
Abstract
Samples from patients with rare diseases, such as primary immunodeficiencies, are often limited, which hampers careful analysis of the pathomechanisms involved in immune cell dysregulation. To overcome this issue, induced pluripotent stem cells (iPSCs) represent an almost inexhaustible cell source and thus provide an excellent opportunity to generate disease models for rare diseases and to validate new therapeutic approaches. To obtain a better understanding of primary immunodeficiencies associated with the interleukin (IL)-10 signaling pathway, for example, very-early-onset inflammatory bowel disease (VEO-IBD), we generated genetic knockouts (KOs) of IL-10RA (IL-10 receptor α-chain) and IL-10RB (IL-10 receptor β-chain) as well as the downstream targets of the IL-10-receptor (IL-10R) signal transducers and activators of transcription (STAT)1 and STAT3 via an sgRNA (single-guide RNA)-CRISPR-Cas9-expressing lentiviral system. IL-10 signaling-associated KO models and a VEO-IBD patient-derived iPSC clone were differentiated into macrophages for disease models. IL-10R- or STAT3-deficient disease models showed no IL-10-induced BCL3 or SOCS3 expression, whereas lipopolysaccharide (LPS) stimulation induced IL-10R independently of BCL3 and SOCS3 expression. Cytokine secretion profiles from iPSC-derived macrophage disease models showed that IL-10 was involved in many inflammatory cytokine secretions, which indicated formation of both anti- and proinflammatory macrophage phenotypes. Macrophage-secreted cytokines were separated into IL-10R- and STAT3-dependent (IL-6, TNF-α), or into IL-10R-, STAT1-, and STAT3-dependent cytokines (CCL2, CXCL10). Importantly, lentiviral correction restored IL-10-mediated regulation of LPS-induced cytokine secretion in corrected IL-10RB, STAT1, and VEO-IBD patient-derived disease models. Furthermore, treatment of IL-10RB-deficient macrophages with anti-inflammatory small molecules (SB202190, filgotinib) reduced proinflammatory cytokine secretion patterns. Taken together, the described iPSC KO models gave new insights into the pathomechanisms of immune cell dysregulation and served as model systems to test potential therapeutic approaches, including lentiviral gene therapy and targeted small-molecule treatment.
Introduction
In Europe, 2.1
In recent years, it has become increasingly evident that VEO-IBD patients have a genetic defect that inhibits anti-inflammatory interleukin (IL)-10 signaling. 6 The IL-10 receptor (IL-10R) is a heterotetramer consisting of two IL-10R-specific and IL-10 binding IL-10 receptor α-chain (IL-10RA) subunits, and two constitutively expressed IL-10 receptor β-chain (IL-10RB) subunits that are responsible for signal transduction. 11,12 Binding of IL-10 to its receptor triggers anti-inflammatory signaling via IL-10R downstream targets, signal transducers and activators of transcription 3 (STAT3) and suppressor of cytokine signaling 3 (SOCS3). Secretion of chemoattractant chemokines, as well as the activity of monocytes, macrophages, and dendritic cells, is decreased, which leads to reduced secretion of proinflammatory chemokines and promotes the production of anti-inflammatory molecules. 13
In addition to an IL-10 cytokine mutation causative for VEO-IBD, 8 the investigations of Glocker et al. showed that deficiencies in IL-10RA or IL-10RB also led to VEO-IBD. 6 Immunosuppressive and anti-inflammatory therapies were found to be ineffective for many of these patients, and the only curative treatment that successfully restored the anti-inflammatory response (AIR) in an IL-10RB-deficient patient was allogeneic hematopoietic stem cell transplantation, which is an invasive therapeutic intervention.
Besides IL-10R defects, which interfere with the IL-10-mediated anti-inflammatory effect, defects in the IL-10R downstream targets STAT1 or STAT3 are also associated with severe immune diseases. 3 STAT1 deficiencies can lead to immunodeficiency 31A, immunodeficiency type 31B, or Mendelian susceptibility to mycobacterial disease (MSMD). 14 –16 STAT3 stimulates expression of AIR factors (e.g., B cell lymphoma 3-encoded protein [BCL3]) and thus suppresses expression of proinflammatory genes, 17 and mutations in STAT3 can lead to diseases such as HIES1 (hyper-IgE recurrent infection syndrome 1) 18 or ADMIO1 (autoimmune disease, multisystem, infantile-onset 1). 19
Since patient material for rare diseases is often limited, it is difficult to analyze the pathomechanisms in immune cell dysregulation. An interesting solution to this challenge may be induced pluripotent stem cells (iPSCs) as they provide an almost inexhaustible cell source and as they can be obtained by reprogramming somatic cells from patients 20 by the overexpression of the so-called Yamanaka transcription factors (octamer-binding transcription factor 4, sex determining region Y-box 2, Kruppel-like factor 4 and C-MYC). 21,22
Due to their highly proliferative potential and their ability to differentiate into any cell type, iPSC models have great potential for disease modeling and the investigation of new therapeutic approaches. 23 iPSCs have been applied in disease modeling of PIDs, as, for example, Chang et al. showed for patient-derived iPSCs in the severe combined immunodeficiency context 24 or in our group by Dreyer et al. or Klatt et al. for X-linked chronic granulomatous disease. 25,26 Furthermore, iPSCs can be genetically modified by retroviral vectors or alternatively with CRISPR-Cas9 technology, which provides the opportunity to introduce disease-specific mutations. 26,27 Subsequently, a differentiation into disease-affected target cells can be used to test different treatment options, for example, gene therapeutic approaches, or to obtain new information about disease-affected signaling pathways. For in vitro disease modeling of PIDs, and particular IL-10 signaling pathway-associated defects, hematopoietic differentiation into macrophages is important for the analysis of the IL-10-mediated immune response. 28 Hematopoietic differentiation protocols are based on monolayer (2D) or spheroid/embryoid body (3D) cultures. In addition to the differentiation of iPSCs via embryoid body (EB) formation and the myeloid cell forming complex (MCFC) to generate monocytes and macrophages, 29 a new forward programming protocol to produce hematopoietic progenitor cells (HPCs) through endothelial-to-hematopoietic transition is a promising approach to generate disease models. 30
In the present study, we aimed to further improve disease modeling for PIDs, to characterize the IL-10 signaling pathway and associated diseases, as well as to investigate potential treatment options by using an RNA-guided CRISPR-Cas9 system to introduce knockouts (KOs) in the IL-10 signaling pathway-associated IL-10RA and IL-10RB chains as well as in the downstream targets STAT1 and STAT3. To analyze the different IL-10 signaling defects, the iPSC KO models and a patient-derived disease model were differentiated into macrophages. The functionality of the iPSC-derived macrophages was demonstrated and analyzed by phagocytosis ability, phosphorylation, and expression of IL-10R downstream targets (STAT3, pSTAT3, BCL3, SOCS3), as well as impeded cytokine secretion, STAT3 phosphorylation, and induction of gene expression by the disease models. In addition, gene therapeutic approaches were performed by correcting the defective gene with a lentiviral SIN vector during hematopoietic differentiation in selected disease models. As a potential further therapeutic option, we treated IL-10RB-deficient macrophages with anti-inflammatory small molecules (SB202190 and filgotinib) to reduce proinflammatory cytokine secretion patterns of the disease models.
Materials and Methods
Generation of lentiviral CRISPR-Cas9 nuclease and single-guide RNA targeting vectors
With the CCTop program, target sequences for single-guide RNA (sgRNA) were designed for selected genes. The following primer sequences were used for the generation of the corresponding sgRNAs: IL-10RA 1 (5′ CACCGCAGGAGCGCCACTTCATAGC 3′, 5′ AAACGCTATGAAGTGGCGCTCCTGC 3′), IL-10RA 2 (5′ CACCGCTGCGGTAAGGTCATAGGAC 3′, 5′ AAACGTCCTATGACCTTACCGCAGC 3′), IL-10RB 1 (5′ CACCGTTCATTCTGACATTTTCGGG 3′, 5′ AAACCCCGAAAATGTCAGAATGAAC 3′), IL-10RB 2 (5′ CACCGGACTTATAATGTGCAATAC 3′, 5′ AAACGTATTGCACATTATAAGTCC 3′), STAT1 1 (5′ CACCGATTGATCATCCAGCTGTGAC 3′, 5′ AAACGTCACAGCTGGATGATCAATC 3′), STAT1 2 (5′ CACCGTGTAAATGATCATAGACATC 3′, 5′ AAACGATGTCTATGATCATTTACAC 3′), STAT3 1 (5′ CACCGAGAGAACATTCGACTCTTGC 3′, 5′ AAACGCAAGAGTCGAATGTTCTCTC 3′), STAT3 2 (5′ CACCGAGATTGCCCGGATTGTGGCC 3′, 5′ AAACGGCCACAATCCGGGCAATCTC 3′). Sequence-specific oligonucleotides were synthesized by Eurofins Genomics Germany GmbH (Ebersberg, Germany) for the generation of sgRNAs.
