
Abstract
The uridine-rich 7 (U7) small nuclear RNA (snRNA) is a component of a small nuclear ribonucleoprotein (snRNP) complex. U7 snRNA naturally contains an antisense sequence that identifies histone premessenger RNAs (pre-mRNAs) and is involved in their 3′ end processing. By altering this antisense sequence, researchers have turned U7 snRNA into a versatile tool for targeting pre-mRNAs and modifying splicing. Encapsulating a modified U7 snRNA into a viral vector such as adeno-associated virus (also referred as vectorized exon skipping/inclusion, or VES/VEI) enables the delivery of this highly efficacious splicing modulator into a range of cell lines, primary cells, and tissues. In addition, and in contrast to antisense oligonucleotides, viral delivery of U7 snRNA enables long-term expression of antisense sequences in the nucleus as part of a stable snRNP complex. As a result, VES/VEI has emerged as a promising therapeutic platform for treating a large variety of human diseases caused by errors in pre-mRNA splicing or its regulation. Here we provide an overview of U7 snRNA's natural function and its applications in gene therapy.
Introduction
Many human diseases are caused by mutations in the genomic sequence that alter RNA splicing, impairing the production of a functional protein. The ability to target aberrant RNA provides an opportunity to modulate splicing and potentially treat numerous genetic disorders. Early strategies for modulating splicing focused on the use of antisense oligonucleotides (AONs) to interfere with important splice sites or exon regulatory elements. 1 –6 The effectiveness of AONs in altering splicing patterns was first demonstrated by Dominski and Kole. 7 They showed that AONs targeted against β-globin premessenger RNA (pre-mRNA) could correct the aberrant splicing patterns which cause β-thalassemia. Since this landmark publication, AONs for splicing modulation have been studied and used to treat patients with several genetic disorders.
AONs efficiently compete with the spliceosome for recognition of splicing motifs, which often results in exclusion of the targeted exon from the mRNA. However, the effects of AONs are transient due to degradation by endo and exonucleases, meaning that therapies would require regular life-long readministration. 7,8 As an alternative to standard AONs, derivatives of uridine-rich 7 (U7) small nuclear RNA (snRNA) containing optimized Sm protein-binding sites and a modified antisense sequence (termed U7 Sm OPT snRNA) has proved to be promising modulators of pre-mRNA splicing. Expression of U7 Sm OPT snRNA allows for the continuous nuclear accumulation of therapeutic antisense sequences embedded in a stable small nuclear ribonucleoprotein (snRNP) complex. 8,9 This long-term expression of antisense prevents or significantly reduces the need for readministration of the therapy.
Delivery of U7 Sm OPT snRNA via a viral vector such as adeno-associated virus (AAV) enables the selective inclusion or exclusion of a targeted exon in tissues of interest. This technology is also referred to as vectorized exon skipping/inclusion (VES/VEI) and can be used to ameliorate the effects of genetic errors. Although VES/VEI has been explored in many animal models across several diseases, in 2020, this approach has finally entered its first clinical trial using systemic AAV delivery in patients with Duchenne muscular dystrophy (DMD; NCT04240314), one of the most common and severe neuromuscular disorders. This review covers the natural function of U7 snRNA in histone pre-mRNA processing, its development as a splicing modulation tool, and current advancements in VES/VEI, underlining its promise as therapeutic tool for a broad range of diseases.
