerations of duty to non-maleficence, beneficence, and autonomy of patients. Every medical procedure conducted confers a certain risk to patients and yet is performed despite this risk (when the benefits outweigh the risks in the minds of both patients and practitioners). For many debilitating genetic diseases there is no cure, no treatments, and little hope for a positive outcome. For these patients, the potential benefits may outweigh even a higher than ultimately desirable risk. This is precisely why these discussions on safety and efficacy need to include more opinions than just those of the scientific community.
To effectively evaluate the safety of CRISPR/Cas9 base editing, an up-to-date overview of the issues raised in the current literature is required (Table 1).
Base editor variants discussed and important attributes
Y: yes, observed significant increase in off-target effects; N: no significant increase in off-target effects observed compared to background controls; ?: no data available.
ABE, adenine base editor; CBEs, cytosine base editors; PAM, protospacer adjacent motif.
Off-Target Mutagenesis
Detection of all potential off-target effects by CRISPR/Cas9 is currently limited by the high costs of deep sequencing of whole genomes, which is required to detect these very rare events in large cell populations without getting filtered out as sequencing errors. Due to the high costs, many research groups limit their studies to analyzing a subset of sites in the genome with the highest predicted homology to their gRNA sequence. Many different types of software have been developed to facilitate these types of studies, for example: COSMID, CHOPCHOP v2, Cas-OFFinder, CasOT, CRISPRseek, and the more recently created CRISPResso and Crisflash. 40 –46 With so many options, all performing essentially the same task, it is difficult for researchers to select the best tool for their specific needs.
As our understanding has evolved, it has also become apparent that the importance of each nucleotide within the guide sequence varies. 47 In addition, homology is determined by using the human reference genome, which has only recently begun to account for some of the natural variation that occurs between individuals and between ethnic groups. 48 Natural variation is a key factor that will greatly affect the subset of sites that should be sequenced for each individual patient. 49 Lastly, these analyses bias results by only testing the homology of the gRNA sequence, a determinant for the fidelity of the Cas9 component of the system. They do not consider the fidelity of the base editing component, the cytidine deaminase, or adenosine deaminase of current base editors.
Kim et al. recently developed a high-throughput method to analyze the matched and mismatched target activities of spCas9, xCas9, and spCas9-NG by using a library of 26,478 lentivirally integrated target sequences and 78 endogenous target sites in human cells. 50 By utilizing such a large library of target sites, they were able to uncover a number of interesting subtleties about the PAM sequence preferences of each Cas9, uncover nuances in the position effect of mismatches in the guide, and develop new deep-learning models to predict the activity of each at any given target sequence (called: deepxcas9 and deepspcas9-ng).
They investigated the PAM sequence preference of each Cas9 enzyme with an additional 2 nucleotides included downstream (NGGNN) and discovered that although WT spCas9 has no significant preference for any NGGNN, both xCas9 and spCas9-NG had significant and opposite preferences (NGGCN for xcas9 and NGGDN for spCas9-NG). They also characterized new non-NGG PAMs for each of the Cas enzymes included, demonstrating that high-throughput analyses are required to fully characterize the PAM preferences of every Cas enzyme. Through their analysis they re-affirmed the previous findings that xCas9 has a higher sequence fidelity than WT spCas9 and that spCas9-NG is similar to WT.
Despite the differences in overall fidelity, there was an important commonality seen across all Cas9's analyzed: Mismatches in the guide sequence were most tolerated at position 20 (furthest position away from PAM) and gradually decreased closer to the PAM, to a minimum at position 6, after which mismatch tolerance gradually increased again to a maximum at position 1 similar to that observed for positions 10 and 11. This observation suggests that mismatches are better tolerated at positions 1–4 than at positions 5–8 (confirmed by an extensive literature search). This is an important discovery for correctly identifying the most likely off-target candidates not only for a particular guide design but also for designs in which introducing additional mutations is desirable to prevent multiple cas9 cutting events (i.e., for HDR and prime editing techniques, e.g.).