Single-stranded DNA-oligonucleotides were phosphorylated and annealed pairwise with the T4 DNA Ligase (New England Biolabs GmbH [NEB], Frankfurt am Main, Germany) buffer in a thermocycler for 45 min at 37°C followed by 2:30 min at 95°C and cool down at 0.1°C/s to 22°C to generate double-chain DNA-fragments (oligonucleotide duplex). The spleen focus forming virus (SFFV) U3 promoter-driven lentiviral SIN vector, which carries a filler for an sgRNA, a Streptococcus pyogenes-derived Cas9, a peptide cleavage site (T2A), as well as a red fluorescent protein (dTomato) (pRRL.PPT.U6.filler-sgRNA.SF.hspCas9-NLS.T2A.dTomato.pre), 26 was digested with BsmBI at 37°C for 45–60 min, and the isolated fragment was ligated with the sgRNA-specific oligonucleotide duplex (1:500 in H2O) for 60 min at room temperature (RT).
Generation of target-specific reporter cell lines
For the generation of lentiviral reporter vectors, the target sequences for IL-10RA, IL-10RB, STAT1, or STAT3 were directly inserted after starting ATG into the open reading frame of a superfolded green fluorescent protein (sfGFP) reporter cassette. Reporter cassettes were amplified using the SFFV U3 promoter-driven lentiviral vector (LV) with an sfGFP and internal ribosome entry site linked puromycin resistance (Puro) cassette
Cloning of lentiviral correction vectors
Codon-optimized (co) STAT1 cDNA was synthesized by GeneArt/Thermo Fisher Scientific. PCR was performed with the co-cDNA as template and specific primers with AgeI (in the forward primer STAT1: CGGATCCACCGGTGCCACCATGAGCCAG) and SalI (in the reverse primer; STAT1: GGGATCCGTCGACTCAGACGGTGTTCAT) restriction sites. PCR was performed, and purified PCR products and vector backbone of an SIN LV carrying an SFFV-driven transgene cassette including GFP (eGFP), were digested with AgeI and SalI and ligated.
For the generation of the IL-10RB correction vector pRRL.PPT.CBX3.EFS.hIL-10RBco.pre, plasmid was digested with AgeI and SalI to obtain co-cDNA of IL-10RB. Co-IL-10RB was ligated in an AgeI/SalI-digested
Creation and verification of IL-10-associated iPSC KO clones
C14E2 iPSCs (reprogrammed commercial newborn foreskin fibroblasts) were transduced with an RNA-directed CRISPR-Cas9 system (pRRL.PPT.U6.IL-10RAsgRNA.SF.hspCas9-NLS.T2A.dTomato.pre, pRRL.PPT.U6.IL-10RBsgRNA.SF.hspCas9-NLS.T2A.dTomato.pre, pRRL.PPT.U6.STAT1sgRNA.SF.hspCas9-NLS.T2A.dTomato.pre, pRRL.PPT.U6.STAT3sgRNA.SF.hspCas9-NLS.T2A.dTomato.pre) with a multiplicity of infection (MOI) of 10. dTomato-positive cells were sorted on FACSAria Fusion (Becton Dickinson) 4 days after transduction. Positive cells were seeded on irradiated C3H embryonic mouse fibroblasts, and single iPSC clones were picked after at least 10 days, genomic DNA of iPSCs was isolated, and PCR was performed for the respective target gene. The PCR product was ligated into a shuttle vector, transformed into competent bacteria, and sequenced by Microsynth Seqlab–Sequence Laboratories GmbH (Göttingen, Germany) to analyze allele-specific mutations. Anonymized patient materials were used following approval by the Ethics Committtee of the Hannover Medical School.
Generation of macrophages by forward programming
The iPSC differentiation in hematopoietic progenitors is based on the forward programming protocol.
30
iPSC models were transduced with
Phagocytosis assay
Macrophages (5 × 104) were seeded and incubated overnight in hematopoietic differentiation medium (RPMI 1640, 10% FBS, 2 mM glutamine, and 1% penicillin–streptomycin, 100 ng/mL hM-CSF). After 24 h, medium was renewed and 20 μL of pHrodo™ Red E. coli BioParticles™ conjugates (dissolved in RPMI 1640 with 2% HEPES; Thermo Fisher Scientific) was added to the cells and incubated for 4 h. After 4 h, cells were analyzed by flow cytometry with the cytoFLEX (Beckman Coulter, Krefeld, Germany) for pHrodo Red indicator (YG585-42-H).
Quantitative (SYBR Green) polymerase chain reaction
RNA of stimulated iPSC-derived macrophages was isolated using the Direct-zol™ RNA Mini Prep Kit (ZymoResearch, Freiburg, Germany) according to the manufacturer's protocol and reverse transcribed with the QuantiTect Reverse Transcription Kit (Qiagen).
SOCS3 (Primer: 5′ GGAGACTTCGATTCGGGACC 3′, 5′ GAAACTTGCTGTTGTGGGTGACC 3′) and BCL3 (5′ CCACAGACGGTAATGTGGTG 3′, 5′ TATTGCTGTGGTGCAGGGTA 3′) expression was quantified by the QuantiTect SYBR® Green RT-PCR Kit (Qiagen) using the StepOnePlus Real-Time PCR System (Applied Biosystems, Darmstadt, Germany). Expression of SOCS3 and BCL3 was evaluated as ΔΔCt relative to expression of a housekeeper gene β-actin (Primer: 5′ CCTCCCTGGAGAAGAGCTA 3′, 5′ TCCATGCCCAGGAAGGAAG 3′).
Western blot
Western blot analyses were performed to detect STAT3, pSTAT3, STAT1, and α-tubulin expression in iPSCs and macrophages.
For analysis of STAT3/pSTAT3 expression in stimulated macrophages, cells were incubated with IL-10 (20 ng/mL; PeproTech GmbH). After 30 min of stimulation, the cells were washed and detached. Cell pellets were stored at −80°C or resuspended in 30–50 μL lysis buffer (50 mM HEPES, 150 mM NaCl, 50 mM NaF, 10 mM Na4P2O7, 10% glycerine, 1% Triton X-100) with 1 μL of Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific). Protein concentrations were determined by Bradford assay (Bio-Rad). Ten to 15 μg protein was loaded on polyacrylamide-gel. PAGE (polyacrylamide-gel electrophoresis) was performed for 1.5 h and the samples were blotted onto nitrocellulose membranes for 1.5 h at 4°C (400 mA).
Membranes were blocked for 1 h at RT with 5% bovine serum albumin (PAA Laboratories GmbH) or 3% milk (Carl Roth GmbH+Co. KG, Karlsruhe, Germany) and stained for STAT3 (1:2,000; Cell Signaling Technology®), pSTAT3 (1:1,000; Cell Signaling Technology), STAT1 (1:1,000; Cell Signaling Technology), or α-tubulin (1:10,000; Abcam) overnight. Secondary antibody stain (goat-anti-mouse 1:5,000, goat-anti-rabbit 1:5,000; Abcam) was performed with peroxidase-conjugated antibodies according to the manufacturer's description 1 h at RT. Protein bands were detected by the SuperSignal West Pico Chemoluminescent Substrate (ThermoFisher Scientific) with a FusionFX instrument (Peqlab GmbH).
Quantification of cytokine secretion by multiplex assays
Macrophages were seeded on day 21 of culture onto adherent plates for Bio-Plex (5 × 104 cells/96-well) analysis of cytokine secretion on day 22 upon 6 h of IL-10 (100 ng/mL), lipopolysaccharides (LPS; 100 ng/mL), or IL-10 and LPS stimulation. Supernatants were collected for multiplex analyses and stored at −20°C until use. Medium supernatants were analyzed for the presence of 27 different proteins, that is, cytokines, chemokines, and growth factors with the Bio-Plex Pro™ Human Cytokine 27-plex Assay Kit (Bio-Rad). Bio-Plex analysis was performed according to the manufacturer's protocol.
Correction of KO cell lines during hematopoietic specification with an LV
HPCs (CD34+/CD45+) were harvested on day 11 of hematopoietic differentiation and transduced with the LVs
MTT assay
iPSC-derived macrophages were cultured for 24 h with SB202190 (5, 10, 50 μM; Axon MedChem, Groningen, Netherlands) or filgotinib (20, 100, 1,000, 5,000 nM; MedChemExpress Sollentuna, Sweden). MTT reagent (Sigma-Aldrich) was added (1:4) to the cell medium (RPMI, 1% penicillin–streptomycin, 10% FBS, 100 ng/mL M-CSF) with the corresponding inhibitor, and cells were incubated for 3 h. One hundred microliters of SDS (sodium dodecyl sulfate; 20%) was added, and cells were incubated overnight on a shaker at RT. Detection was performed at 540 and 700 nm with the ELISA plate-reader SpectraMax 340PC (Molecular Devises, San Jose).