U7 snRNA Transcription and Assembly
U snRNAs are a family of RNAs with diverse and complex functions. The best-known U snRNAs are those of the spliceosome, which are involved in removing introns from pre-mRNAs during transcription (U1, U2, U4, U5, and U6). The U7 snRNA was first discovered in sea urchins, where it is crucial for histone pre-mRNA 3′ end processing. 10,11 Later, U7 snRNA was identified to have the same role in mice and humans. 12 –15 It is now known that U7 snRNA exists in nearly all animals. 16,17 Like almost all other U snRNAs, the U7 snRNA has its own promoter and is transcribed by RNA polymerase II (pol II). The U7 promoter contains U snRNA-specific elements not found in other genes transcribed by pol II that are essential for its transcription: a distal sequence element responsible for the enhancement of transcription and a proximal sequence element important for specifying the site of transcription initiation. 18,19
U snRNAs do not contain a polyadenylation signal and their 3′ untranslated region is processed through cleavage only. 20 –22 Efficient formation of the 3′ ends of the pre-snRNA transcript requires a 3′ end signal called the 3′ box. 20,22 This signal is only recognized if transcription is initiated from a U snRNA promoter, and replacement with a noncompatible pol II snRNA promoter results in inefficient 3′ end formation. 22,23
Maturation of U snRNAs begins in the nucleus, continues in the cytoplasm, and completes in the nucleus following nuclear reentry. In the nucleus, the U snRNA transcript initially acquires a standard m7GpppG 5′ cap structure for export to the cytoplasm. In the cytoplasm, U7 snRNA associates with five Sm proteins (B/B′, D3, E, F, and G) and two U7-specific Sm-like proteins (Lsm10 and Lsm11), which are important for histone pre-mRNA processing. 24 In contrast, the spliceosomal snRNAs associate with seven Sm proteins (B/B′, D1, D2, D3, E, F, and G).
Assembly of the Sm core stabilizes and protects the snRNA from nuclease degradation and is required for downstream processing of the snRNA. Following heptamer formation, the snRNA 3′ end is trimmed, and the cap structure is hypermethylated to a 2,2,7-trimethyl guanosine (3mG) cap, which functions as a nuclear localization signal. 25 Once in the nucleus, the snRNP reenters cajal bodies for further RNA modification and maturation. 26,27 After maturation, the U7 snRNP localizes to nuclear structures involved in the assembly of the machinery needed for histone pre-mRNA processing (reviewed in Nizami et al. 28 ).
U7 snRNP Structure and Function
The U7 snRNP involved in histone RNA 3′ end processing has three major components: (1) a histone downstream element (HDE)-binding sequence, (2) a Sm/Sm-like binding site, and (3) a 3′ stem-loop that stabilizes the RNA 10 (Fig. 1A). U7 snRNP functions by recognizing histone pre-mRNAs through base pairing to the HDE that follows the histone pre-mRNA 3′ processing site. 12 Once bound, U7 recruits the appropriate cleavage factors essential for histone pre-mRNA processing. 11,29,30 This process relies on both Lsm11 and Lsm10 (Fig. 1A). If these two proteins are exchanged with other Sm proteins, the resulting U7 snRNP molecule can still bind to histone pre-mRNAs, but is unable to process their 3′ end. 31
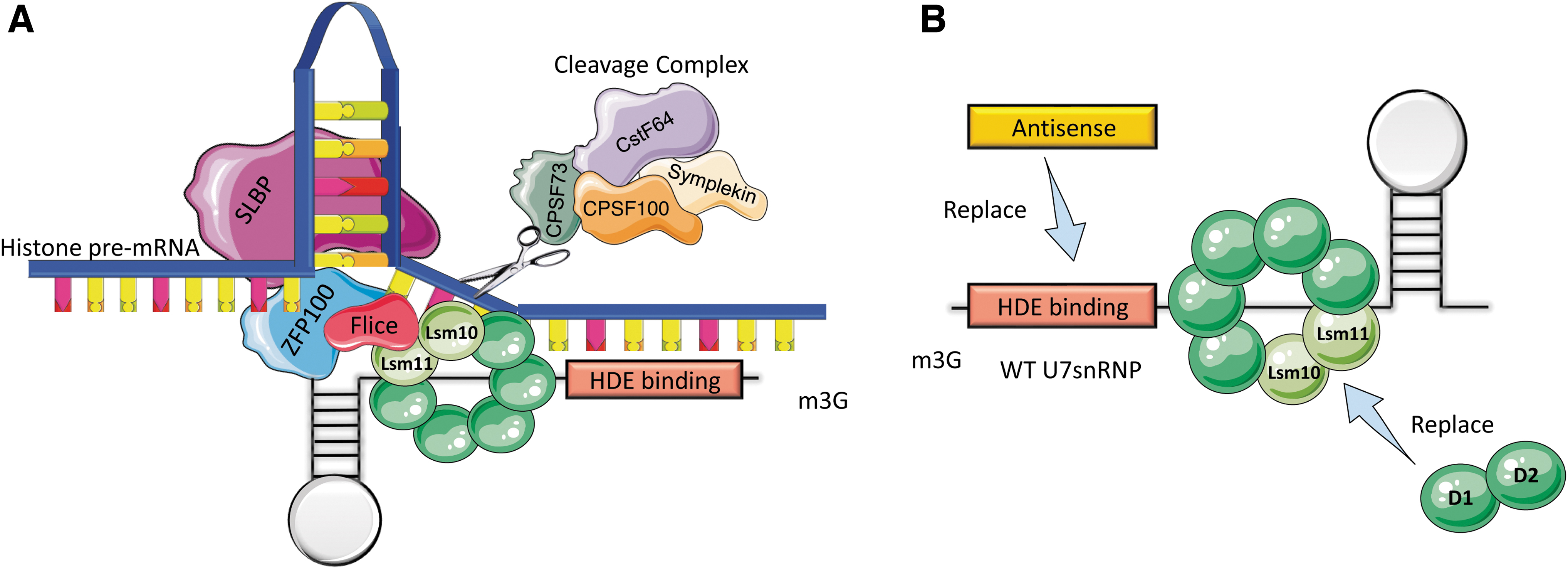