Recently, two papers were published that demonstrated the ability of base editors (BEs) to edit DNA unbiasedly and throughout the genome. 51,52 Zuo et al. used a novel technique, genome-wide off-target analysis by a two-cell mouse embryo injection (coined GOTI) to compare the frequency of SNVs found in the cell progeny of non-injected cells with injected cells from the same embryo. 51 They found that although ABE and WT Cas9 injected embryos had an SNV frequency similar to the baseline mutation rate and no bias toward any particular type of base substitution, the BE3 injected embryos showed a 20 × higher frequency of SNVs, with a strong bias toward G to A and C to T edits (suggesting that the substitutions were induced by cytidine deaminase). 16
They found that none of the off-target SNV sites was shared among the edited embryos, nor did they show high sequence similarity to the gRNA. SNVs were enriched at transcriptionally active loci, particularly in genes with high expression levels (ssDNA is required for APOBEC1 activity). 53 –55 Of the 1,698 total SNVs founds in all BE3 injected embryos (average of 283 per embryo evaluated), 1.53% were found in exons (26 in total) and 0.82% (14 in total) were non-synonymous changes. Of these 26 exonic mutations, 14 affected proto-oncogenes or tumor suppressor genes, a serious concern likely caused by the APOBEC1 bias toward highly expressed genes. Tumor suppressor genes and proto-oncogenes commonly play developmental roles and would be expected to be highly expressed in developing mouse embryos.
Jin et al. used clonally derived primary transformants of rice (Oryza sativa L.), transformed with BE3, HF-BE3, or ABE using Agrobacterium as a means to evaluate genome-wide off-target editing. 52 Overall, they found very similar results to the GOTI experiments: low sequence similarity between the gRNAs utilized and the SNVs found, BE3 and HF-BE3 had a higher frequency of SNVs compared with controls, ABE had a similar frequency of SNVs to the controls, and the SNVs found in BE3 and HF-BE3 samples were significantly biased toward actively transcribed regions when compared with the ABE and control groups.
Based on the results of these two research studies, Doman et al. developed a set of new assays to identify cas9-independent editing by CBEs that are more cost-effective, higher throughput, and less time-intensive. 56
The first assay done in Escherichia coli utilized C•G-to-T•A mutations in the rpoB gene that confer resistance to rifampin, to evaluate the degree of cas9-independent deamination activity for a given CBE based on the frequency of acquired resistance after transfection with a CBE-containing plasmid. At the same time, a second plasmid with a CBE-editable mutation in chloramphenicol acetyltransferase was used to quantify on-target editing based on survival rates with the addition of chloramphenicol. The results of this first assay demonstrated similar levels of Cas9-independent deamination for CDA, APOBEC3A, and APOBEC3B, compared with the original APOBEC1 base editor. 56,57 APOBEC3G and AID base editors, however, exhibited substantially lower Cas9-independent deamination compared with the rest. 56,58
Lastly, they tested a suite of engineered cytidine deaminases, including: APOBEC1 mutants YE1, YE2, EE and YEE, APOBEC1 R33A and APOBEC1 R33A+K34A, APOBEC3A(eA3A), 58,59 and FERNY. 59 –61 The majority of engineered cytidine deaminases tested showed similar levels of rifampin resistance to negative controls: YE1, YE2, YEE, EE, R33A, R33A+K34A, and eA3A (only FERNY was still noticeably higher than control). A second assay was developed to utilize a different set of 5′ base contexts (because of the known different substrate preferences for each cytidine deaminase), by using mutations in the herpes simplex virus thymidine kinase gene as the basis. This assay confirmed the results of the first.
Lastly, they developed an assay in human cells not reliant on whole genome sequencing to avoid the issues of low power to detect rare chance events. To do this they artificially created a “hot spot” for off-target Cas9-independent editing by utilizing the single-strand DNA substrate preference of cytidine deaminase (observed by high levels of editing in actively transcribed areas) and guiding an inactive, dSaCas9 to arbitrary locations in the genome. The binding of dSaCas9 opens an R-loop, exposing the bait: a single-stranded DNA loop, primed for any perpetrating cytidine deaminase to edit. As seen in the previous assays, A3A-BE4 (uses APOBEC3A) showed the highest frequencies of editing in these artificial hot spots.