Small-molecule treatment of IL-10RB disease models
iPSC-derived macrophages were stimulated for 6 h with LPS (100 ng/mL) and filgotinib (5,000 nM) or SB202190 (10 μM). Supernatants were collected and analyzed by multiplex analysis. Further methods can be found in the Supplementary Data.
Results
Generation of IL-10 signaling pathway-associated disease models with an RNA-guided CRISPR-Cas9 system
To generate IL-10-associated KO iPSC models, different targets from the IL-10 signaling pathway were selected. Targets were either associated with difficult-to-treat diseases or with important downstream functions in the immune response. Since it was described that defects in IL-10RA and IL-10RB can lead to VEO-IBD, both receptor chains were selected as targets for the generation of KO iPSC models. 6 STAT1 and STAT3, direct downstream targets of the IL-10R, were also selected for the generation of KO models, as they are associated with diseases of the immune system and regulate important functions of the immune response.
Specific sgRNA-directed CRISPR-Cas9 LVs were created to generate IL-10RA, IL-10RB, STAT1, and STAT3 KO iPSCs (Supplementary Data). Two specific sgRNA sequences were generated for each target with the CCTop—CRISPR-Cas9 target online predictor. 31 Those sgRNAs that were predicted to have few off-target sites and to bind the specific gene in the 5′ region of the exon were integrated into a lentiviral CRISPR-Cas9 vector system. A dTomato fluorescence protein cassette linked via a 2A target site to the Cas9 coding sequence was included to detect the LV expression (Fig. 1A). Effectiveness of the different sgRNAs was determined by the transduction (MOI of 0.3) of a target gene-specific reporter cell line (Supplementary Fig. 1A).
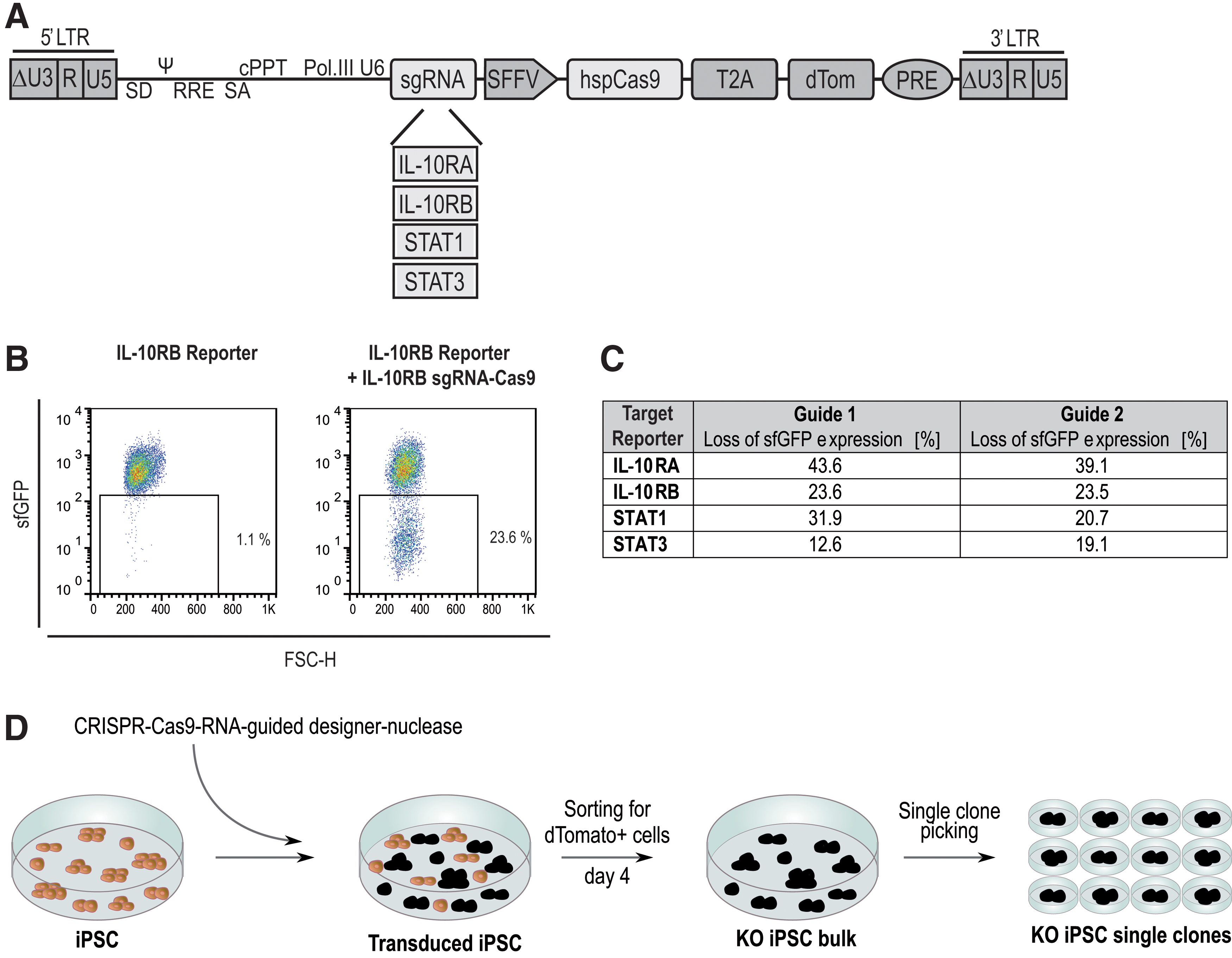
Generation of KO iPSCs by CRISPR-Cas9 RNA-guided designer nucleases.
The KO efficiency of individual sgRNAs was estimated by the guided CRISPR-Cas9 nuclease-mediated loss of the reporter cell lines sfGFP fluorescence signal (Fig. 1B and Supplementary Fig. 1B). After transduction with the different sgRNAs, the loss of the fluorescence signal was measured. The reporter cells showed fluorescence signal losses between 12.6% and 43.6% (Fig. 1C). The sgRNA with greater potential was used to generate iPSC KO models. Individual KO clones were generated from a human C14E2 iPSC clone (Fig. 1D and Supplementary Fig. 2), which was derived from healthy human fibroblasts and was transduced with the sgRNA-directed CRISPR-Cas9 vectors.
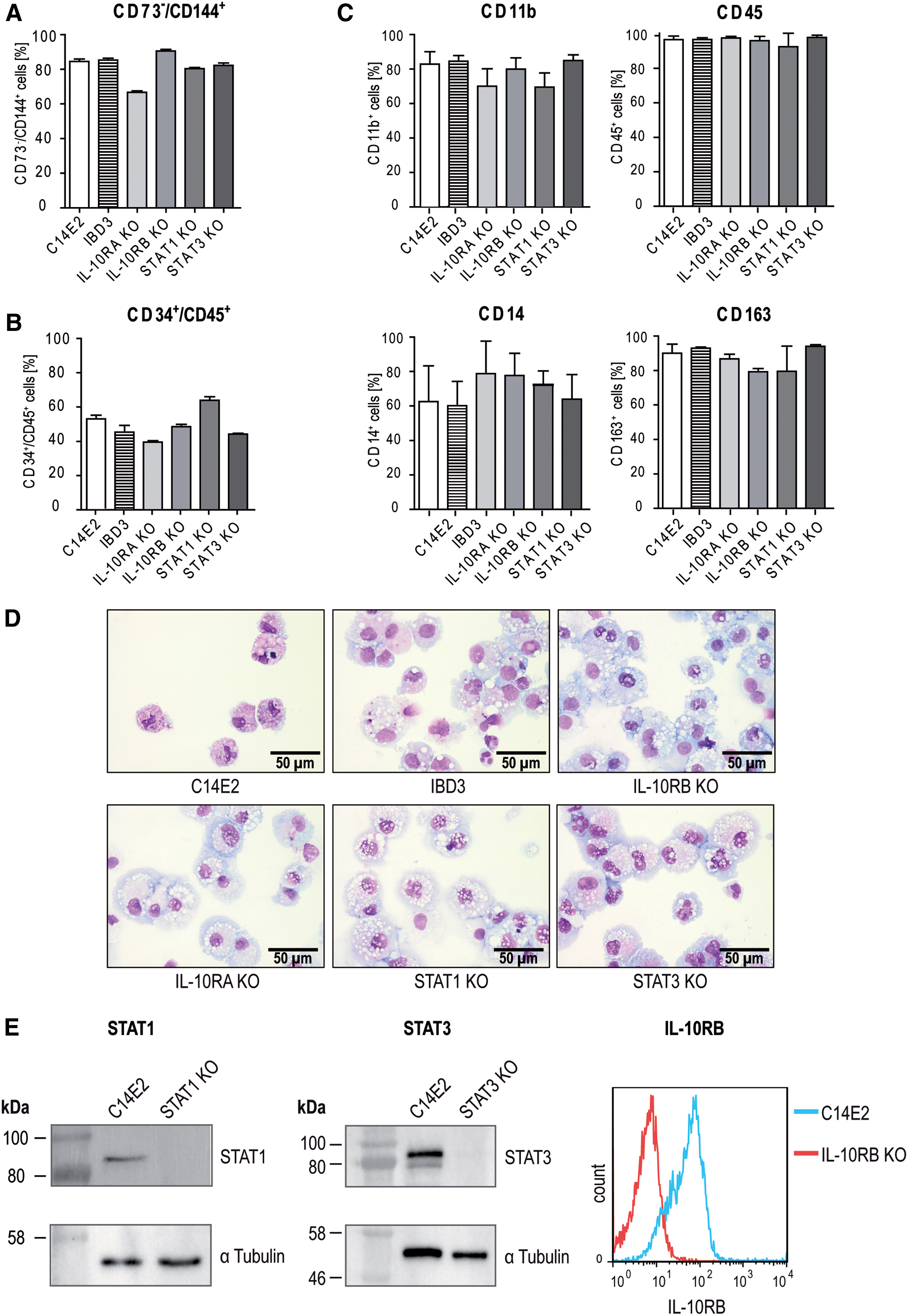
Differentiation of iPSC disease models into macrophages.