Structure of wild-type U7 snRNP and processing of histone pre-mRNA 3′ end processing.
U7 Sm OPT snRNA as a Splicing Modulation Tool
The ability to target aberrant pre-mRNA splicing could provide treatments for a variety of genetic disorders caused by alteration of typical splicing patterns. The spliceosome carries out pre-mRNA splicing by recognizing important conserved sequence elements (also referred to as exon definition elements) located at intron-exon junctions: the 5′ donor (GT), the 3′ acceptor (AG) splice sites, and the branch point sequence near the 3′ splice site of the intron (Fig. 2A).

The classical spliceosome machinery and its interaction with U7 sm OPT.
In addition, the spliceosome's ability to recognize a specific exon depends on the exon's own definition sequences and the relative strength of surrounding sequences/elements known as splicing regulatory elements (SREs). SREs promote exon recognition and selection of splice sites (reviewed in Cartegni et al. 32 ). Binding of regulatory proteins and the added effects of positive and negative elements determines whether the spliceosome will recognize and include/exclude an exon (reviewed in Cartegni et al. 32 , Matera and Wang 33 , Smith and Valcárcel 34 ). Positive elements are exonic splicing enhancers (ESEs) and intronic splicing enhancers (ISEs). Negative elements are exonic splicing silencers (ESSs) and intronic splicing silencers (ISSs) (Fig. 2C).
Replacement of the wild-type U7 Sm-binding sequence with the consensus Sm-binding sequence found in the major snRNAs creates the U7 Sm OPT snRNA, which can associate with all the Sm proteins of spliceosomal snRNAs. 31 The resulting modified snRNP is more efficient at accumulating in the nucleus than its wild-type counterpart and is no longer able to induce cleavage of histone pre-mRNAs. 8,9,31,35 Further, by replacing the intrinsic HDE-binding sequence of the U7snRNA with a custom antisense sequence (Figs. 1B and 2B), the splicing pattern of a target pre-mRNA can be modified.
U7 Sm OPT snRNAs usually contain one antisense sequence and thus are termed “single-target” constructs (Fig. 3A). However, improved efficiency can be obtained with the “double-target” U7 Sm OPT snRNA constructs, which carry two tandem antisense sequences targeting two different splice sites (3′ acceptor splice site and 5′ donor splice site) 36 (Fig. 3B). Double-target constructs induce removal of the targeted exon (exon skipping). Additionally, a 5′ or 3′ tail can be added to the antisense sequence to form a “bifunctional” U7 Sm OPT snRNA (Fig. 3C). Such tails carry established ESE, ESS, ISE, or ISS sequences to provide binding sites for splicing enhancer or silencer factors to improve the likelihood of the desired splicing event occurring (for reviews see Meyer and Schümperli 37 , Benchaouir and Goyenvalle 38 ). Taken together, the ability to exchange sequence elements as desired makes U7 Sm OPT snRNAs a stable, antisense-carrying molecule that can be used as a therapeutic for a variety of human diseases. Importantly, the small size of U7 expression cassettes (U7 promoter and U7 Sm OPT snRNA sequence) allows the use of AAV vectors for delivery to target cells or organs. Using this system, the U7 snRNA will be continuously expressed in the nucleus of transduced cells and does not have to be readministered for an extended period.