Using this assay, 14 different cytidine deaminase-BE4max constructs were tested in total and of these R33A-BE4, YE1-BE4, YE2-BE4, EE-BE4, YEE-BE4, and R33A+K34A-BE4 showed the most promise. Both R33A-BE4 and YE1-BE4 maintain the best balance of high on-target and low off-target efficiency. Lastly, they demonstrated that the YE1 fused to an engineered cas9 such as Cas9-NG maintained efficient editing capabilities, expanding the number of targets available to be safely edited in therapeutic contexts.
These reports show that it is imperative to consider biological function, including all potential substrates, of a base-substituting enzyme and devise a custom technique to evaluate the potential off-target effects based on this knowledge. We explore in the next section how this is a critical consideration in base editor design.
Transcriptome Effects
The type of CRISPR system most commonly used by researchers is the type II CRISPR/Cas system using Cas9. Cas9 has evolved to scan dsDNA for homology to the gRNA sequence and PAM (NGG for Cas9), separate the strands locally by creating a RNA:DNA duplex, and create DSB 3 nucleotides away from the PAM site. 5 Cas9 evolved to use only DNA as a substrate, and therefore should not be able to directly affect the transcriptome. However, the APOBEC1 used in the cytidine deaminase base editors were originally characterized for their RNA editing activity. 61 –63 Endogenous expression and overexpression of APOBEC1 has previously been shown to widely edit mRNA in different cell types. 64 –67
Grünewald et al. point out that the base editor used in ABE evolved from the E. coli TadA adenosine deaminase, which functions to deaminate the adenine 34 in the transfer (t)RNAArg2. 61,68,69 The ABE was not evolved to effectively target DNA whereas it specifically avoided binding RNA. 18 In their experiments, Grünewald et al. found that both types of base editors widely affect the transcriptome of edited cells, regardless of the Cas9 variants (high-fidelity or otherwise) fused to them. They used RNA sequencing from total RNA extracted from HepG2 cells that were successfully transfected with BE3 or a negative control nCas9-UGI-NLS (no APOBEC1) to evaluate the extent of RNA editing by cytidine deaminase. 61
They found that 99% of the transcriptome alterations were C to U changes, which were distributed throughout the transcriptome, and that the frequency of specific alterations varied greatly (associated with APOBEC1 motif preferences in RNA). Ninety-eight percent of these alterations showed no evidence of editing in the corresponding DNA sequences. The results were very similar when they analyzed ABEmax.
Next, they evaluated the effects of adding 16 different substitutions in APOBEC1 that were previously reported to reduce RNA editing. 61,70 –74 They identified two variants, BE3-R33A and R33A+K34A-BE3 (“SECURE-BE3 variants”), that were able to significantly decrease the RNA editing ability of the CBEs from tens of thousands to only hundreds and less than 26, respectively, without significantly compromising the efficiency of on-target DNA editing. 61 Thorough structural and functional characterization of all potential deaminases to be incorporated into the base editors would greatly aid in the development and analysis of future base editors.
Zhou et al. also recently analyzed the off-target RNA editing of the CBEs and ABEs. They also found tens of thousands of RNA edits that were induced in cells transfected with CBEs and ABEs. 51 As might be expected, they found a correlation between high expression of the deaminases and higher levels of RNA mutagenesis; and cells with lower expression had much less RNA editing. Zhou et al. used their analysis to test a variant of BE3, BE3W90Y/R126E, that includes two mutations shown to reduce the hydrophobicity and binding affinity for DNA, theoretically increasing editing specificity and reducing RNA activity. 19 They found that RNA off-target editing using this base editor was reduced to the negligible amounts observed in control cells, whereas on-target DNA editing efficiency was maintained.