Transduced cells were sorted for the dTomato signal of the LVs 4 days after transduction. Single clones were picked from the dTomato-positive iPSC bulk population. To characterize the two allele-specific CRISPR-Cas9-induced mutations by sequencing, PCR on the gene of interest was performed, followed by subcloning of the PCR product into a shuttle vector. To generate the STAT3KO clone, two transductions with two different sgRNAs were necessary to create two different nonsense mutations (Supplementary Fig. 3A).
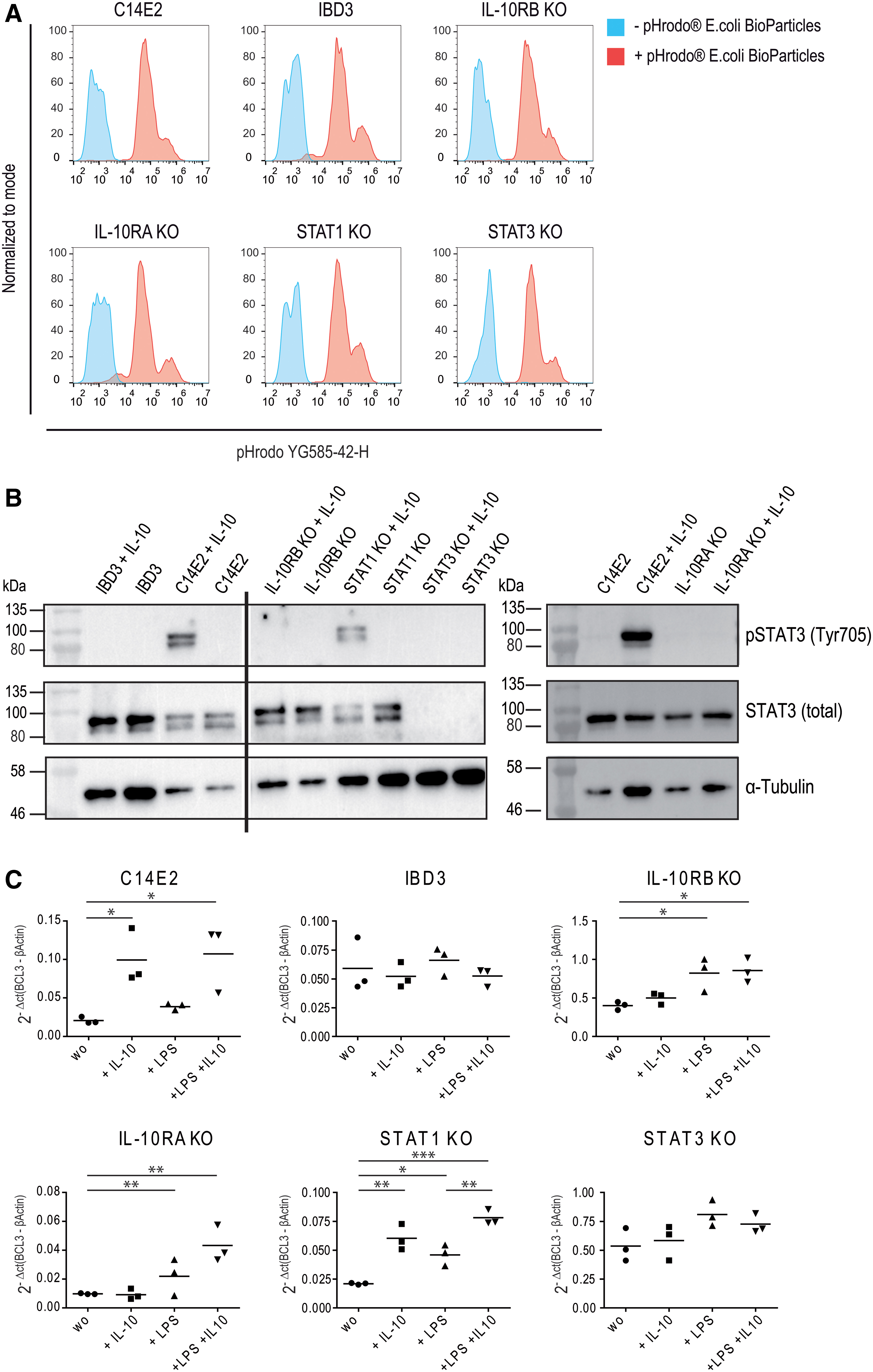
Investigation of phagocytosis ability, STAT3 phosphorylation, and expression of IL-10R downstream target BCL3 in macrophage disease models.
Differentiation of iPSC disease models into functional macrophages
To produce large amounts of hematopoietic cells, a forward programming system based on inducible and regulated overexpression of the transcriptional regulators SCL, LMO2, GATA2, and ETV2 was used. 30 The expression of these transcriptional regulators strongly improves the generation of hemato-endothelial progenitor cells (HEPs) by gradual hemato-endothelial differentiation and HPC through endothelial-to-hematopoietic transition. The established KO iPSCs were transduced with the LVs of the forward programming system and during hemato-endothelial differentiation from day 0 to 7 (Phase I), HEPs (CD73−/CD144+) were produced (Fig. 2A and Supplementary Fig. 4A).

Cytokine secretion of IL-10 pathway-associated disease models.
This was followed by endothelial-to-hematopoietic transition and the production of hematopoietic progenitor CD34+/CD45+ cells from day 7 to 11 (Phase II), as shown by flow cytometric analyses on day 11 (Fig. 2B and Supplementary Fig. 4B). We further improved the previously described protocol by an additional monocyte and macrophage specification phase (Phase III) from day 11 to 15, and obtained fully mature macrophages (CD45+/CD11b+/CD14+/CD163+) after terminal differentiation (Phase IV, day 15 to 22) (Fig. 2C and Supplementary Fig. 5).