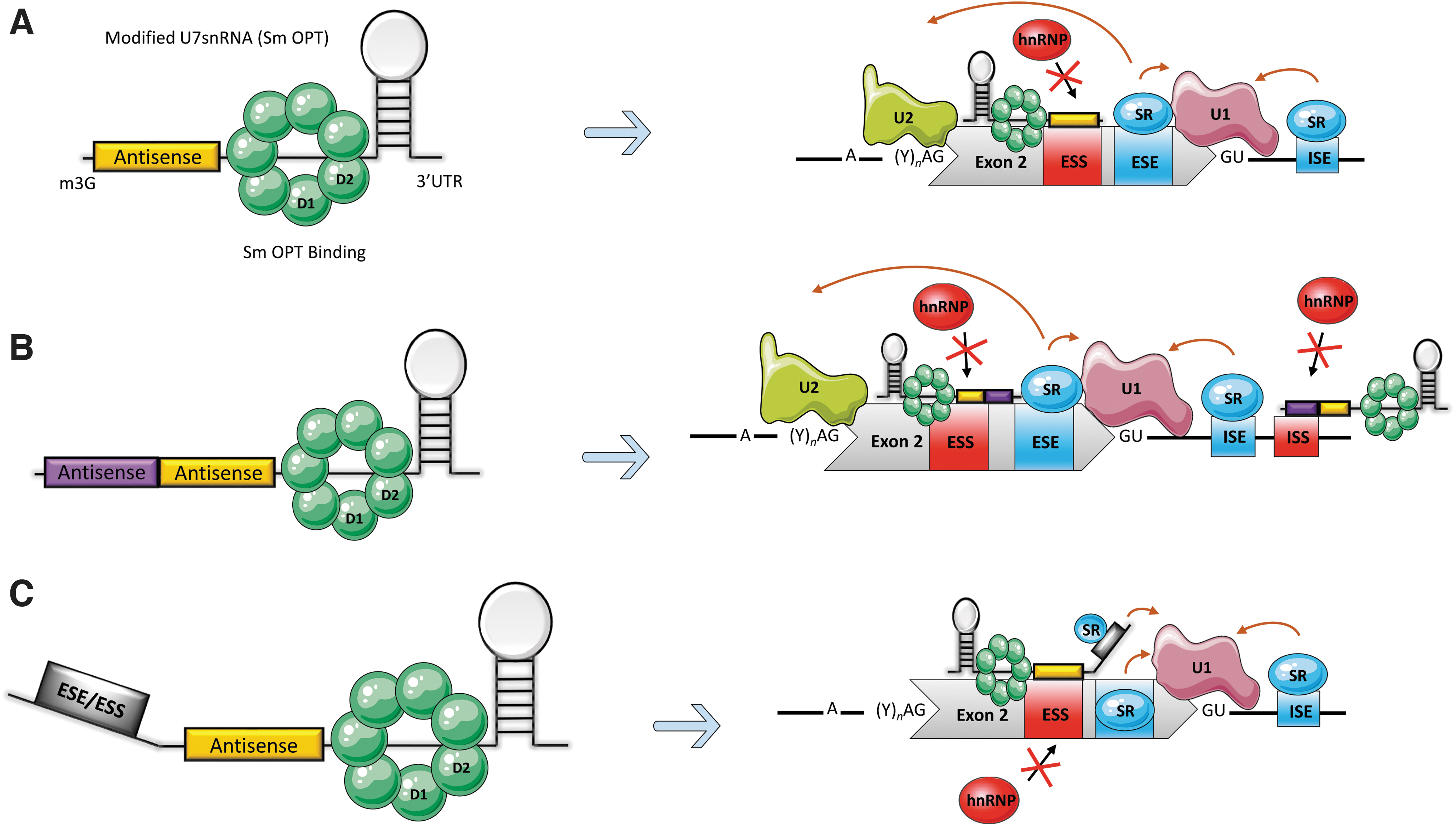
Modified U7 snRNA as a splicing modulator and its derivatives.