Next, they evaluated the effectiveness of a base editor that has APOBEC1 replaced with human APOBEC3A (reported to have no RNA binding activity) and observed a significant reduction in RNA editing. 59,75 Lastly, they tested the introduction of two point mutations, R128A and Y130F, into the APOBEC3A component of this base editor and were able to further reduce RNA editing to the baseline levels seen in the control cells. 75 –77 Finally, they sought to engineer an ABE variant with reduced RNA editing as well. They tested two TadA mutants, D53E and F148A, and found that although both variants reduced RNA editing, ABE7.10F148A showed a complete reduction of RNA editing back to baseline. 68,69,78,79 This particular ABE variant warrants rigorous validation, as it has the potential to demonstrate both no RNA editing and no off-target DNA editing.
Grünewald et al. recently expanded on the success of the SECURE-BE3 variants by evaluating the substitution of rAPOBEC1 for one of several different cytidine deaminases. 80 They included base editors fused with APOBEC3A (hA3A), enhanced A3A (eA3A), human activation-induced cytidine deaminase (hAID), and sea lamprey Petromyzon marinus cytidine deaminase (pmCDA1) in their RNA editing evaluations. 59,75,81 Of these base editor constructs, they found that both pmCDA1-BE3 and hAID-BE3 reduced RNA editing to levels comparable to the negative control. Of these two, pmCDA1-BE3 possessed the higher on-target editing efficiency. They coined this base editor construct as “Target-AID.” To compare with their previous two SECURE-BE3 variants: eA3A-BE3, hAID-BE3 and Target-AID all had lower RNA C-to-U edits than BE3R33A, and hAID-BE3 and Target-AID were comparable to BE3R33A/K34A.
To create a novel ABEmax variant, they first removed the E. coli WT TadA monomer (ABE harbors a heterodimer with one engineered variant and one WT), hypothesizing that the WT TadA may recognize and bind its target motif in RNA. Consistent with this hypothesis, they reviewed their previous data and discovered that the most efficiently edited adenines were contained in a CU
Overall, these findings raise the optimism that the problem of off-target transcriptome editing by base editors will be overcome by novel base editor variants and by advancements in high-throughput screens for off-target DNA and RNA editing.
On-Target Mutagenesis: Undesired Editing in Neighboring Nucleotides
Currently, base editors have a roughly 5 nucleotide window of editable DNA within the 20 nucleotide length protospacer (DNA target sequence homologous to the spacer sequence in the gRNA). 16,18,19,82 Within this editing window, all potential target nucleotides can be edited at varying levels depending on their position in relation to the PAM and motif preferences of the deaminases. This poses a particular issue in cases where substitutions in the neighboring nucleotides result in nonsynonymous changes. Researchers have developed a base editor called “BE-plus” that expands this editing window to roughly 12 nucleotides. 83 This editor has important potential applications in gene engineering for generating gene knockouts; however, the utility for gene therapy is more limited because of the increased likelihood of inducing on-target mutagenesis.
Previously, the size of the editing window was optimized by modifying the length of the flexible linker between the Cas9 domain and the deaminase domain. 16 In doing this, they found that both the editing window and the efficiency were altered by the linker length (both reduced with a shorter linker). A 16-amino-acid long linker was a suitable compromise between efficiency and editing window size, and it is currently used in all the base editors. However, more recently, researchers engineered a new, rigid, proline-rich linker for BE3 that reduced the size of the editing window by 40% to positions 14–16, while retaining more than 90% of the editing efficiency. 84 These changes were likely the result of more precise and rigid positioning of the deaminase domain on the target sequence. 84
A second approach to limiting bystander mutations is through utilizing the motif preferences of different cytidine deaminase enzymes; for example, APOBEC3G strictly edits the second C in a CC motif and APOBEC3A preferentially edits TCR motifs (more stringent with the addition of the N57G mutation, eA3A-BE3). 56,58,59 Most recently, Zhang et al., increased the efficiency and editing window of the base editors by adding a non-sequence-specific single-stranded DNA-binding domain from Rad51 between the cas9 and the cytidine deaminase. When combined with eA3A (hyeA3A-BE4max), this creates a highly efficient base editor, with a wide editing window and strict specificity to TCR motifs. 85 These improved base editors are only the beginning but are a very exciting advancement in increasing the safety of CRISPR/Cas9 base editing in terms of on-target mutagenesis.