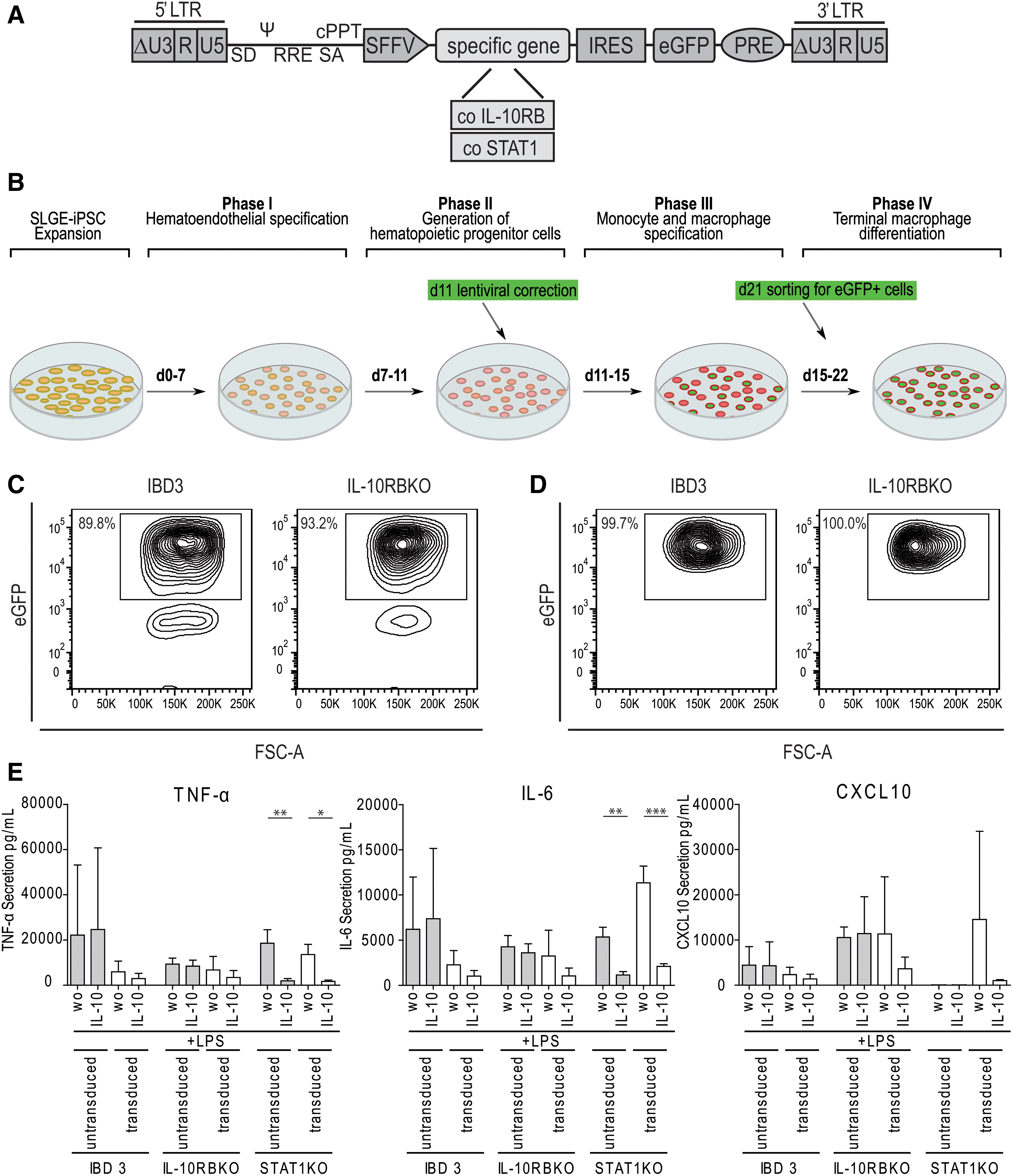
Lentiviral correction of iPSC-derived disease models.
Pappenheim staining of the iPSC-derived CD45+/CD11b+/CD14+/CD163+ cells demonstrated typical macrophage morphology (e.g., vacuolated cytoplasm) (Fig. 2D, Supplementary Data). Furthermore, the vector copy number (VCN) of the RNA-guided CRISPR-Cas9 vector was determined in the individual KO clones (Supplementary Fig. 3B). Although the VCNs were different in iPSC KO clones, no major differences were found in the following vector expression analyses. Quantitative polymerase chain reaction (qPCR) analyses with primers to detect dTomato showed only minimal residual expression of the Cas9 vector in iPSCs and during hematopoietic differentiation (Supplementary Fig. 3C, D), which indicated efficient silencing of the SFFV U3 promoter-driven Cas9 vector.
Next, we verified the generated KOs of IL-10RB, STAT1, and STAT3 on the protein level in iPSCs and differentiated hematopoietic cells. Western blot analysis showed that the STAT1 protein could not be detected in STAT1KO iPSCs, and that the STAT3 protein could not be detected in STAT3KO iPSCs in comparison with healthy C14E2 cells. IL-10RBKO cells were analyzed by flow cytometry on day 14 during hematopoietic differentiation, and respective hematopoietic cells showed a strongly reduced IL-10RB expression in comparison with hematopoietic cells derived from the healthy C14E2 control (Fig. 2E).
KO iPSC-derived macrophage disease models showed functional phagocytosis and KO-related dysregulation in the IL-10 signal pathway
To analyze the functionality of the different iPSC-derived KO macrophages and to determine the influence of the KOs on phagocytosis ability, macrophages were incubated with pH-sensitive pHrodo Red E. coli BioParticles conjugates. Within phagosomes, the pHrodo Red indicator (YG585-42-H)-conjugated particles become fluorescent due to the acidic pH value and indicate functional endocytosis and phagocytosis. After 4 h of incubation, flow cytometric analyses showed successful phagocytosis and a comparable uptake of the particles for all iPSC-derived macrophages, including the macrophages generated from the healthy iPSC clone (C14E2), a VEO-IBD patient clone (IBD3) with a nonsense mutation in the IL-10RB gene, and all KO settings (Fig. 3A).
Several anti-inflammatory processes in the IL-10 signaling pathway are regulated by STAT3, whose phosphorylation and activation are initiated by the binding of IL-10 to IL-10R. To characterize STAT3 functionality in the different disease models, the macrophages were stimulated with IL-10, and the STAT expression and phosphorylation were investigated by Western blot. IL-10-stimulated IL-10R-deficient macrophages showed STAT3 expression but no STAT3 phosphorylation. As expected, STAT3 was not detected in STAT3KO macrophages. In contrast, C14E2 and STAT1KO macrophages expressed the 79 and 86 kDa STAT3 isoforms, both of which were efficiently phosphorylated after IL-10 stimulation (Fig. 3B). Taken together, the iPSC-derived KO macrophages maintained functional phagocytosis but showed the expected deficiencies within the IL-10R/STAT3/STAT1 pathway.
IL-10R- and STAT3-dependent BCL3 and SOCS3 expression in KO iPSC-derived macrophages
Next, we investigated anti-inflammatory downstream factors in our disease models. BCL3 and SOCS3 suppress the production of proinflammatory mediators such as TNF-α and are crucial for the AIR. 32,33 Both genes are regulated by STAT3 phosphorylation after IL-10 induction. To investigate how the different KOs affect IL-10R downstream signaling, the expression of the anti-inflammatory mediators BCL3 and SOCS3 was examined. The disease models were stimulated with IL-10, LPS, or both, and BCL3 and SOCS3 expression levels were determined using qPCR. IL-10 led to a significantly higher expression of BCL3 and SOCS3 in the healthy control (C14E2) and STAT1 KO macrophages, whereas IL-10R- or STAT3-deficient macrophages showed no induction of BCL3 (Fig. 3C) or SOCS3 (Supplementary Fig. 6) RNA after IL-10 stimulation. Interestingly, LPS clearly induced the expression of BCL3 in IL-10RAKO, IL-10RBKO, and STAT1KO iPSC-derived macrophages and SOCS3 expression independently of IL-10R in the iPSC-derived macrophages. This indicated that IL-10- and not LPS-mediated BCL3 and SOCS3 expression was IL-10R- and STAT3-dependent.
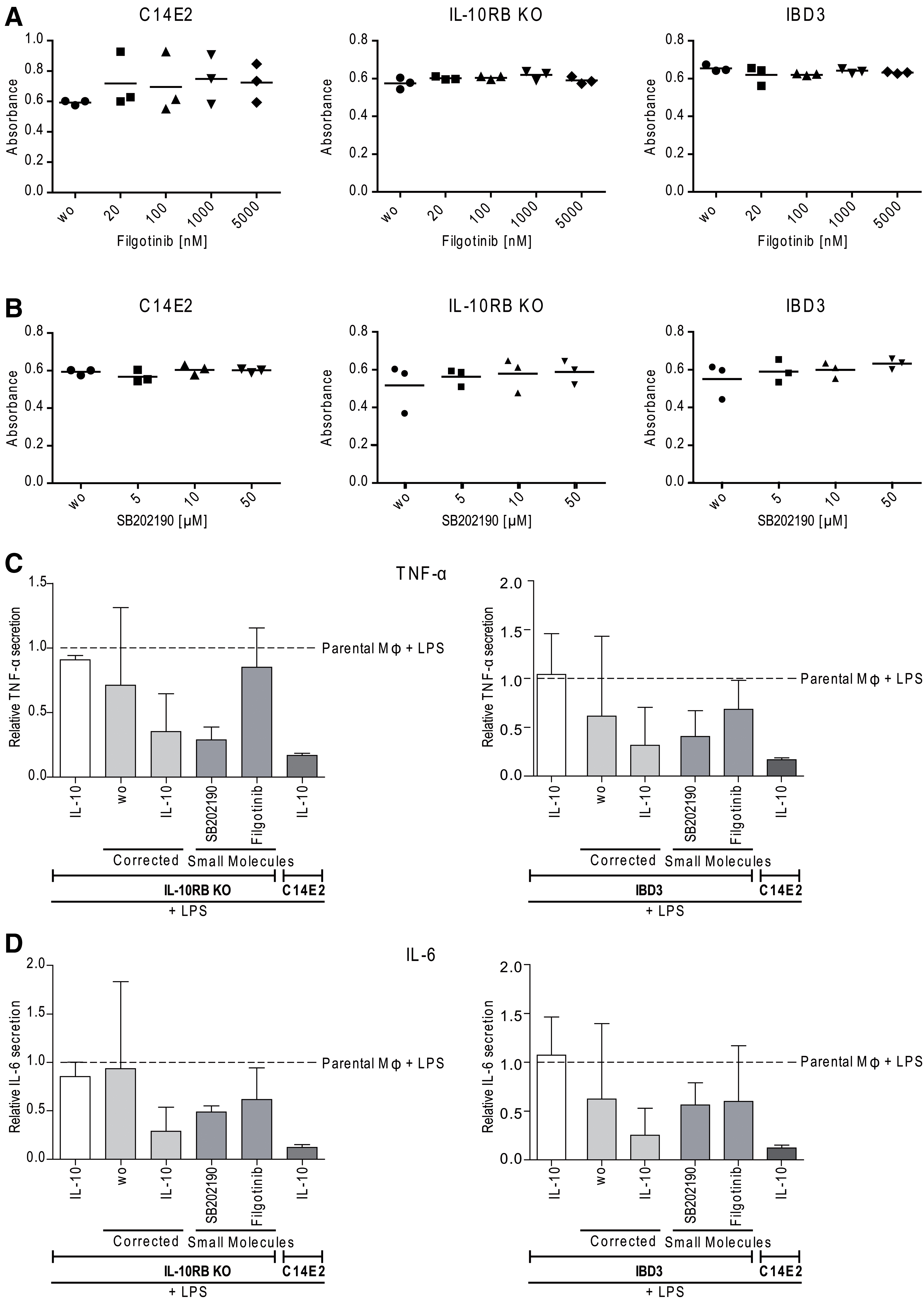
SB202190 and filgotinib treatment of IL-10RB disease models.