For the gRNA to bind, a PAM site is also required, and this site determines the target nucleotide's position within the gRNA and, consequently, the efficiency with which it is edited (also the efficiency with which neighboring nucleotides are edited as well). In reality, elimination or relaxation of the PAM site requirement is necessary to go hand-in-hand with narrowing the editing window. Without this, the number of potential targeting sites will be greatly reduced.
Base editors developed from the Cas9s of different species, Streptococcus aureus for example (as opposed to the most commonly used Streptococcus pyogenes, spCas9), have helped expand the range of PAM site sequences that can be targeted. SaCas9 naturally accepts a NNGRRT PAM; however, variants of this Cas9 can also accept NNNRRT PAM sites. 19 Base editors have also been fused to Cpf1 (Cas12a), an alternative Cas protein that possesses several key differences from Cas9: Requiring a T-rich PAM (TTTV), the gRNA is shorter and the nuclease cleavage site is distal as opposed to proximal-like Cas9. 86 It is important to note, however, that the PAM sequence for spCas9, NGG, is the most abundant, non-engineered PAM sequence in the human genome that has been found to date. 19
Hu et al. used Phage Assisted Continuous Evolution (PACE) to evolve a form of the spCas9 base editor, xCas9, that has far greater PAM site flexibility. 82 XCas9 is reported to accept NG, GAA, and GAT PAM sequences to various levels. Since the conception of xCas9, researchers have been able to engineer new Cas9 variants that accept NG PAM sites (SpCas9-NG) and outperform the previous efficiency limitations of xCas9. 87,88 Using sophisticated modifications to the PACE system that originally evolved xcas9, Miller et al. recently evolved three new more flexible PAM-tolerating Cas9s: spCas9-NRRH, -NRTH, and -NRCH. Each of these adds greatly to the number of mutations that cannot just be targeted, but be targeted with maximal efficiency and minimal on-target undesirable editing. 89
These new and promising advancements demonstrate that with dedicated scientists and a fair bit of ingenuity, we can hope to overcome the limitations that are holding back CRISPR/Cas9 base editing therapies.
Recently, Richter et al. evolved a new variant of ABE7.10, coined ABE8e: specifically developed to improve deamination kinetics, a key limiting factor for any ABE fusion to Cas enzymes with decreased resident times on DNA substrates (e.g., this includes: SaCas9, SaCas9-KKH, SpCas9-NG, and CP-Cas9). 90 ABE8e substantially increases the efficiency of adenine base editing with these Cas enzymes (590-fold improved deamination kinetics), increases editing at the boundaries of the activity window, slightly expanding the window for some Cas fusions, and increases processivity (i.e., likelihood of editing multiple adenines within the window) with the drawback of increasing off-target DNA and RNA editing. RNP delivery was shown to dramatically reduce the on-target:off-target DNA editing ratio up to 1,300-fold.
Introduction of the additional mutation, ABE8e(TadA-8e V106W), reduced the levels of transcriptome editing down to similar levels that were observed for ABE7.10, which are still higher than with WT Cas9. The increased deamination dynamics of ABE8e lends extremely well to therapeutic applications where the goal is a short burst of editing followed by rapid degradation of the editor (e.g., RNP delivery with a lipid nanoparticle or other short-lived delivery vessel). The widened editing window and increased processivity, however, may be an issue depending on the specific target sequence and the off-target editing of DNA and RNA will likely need to be reduced for this particular editor, potentially by incorporating the mutations discussed earlier.
New variants such as ABE8e greatly expand the potential of base editing, but they will require refinements to become ultimately safer for therapeutic applications.
CRISPR/CAS9 Immunogenicity Concerns and Potential to Mitigate Risks
Immunogenicity is a significant safety concern for protein-based therapies in that they can trigger unwanted immune responses against themselves. 91 Complications associated with severe immune responses and inflammatory toxicities can be life-threatening and likely more so when we consider treating patients with severe genetic diseases, who may be at higher risk of complications. It is important to recall that in the past the field suffered a major setback when a patient, Jesse Gelsinger, died from an inflammatory response during a clinical trial. 92 Moreover, treatment plans for patients who do develop a severe immune response are likely be more complex, overall less effective, and eliminate the possibility of repeated doses. 91 Early prediction of patients at an increased risk for developing a severe immune response and developing modified treatment plans will be challenging, but this could ultimately be a rewarding approach for many new protein-based therapeutics.
Antibodies against both SaCas9 and SpCas9 have been detected in human blood serum in 78% and 58% of tested study participants, respectively. 93 In addition, anti-SaCas9 T cells and anti-spCas9 T cells have also been detected (78% and 67%, respectively). Other research groups have observed relatively similar results by using different detection methods and different populations. 94 –96 This observation is not necessarily surprising given the commonality of S. aureus and S. pyogenes colonization in the general human population. What is more difficult to determine is what this pre-immunity means for saCas9- and spCas9-based gene therapies.
A very recent study evaluated the therapeutic effect of SaCas9 pre-immunity to an adeno-associated viruses (AAV)-delivered CRISPR therapy targeting the liver cells in mice. 97 In this study, Li et al. pre-immunized mice subcutaneously with SaCas9 protein 1 week before delivering an AAV-CRISPR gene therapy (by intraperitoneal injection). Successful gene editing was detected in the liver in both pre-immunized mice and control (non-pre-immunized) mice; however, those that were pre-immunized also developed a strong cytotoxic CD8+ T cell response. After 12 weeks, the percentage of successfully gene-edited hepatocytes was substantially reduced in pre-immunized mice; characterized by hepatocyte apoptosis, loss of recombinant AAV genomes, and compensatory liver regeneration.
This indicates that pre-immunity to Cas9 may play a significant role in modulating the effectiveness of gene editing. One key note by the authors was that subcutaneous immunization of SaCas9 protein 1 week before therapeutic delivery does not necessarily mimic the pre-immunity observed in the general human population. In addition, this outcome might not occur in non-regenerative tissues, and it thus needs to be explored further. Predicting human immune responses is a major challenge for model organism studies, and ideally new studies in larger model organisms may help to shed light on this issue. However, as Crudele and Chamberlain point out, regardless of the results in model organisms, there should be extreme caution in the therapeutic design and careful selection of target diseases/tissues (the immune privileged eye, e.g.) in the initial human clinical trials for CRISPR/Cas9 therapies. 98
There have been a number of suggestions on how to approach the safety concerns surrounding immunogenicity and CRISPR gene therapies. Immune suppression using corticosteroids, for example, as is already utilized in AAV gene augmentation therapies, may be an effective way to minimize inflammation caused by reactivation or initial development of anti-Cas9 T cells after delivery. 98 In addition, short-term, highly localized exposure to the Cas9 protein may help reduce the chances of a severe immune response being mounted. For example, Li et al. noted a lag in the time before a CD8+ T cell response was detected, suggesting that the prolonged expression of the Cas9 genes on the viral vector may have contributed to the response. 97,98 Li et al. also suggest investigating the adoption of more exotic Cas9 variants (such as those discovered in hot springs, or re-engineering of novel Cas9 variants), which likely would be less immunogenic in humans. 97
Lastly, another suggestion has been to shift the goal toward treating patients at a younger age, when they are less likely to have been previously exposed to S. aureus and S. pyogenes and may not have a fully developed immune system. 93 This is an interesting suggestion in that for many genetic diseases, there are significant benefits in early treatment (e.g., developmental diseases); however, it also poses the added ethical challenges and risks of clinical trials in young children.
Delivery methods also play a pivotal role in considering the potential for immune responses for the development of gene-based therapeutics. For example, pre-existing immunity and de novo immune responses against viral-based delivery methods complicate treatments, reduce accumulation of the gene therapy products at the target tissue site, and may reduce the number of patients who can safely receive the treatment. 99 In light of this concern, many researchers have been inspired to seek novel delivery strategies that may mitigate immunogenicity concerns, increase bioaccumulation of gene products at the target site while reducing toxicity, and support the goal of short, highly effective bursts of gene editing.