IL-10R- and STAT3- or IL-10R-, STAT1-, and STAT3-dependent cytokine secretion in IL-10 signaling pathway-associated disease models
To further characterize the generated KO macrophage disease models, inflammatory cytokine secretion was analyzed after 6 h of IL-10, LPS, or IL-10 and LPS stimulation by multiplex immunoassays. The medium supernatants of the macrophages were investigated for secreted immunomodulatory cytokines (CCL2, CCL3, CCL4, CCL11, CXCL8, CXCL10, FGFβ, G-CSF, GM-CSF, IFN-γ, IL-1β, IL-1RA, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12[p70], IL-13, IL-15, IL-17A, PDGF-bb, CCL5, TNF-α, VEGF) that are involved in anti-inflammatory (e.g., IL-1RA, IL-4, IL-10, IL-13) or proinflammatory (e.g., CXCL10, IL-1β, IL-2, IL-6, IFN-γ, TNF-α) immune responses.
To simplify visualization, all data are summarized in a heatmap, which clearly shows a lack of cytokine stimulation in unstimulated macrophages. In contrast, stimulation with LPS induced secretion of almost all analyzed cytokines (21/27). Interestingly, the addition of IL-10 did not suppress LPS-induced cytokine secretion in IL-10R- or STAT3-deficient, as well as in the patient (IBD3) macrophage disease models (Fig. 4A). In more detail, the LPS-induced secretion levels of, for example, TNF-α and IL-6 were significantly reduced in healthy and STAT1KO clones by the addition of IL-10. This effect seemed to be IL-10R and STAT3 dependent as no IL-10-induced reduction was detected in IBD3, IL-10RBKO, IL-10RAKO, or STAT3KO macrophages. This effect was also seen for IL-2, CXCL8, GM-CSF, and CCL5 (Fig. 4B and Supplementary Fig. 7).
The second group of LPS-induced cytokines, which are IL-10R, STAT1, and STAT3 dependent, was identified by multiplex analysis. These include IL-4, IL-5, IL-9, IL-15, IL-17A, FGFβ, IFN-γ, CCL2, CXCL10, and VEGF (Fig. 4C and Supplementary Fig. 7).
VEO-IBD-associated findings described increased proinflammatory cytokine secretion (IL-6, CCL5, TNF-α) upon LPS stimulation, which could not be decreased by IL-10 stimulation. 6 The analyses of our VEO-IBD-associated KO models (IL-10RAKO, IL-10RBKO) reflected these findings as the LPS-induced secretion of, for example, IL-6, CCL5, and TNF-α could not be regulated by additional IL-10 stimulation. In contrast to this, in our healthy macrophages (C14E2), all investigated cytokines were induced in an LPS-dependent manner and additional IL-10 stimulation suppressed the secretion of most cytokines. Moreover, our macrophage disease models showed that some cytokines in our model are IL-10R and STAT3 dependent, but also indicated STAT1 as an important factor in the IL-10-mediated immune response, which seemed to be necessary for control of several proinflammatory cytokines.
Reconstitution of the immune response by lentiviral gene correction of the KO iPSC-derived macrophages
After a distinct immunological dysregulation was detected in the generated KO-settings, we aimed to investigate therapeutic options. As novel therapeutic approaches for PID associated with the IL-10 signaling pathway are highly desirable, genetic correction by LVs was tested in the KO models. Since IL-10RB defects are associated with VEO-IBD and STAT1 defects with MSMD, we generated an LV harboring the codon-optimized form of IL-10RB or STAT1, and an eGFP fluorescence signal (Fig. 5A). On day 11 of the hematopoietic differentiation protocol, hematopoietic cells were transduced with the specific lentiviral gene-therapy vector (Fig. 5B). HPCs were further differentiated into macrophages and sorted for the eGFP signal of the LV on day 21 (Fig. 5C). All eGFP+-sorted macrophages expressed the correction vector with eGFP in the reanalysis (Fig. 5D).
We examined the influence of lentiviral correction on the cytokine secretion level, and corrected disease models were compared with IBD3, IL-10RBKO, and STAT1KO macrophages after LPS or IL-10 and LPS stimulation. The LPS-induced secretion of the proinflammatory cytokines TNF-α, IL-6, and CXCL10 was lower in LV-corrected IL-10RB-deficient macrophages after LPS stimulation than in the respective uncorrected IBD3 disease model. Furthermore, the cytokine secretion was reduced by additional IL-10 stimulation in IL-10RBKO and IBD3 iPSC-derived macrophages (Fig. 5E). The IL-10-mediated reduction of LPS-induced cytokine secretion was also seen for IL-1β, IL-2, IL-4, IL-5, IL-9, IL-15, IL-17, FGFβ, G-CSF, GM-CSF, IFN-γ, CCL2, PDGF-bb, and VEGF, but no effect on IL-1RA, IL-7, IL-12(p70), IL-13, CCL11, and CCL3 secretion was detectable (Supplementary Fig. 8). Also, LV correction of the STAT1 disease model restored CXCL10 secretion upon LPS stimulation (Fig. 5E).
In general, multiplex analyses revealed the reconstitution of the anti-inflammatory immune response by correction of IL-10RB and STAT1 deficiency. Furthermore, IL-10 stimulation of lentivirally corrected IL-10RBKO and IBD3 macrophages led to reduced LPS-induced secretion of many proinflammatory cytokines.
Reduction of anti-inflammatory immune response by SB202190 or filgotinib treatment of KO macrophage disease models
Small molecules are an alternative treatment option to alleviate the symptoms of severe inflammation of the intestine. We tested two small molecules, SB202190 and GLPG0634 (Filgotinib), for their capacity to reduce the proinflammatory immune response in the IL-10RB disease models. SB202190 can inhibit p38 mitogen-activated protein kinase (MAPK) and therefore suppress TNF-α expression. 34 Filgotinib impedes inflammatory cytokine secretion by inhibiting Janus kinases JAK1, JAK2, JAK3, and TYK2 in a dose-dependent manner. 35 The effects of the different inhibitors were investigated on C14E2, IBD3, and IL-10RBKO macrophages, to analyze and directly compare the patient-derived macrophages with the corresponding KO model and healthy control.
First, an MTT assay was performed with the disease models to exclude that the small molecules may affect cell viability. Treatment of macrophages with different SB202190 or filgotinib concentrations demonstrated no influence on macrophage viability with the tested doses (Fig. 6A, B).
Next, the effects of SB202190 and filgotinib on the inhibition of inflammatory mediators were investigated by multiplex analyses. We observed reduced proinflammatory TNF-α, IL-6, and CCL5 secretion when using SB202190. Filgotinib also markedly reduced IL-6 secretion from healthy C14E2 macrophages similarly to SB202190, but did not alleviate proinflammatory IL-6 or TNF-α secretion in IBD3 and IL-10RB KO macrophages (Fig. 6C, D). However, a strong reduction of CCL5 secretion was observed by the treatment with filgotinib (Supplementary Fig. 9). Also, stimulation with SB202190 or filgotinib resulted in markedly reduced secretion of CXCL10, IL-1RA, IL-10, IL-13, and IFN-γ in the disease models. Furthermore, SB202190 reduced IL-2 secretion, and filgotinib reduced CCL2 secretion.
Moreover, it became evident that SB202190 and filgotinib had no effect on LPS-induced secretion of the cytokines IL-1β, IL-4, IL-5, IL-7, IL-9, IL-12, IL-15, IL-17, FGFβ, G-CSF, GM-CSF, CCL3, PDGF-bb, or VEGF in C14E2- or IL-10RB-deficient macrophages (Supplementary Fig. 9). Overall, the treatment of IBD3 and IL-10RBKO macrophage disease models with filgotinib or SB202190 reduced the secretion of tested inflammatory cytokines. Whereas filgotinib downregulated the secretion of CCL5 more efficiently than SB202190, SB202190 reduced the secretion of, for example, TNF-α (in IBD3 macrophages 6.7 times, in IL-10RBKO macrophages 3 times) and IL-6 (in IBD3 macrophages 1.5 times, in IL-10RBKO macrophages 1.9 times) and showed anti-inflammatory effects on more cytokines than filgotinib.
By comparison of the effectiveness of gene therapy approaches with small-molecule approaches, we saw that lentiviral correction and SB202190 equally reduced TNF-α and IL-6 secretion in IL-10RB-deficient macrophages. In contrast, stimulation with filgotinib only slightly reduced IL-6 secretion and appeared to be ineffective with regard to TNF-α (Fig. 6C, D). Concerning CXCL8 secretion, it was shown that lentiviral correction was the best way to reduce LPS-induced secretion by IL-10, which was also observed in healthy controls (Supplementary Fig. 10). In this case, both the small molecules showed no effect on CXCL8 reduction. In contrast, CCL5 secretion was most effectively decreased by the use of filgotinib, followed by lentiviral correction (Supplementary Fig. 10).
The analyses of our macrophage disease models revealed that the typical macrophage-secreted cytokines TNF-α, IL-6, and CXCL8 were more effectively regulated by gene therapy in IL-10RBKO or VEO-IBD macrophages than by filgotinib or SB202190 treatment.
Discussion
In this study, we exploited the potential of iPSCs to form a nearly inexhaustible cell source and to be differentiated into macrophages for the generation of IL-10 signaling pathway-associated human iPSC KO models induced by an sgRNA-CRISPR-Cas9 carrying LV. To gain more insight into the role of the individual genes of this important anti-inflammatory signaling pathway, we successfully generated KOs for IL-10RA, IL-10RB, STAT1, and STAT3. KOs were verified in iPSCs on the genomic level by sequencing and additionally on the protein level for STAT1, STAT3, and IL-10RB (Fig. 2). Genomic analysis and functional phenotype of IL-10RAKOs clearly showed IL-10RA deficiency in the IL-10 signaling cascade by a lack of STAT3 phosphorylation upon IL-10 treatment. Defective STAT3 phosphorylation was also observed for STAT3KO, IL-10RBKO, and the IBD3 patient-derived macrophages.
Importantly, irrespective of the VCN, the sgRNA-CRISPR-Cas9 LV was silenced in all established disease models (Supplementary Fig. 3). The silencing of the SFFV U3 promoter is consistent with observations of vector silencing in iPSCs 36,37 and was desired to circumvent potential toxic or off-target effects of continuous Cas9 expression, which might have affected iPSC behavior or hematopoietic differentiation. 38
IL-10 has a crucial anti-inflammatory role in maintaining the intestinal mucosal homeostasis. 39 This function is fulfilled by cells of the innate immune system, such as macrophages, 40 which play a key role in the pathophysiology of IL-10-pathway-dependent diseases. Macrophages can not only secrete IL-10, but also mainly mediate the anti-inflammatory effect, especially in gut homeostasis, by the binding of IL-10 to IL-10R and conditioning of the macrophages to an immunosuppressive phenotype. 28,41 IL-10R deficiencies in macrophages of the intestinal lamina propria were shown to cause severe spontaneous colitis. 28,40 Due to these important features, we focused on the investigation of iPSC-derived macrophages.
The use of the forward programming system allowed us to generate high numbers of macrophages. This protocol enabled us to show that the different KOs did not influence hematopoietic differentiation at the level of HEPs or CD34+/CD45+ HPCs. Furthermore, the forward programming system was used for disease modeling for the first time. The generated macrophages showed functional phagocytosis and secreted cytokines upon stimulation (Figs. 3 and 4). Functional IL-10R downstream signaling was demonstrated by STAT3 phosphorylation and BCL3 and SOCS3 expression according to the disease model after IL-10 treatment (Fig. 3C and Supplementary Fig. 6).
Using the disease models, we investigated which components of the IL-10 pathway are necessary for the anti-inflammatory signaling leading to BCL3 and SOCS3 expression. It was shown that the AIR BCL3, which suppresses the production of LPS-induced TNF-α, is expressed by STAT3 after IL-10 stimulation. 32 Our observations in the KO and patient-derived macrophage disease models confirmed the earlier findings (Fig. 3C). We showed that a defect in one of the IL-10R chains or in STAT3 led to loss of IL-10-induced BCL3 expression, and thus, the products of these genes have an essential role in BCL3 expression. Furthermore, in macrophages, BCL3 plays an important role in the development of endotoxin tolerance by LPS-induced BCL3 expression through TLR-4. 42,43
Interestingly, our human iPSC-derived macrophage disease models showed the LPS-induced BCL3 expression to be independent of IL-10R. These findings are also supported by Mukhopadhyay et al., who described an LPS-induced BCL3 expression in human iPSC-derived macrophages, and by Baillie et al. who observed the same in human monocyte-derived macrophages (Fig. 3C). 44,45 Moreover, our KO models provide an excellent opportunity for the analysis of other STAT3-dependent AIR factors, such as ETV3 (ETS translocation variant 3) or ZFP36 (zinc finger protein 36 homologue), 32 to more deeply study IL-10 downstream anti-inflammatory signaling. The analyses of our KO models showed that in addition to BCL3 expression, IL-10-induced SOCS3 expression in human iPSC-derived macrophages is IL-10R and STAT3 dependent, whereas LPS-induced SOCS3 expression occurred independently of IL-10R. These results support the observation of Mukhopadhyay et al., who detected both IL-10- and LPS-induced SOCS3 expression in macrophages and the loss of IL-10-induced SOCS3 expression in patient-derived IL-10RB-deficient macrophages. 45 Our KO models were also used to show that LPS-induced SOCS3 expression occurred entirely independent of the LPS-induced macrophage-intrinsic IL-10 secretion and IL-10R signaling. In conclusion, these results emphasize the complex functionality of our disease models and show that BCL3 and SOCS3 can be induced by IL-10 and LPS in human iPSC-derived macrophages.
After observing the influence of IL-10 signaling on gene expression of BCL3 and SOCS3 in our macrophage disease models, we further investigated the influence of IL-10 signaling defects on the secretion of inflammatory mediators. By analyzing the cytokine secretion of the disease models, we observed that LPS stimulation of healthy iPSC-derived macrophages resulted in a strong inflammatory response, which was suppressed by additional IL-10 stimulation (Fig. 4 and Supplementary Fig. 7). These findings are consistent with other reported studies describing increased expression of proinflammatory cytokines, such as TNF-α, IL-6, CXCL8, CCL5, and CXCL10, upon LPS stimulation for primary or cell line-derived monocytes or macrophages. 46 –49
In monocytes and macrophages, it became evident that IL-10 stimulation suppressed the expression of these proinflammatory mediators on the transcriptional level. 50 –53 In many VEO-IBD patients with defects in the IL-10RA or IL-10RB subunits of the IL-10R, IL-10-mediated suppression of the LPS-induced inflammatory immune response is lacking, as shown by increased TNF-α secretion in peripheral blood mononuclear cells or iPSC-derived macrophages. 6,45
Furthermore, high secretion levels of IL-1, IL-2, IL-6, and CCL5 were detected in these patients. In the IBD context, increased IL-6 secretion in intestinal macrophages, 54 high CXCL8 levels in inflamed colonic tissue of IBD patients, 55 increased expression of CXCL10 in biopsies and intestinal epithelial cell lines, 56 and elevated CCL5 expression have also been reported. 57,58 Interestingly, the analysis of the cytokine secretion of our disease models has clearly confirmed these IBD-associated findings. Healthy macrophages secreted proinflammatory cytokines (IL-1β, IL-2, IL-5, TNF-α, IL-6, IL-7, CXCL8, IL-9, IL-12p70, IL-15, IL-17A, CCL2, CCL3, CCL4, CCL11, CXCL10, G-CSF, GM-CSF, IFN-γ, PDGF-bb, CCL5), and also cytokines with anti-inflammatory effects (IL-1RA, IL-4, IL-13, IL-10, VEGF, FGFβ) in an LPS-dependent manner (Fig. 4 and Supplementary Fig. 7). While additional IL-10 supplementation led to significant suppression of cytokine secretion (except for IL-1β, IL-7, IL-12[p70], IL-13, CCL11, G-CSF, CCL3, and PDGF-bb) in healthy macrophages, the cytokine secretion patterns of the IL-10 signaling pathway associated disease models were very different.