Mode of Delivery and Strategies to Reduce the Time That CRISPR is Active in Cells
One way to minimize the chances of mutagenesis caused by gene editing, regardless of the technology used, is to limit the number of cells exposed and limit the time of exposure. Currently, the most commonly used delivery system for any gene therapy technology involving gene editing or gene augmentation uses AAV. 100 –102 As has been previously demonstrated, AAV are capable of prolonged gene expression in vivo. 100 –102 This delivery strategy is ideal for gene augmentation approaches, but it poses a major issue for the application to CRISPR/Cas9 gene therapies. The longer CRISPR/Cas9 is expressed in the body, the greater the chance of an off-target mutagenesis event occurring. Newer strategies for delivering therapeutic mRNA in vivo, such as lipid nanoparticles for example, do not maintain long-term expression and therefore may be more synergistic with CRISPR therapeutic technologies. 103 –105
The form that the CRISPR/Cas9 is delivered in is another important consideration for therapeutic designs, as it directly affects the duration of cell exposure to gene editing. As Zhou et al. noted, the amount of RNA editing they observed increased in cells with high levels of base editor expression. 106 The ideal strategy for many applications would be a very rapid burst of gene editing, quickly followed by degradation of the ribonucleoprotein. This strategy heavily favors delivery of the ribonucleotide protein itself or the mRNA sequence instead of vector-based approaches, in which the CRISPR gene sequence can be actively transcribed for a prolonged period. It is important to note that many of the studies of off-target mutagenesis in DNA and RNA mentioned earlier delivered the base editors in plasmid form. 51,52,61,106 More recently, Doman et al. did include analysis of ribonucleoproteins and observed much lower off-target DNA editing with this form of BE. 56
Another strategy to consider is targeted organ and/or cell-type (target specific) therapeutics and localized administration techniques. These delivery strategies are designed to specifically target only the involved tissues, thereby limiting the number of cells exposed to any potential off-target editing, reducing the concerns about the germline transmission of edited genes, and improving efficacy by concentrating the therapeutic in the affected tissue/organ. This is an active area of research for both viral and non-viral delivery approaches. 107 –112
An exciting novel approach was recently demonstrated by Li et al. by using a semiconducting polymer brush (SPPF) vector for delivery of the CRISPR/Cas9 cassette. 113 This delivery strategy allows for near-infrared window (NIR-II) real-time imaging, and guidance of the complex in vivo and on NIR laser irradiation is capable of inducing localized gene editing at the target destination. Using this technology, the Cas9 can be targeted and visualized in the body, but the laser irradiation acts similar to a remote control to turn editing on when it reaches its destination. This exciting area of research may yield additional novel strategies to help mitigate safety concerns associated with gene editing.
CONCLUSION
Gene editing may provide new therapeutic options for patients suffering from incurable diseases. It is vital that the opinions of patients and the public be considered when deciding how and when to transition CRISPR/cas9 base editing therapies to patients. Establishing the safety criteria for gene editing is complex and requires extensive data, such as next-generation sequencing, to detect on- and off-target editing. In our discussion, we identified four major safety aspects: (1) off-target mutagenesis, (2) on-target undesirable effects, (3) transcriptome effects, and (4) delivery and immunogenicity, which must be assessed to define the specific parameters for safe gene editing. There needs to be a consensus on the thresholds to determine when gene editing is deemed safe. Defining tangible, quantitative measures of safety would provide baseline standards for safe and effective gene editing to help keep patients safe throughout the world.
Footnotes
Authors' Contributions
T.M.C. designed the article. All authors wrote the article. All authors designed and prepared the table. All authors have reviewed and approved this article before submission.
Acknowledgments
The authors thank members of the Ross Lab, Siyue Yu and Jafar Hasbullah, for their helpful discussions and suggestions in the preparation of this article. C.J.D.R. is a Michael Smith Foundation for Health Research Scholar.
Disclaimer
This article has been submitted solely to this journal and is not published, in press, or submitted elsewhere.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by funding from Genome BC Sector Innovation Program, the Nanomedicine Innovations Network (NMIN), the UBC Faculty of Pharmaceutical Sciences, and the UBC VP Research Strategic Research Opportunity funding.