Based on our disease models, it became evident that in addition to STAT3, STAT1 is also an important part of the immune response and, as described in the literature, plays a major role in the control of IL-17A, 59 IFN-γ, 60 IL-4, 61 CXCL10, 62 FGFβ, 63 and VEGF. 64 This outcome led us to define IL-10R-/STAT3-dependent (i.e., IL-2, IL-6, CXCL8, GM-CSF, CCL5, TNF-α) and IL-10R-/STAT3- and STAT1-dependent (i.e., IL-4, IL-5, IL-9, IL-15, IL-17A, FGFβ, IFN-γ, CCL2, CXCL10, VEGF) cytokine secretion in our KO and patient-derived cell models. In addition, patient-derived macrophages behaved like IL-10RBKO macrophages in their secretion of inflammatory factors and this demonstrates how well the established KO disease models correlate with patient-based models.
To further characterize the generated macrophage disease models, cytokine secretion patterns were additionally used to investigate whether the macrophages generated by forward programming have microbicidal M1 or immunosuppressive, wound healing, and tissue repair M2 (M2a, M2b, M2c, M2d) phenotypes. 65 –68 We showed that both M1-specific (TNF-α, IL-6, CCL2, CCL3) and M2-specific cytokines (IL-1RA, IL-10, CCL5, CXCL10, VEGF) were secreted. Accordingly, the iPSC-derived macrophages either did not belong to a defined M1 or M2 phenotype, or we generated a mixed population of both M1 and M2 macrophages (Fig. 4 and Supplementary Fig. 7). The fact that macrophages, generated by aid of the forward programming differentiation protocol, showed M1 and M2 phenotypes was also observed by Lange et al. in flow cytometric analyses. 30 Also, iPSC-derived macrophages, which were generated by EB/MCFC formation and the exposure to IL-3 and M-CSF, could not clearly be assigned to the M1 or M2 phenotype after LPS stimulation, which supports our observation. 29
Currently, there is no available specific gene therapy or effective small-molecule treatment for VEO-IBD patients. Therefore, new therapeutic approaches are desirable, as the only therapy that successfully cures VEO-IBD is allogeneic stem-cell transplantation. 6 Alternatively, young VEO-IBD patients receive similar treatment as adult IBD patients, which includes anti-inflammatory agents, anti-TNF-α monoclonal antibodies, immunomodulators, or biologics. Unfortunately, these therapies only show a relatively poor response in VEO-IBD patients. Therefore, we used the disease models (IL-10RBKO, IBD3, STAT1KO) to investigate the possibility to correct the inflammatory phenotype by gene therapy or whether the proinflammatory cytokine secretion pattern can be reduced by anti-inflammatory small molecules (SB202190, filgotinib).
To resolve the specific defects, we transduced HPCs from the forward programming protocol with codon-optimized lentiviral correction vectors and investigated positively transduced cells by detailed cytokine secretion profiling. Encouragingly, IL-10RBKO and patient-derived (IBD3) macrophages showed a reduced secretion of proinflammatory cytokines, such as TNF-α, IL-6, and CXCL8 in LPS-stimulated corrected cells, and even more importantly, reconstituted the anti-inflammatory effect of IL-10 to reduce inflammatory cytokine secretion (Fig. 5E and Supplementary Fig. 8).
By lentiviral correction of the STAT1KO cells, we were able to restore the defective STAT1-dependent CXCL10 secretion after LPS stimulation and re-establish the IL-10-mediated downregulation of CXCL10 (Fig. 5E). STAT1-deficiency is not associated with VEO-IBD in patients, but does occur in MSMD. 14 Thus, the STAT1KO model can be used as an MSMD model to investigate the IFN-γ signaling pathway and therefore to develop new therapeutic approaches. 69
In addition to gene therapy, small molecules are an interesting approach for treating inflammatory diseases. Therefore, we tested anti-inflammatory small molecules (filgotinib, SB202190) in our iPSC-derived macrophage disease models. Filgotinib is already used in various clinical studies, for example, for IBD (UC, CD), 70 and showed a biological and clinical remission of disease symptoms in CD patients. 71,72 Filgotinib inhibits JAK1, 35 which plays an essential role in signaling from proinflammatory cytokine receptors such as IL-2, 73 IL-6, 74 and IL-12. 75 In contrast, SB202190 inhibits p38α and p38β. 34,76 LPS stimulation regulates NF-κB via p38 MAPK and stimulates the production and expression of proinflammatory mediators such as TNF-α. 77 Since p38 MAPK is involved in many inflammatory processes, inhibition of p38 MAPK is an attractive target to balance pro- and anti-inflammatory immune responses.
Neither of the small molecules had a negative influence on cell viability in our analyses (Fig. 6A, B). Furthermore, we were able to confirm the anti-inflammatory effect of both small molecules in our IL-10R disease model (Fig. 6C, D, and Supplementary Fig. 9). SB202190 notably reduced TNF-α and IL-6 secretion, while filgotinib had a stronger impact on CCL5 reduction.
However, we observed that gene-therapeutic correction more efficiently downregulated the typical macrophage-secreted cytokines TNF-α, IL-6, and CXCL8. This might also be due to the fact that the optimal therapeutic dose of the small molecules needs to be further tuned. For future therapeutic approaches, these results might suggest the simultaneous use of the two small molecules to achieve a possible synergistic effect for reducing proinflammatory cytokine secretion or a combined therapy starting with the treatment of a VEO-IBD patient with a small molecule before gene therapy or allogeneic HSC transplantation. Overall, these analyses show that our disease models are suitable for analyzing potential small molecules for anti-inflammatory treatments.
In summary, we have established IL-10RA, IL-10RB, STAT1, and STAT3 KO models as well as a VEO-IBD patient-derived IL-10RB-deficient disease model, which are useful tools to gain a better understanding of the pathogenesis of IL-10-pathway-associated immunodeficiencies, including VEO-IBD. Through our promoter choice for the sgRNA-CRISPR-Cas9 lentiviral system, we reduced potential toxic influences of continuous Cas9 expression as the SFFV promoter was shown to be silenced in iPSC.
For the production of iPSC-derived macrophages, we used the forward programming system for the first time in the context of disease modeling and were able to generate functional macrophages that showed both anti- and proinflammatory phenotypes. Analyses of the IL-10R downstream targets BCL3 and SOCS3 showed that the IL-10R and STAT3 are necessary for IL-10-mediated expression, and that both can be activated independently of the IL-10R by LPS.
The cytokine secretion patterns of the macrophage disease models were classified into two different groups and showed that IL-10 is involved in many pro- and anti-inflammatory processes in LPS-activated human iPSC-derived macrophages. Furthermore, we demonstrated that IL-10R and STAT3 defects resulted in a strong proinflammatory cytokine secretion pattern and showed that in many cases besides IL-10R and STAT3, STAT1 is essential for IL-10-regulated cytokine secretion. Through gene-therapeutic approaches and small molecules, we obtained initial proof-of-concept for the correction of the disease models and efficiently reduced their strong proinflammatory secretion patterns.
Footnotes
Authors' Contributions
Conceptualization: A.S.; Methodology: D.H., J.S., L.L., and A.S.; Investigations: J.S., P.V.B., and C.S.F.; Resources: L.L., D.H., and A.S.; Writing the original draft: J.S. and A.S.; Writing—Review and editing: A.S., C.S.F., D.H., L.L., J.S., M.M., and P.V.B.; Visualization: J.S.; Supervision: A.S.; Funding Acquisition: A.S.
ACKNOWLEDGMENTS
We thank Kerstin Beushausen and Jana Keil for the excellent technical assistance. Technical support for vector design was given by Dr. Johannes Kühle (formerly Hannover Medical School). Parts of this work were part of the dissertation of Johanna Sens (2020, Hannover Medical School).
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by grants from the Deutsche Forschungsgemeinschaft [SFB738, Cluster of Excellence REBIRTH (EXC 62/2)], the REBIRTH Center for Translational Regenerative Medicine funded through the State of Lower Saxony (MWK: ZN3440), and the Bundesministerium für Bildung und Forschung (BMBF; i.e., PidNet and iMAC consortiums). Moreover, this project received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement Nos. 755170, 666908, and from the European Research Council (ERC) under grant agreement No. 819531. Cells from an IL-10RB-deficient VEO-IBD patient were contributed by Prof. Dr. Christoph Klein (formerly Children's Hospital, Hannover Medical School, now at Dr von Hauner Children's Hospital, Munich).
Supplementary Material
Supplementary Data
Supplementary Table 1
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Figure 10
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.