Other modified U snRNAs have also been explored to mediate exon skipping/inclusion. 39 –41 Technically, the antisense sequence can be incorporated into any of the U snRNAs. However, this may lead to competition with the wild-type counterparts, thus interfering with natural function. Therefore, the major U snRNAs may be less suited for therapeutic modification. Since the Sm-binding site of the U7 Sm OPT snRNA has been modified to eliminate U7 snRNA's natural function, and due to the lower expression levels of the U7 snRNA compared to the major spliceosomal U snRNAs, U7 snRNA has a more appealing safety profile than other U snRNAs.
Among the other U snRNAs, the one that has been most tested for splicing modulation is U1 snRNA due to its higher levels of expression; for example, U1 has a sixfold higher natural expression than wild-type U7. 31 Despite this, modified U1 constructs are often less efficient than U7 Sm OPT snRNAs, and replacing the U7 promoter with the U1 does not significantly increase the levels of snRNA or splicing. 39,40 Modified U6 snRNA constructs have also been used; however, they are not as efficient possibly because U6 snRNA alone is unable to access splice sites as it relies on U4 snRNA for snRNP formation. 39 To more efficiently reach target cells, these modified U snRNAs tools have been delivered using both lentiviral 42,43 and AAV vectors 44 (reviewed in Benchaouir and Goyenvalle 38 ).
Use of the U7 snRNA Sm OPT Antisense System to Treat Human Disease
As demonstrated in Table 1, U7 snRNAs have been used in several disorders.
Overview of uridine-rich 7 small nuclear RNA studies to treat various diseases a
Nonexhaustive list.
ALS, amyotrophic lateral sclerosis; BP, branching point; DM1, myotonic dystrophy type 1; DMD, Duchenne muscular dystrophy; DMPK, dystrophia myotonica protein kinase; dup2, duplication of exon 2 of DMD; dysf, dysferlin; GAA, acid α-glucosidase; GRMD, golden retriever muscular dystrophy; hDMD, humanized DMD; ICV, intracebroventricular; IRES, internal ribosome entry site; IV, intravenous; LGMD 2D, limb girdle muscular dystrophy α sarcoglycan; mdx, muscular dystrophy x linked; NA, not available; OCRL 1, inositol polyphosphate 5-phosphatase OCRL gene; PTC, premature termination codon; RyR1, type 1 ryanodine receptor; SMA, spinal muscular atrophy; SMN2, survival of motoneurons 2; SOD1, Cu/Zn superoxide dismutase 1; SR, stratum radiatum; TREM 2, triggering receptor expressed on myeloid cells 2; U7, uridine-rich 7; UTR, untranslated region.
β-thalassemia
Gorman et al. were the first to prove that U7 snRNA's natural function of complementary base-pairing to histone pre-mRNA could be reprogrammed to function in an AON-like manner. 8 They used U7 Sm OPT snRNA constructs to promote exon exclusion in β-thalassemia. β-thalassemia occurs when a point mutation in the β-globin gene creates a novel 5′ splice site and activates a cryptic 3′ splice site in intron 2. This results in the inclusion of an aberrant “exon” (an intron fragment containing an in-frame stop codon) in the β-globin mRNA and consequently leads to a loss of β-globin protein expression. Different U7 Sm OPT snRNAs containing single-target sequences complementary to aberrant 5′ or 3′ splice sites were able to induce exon skipping. This led to the restoration of correct β-globin mRNA splicing and the corresponding protein in patient-derived cell lines. 8
Human immunodeficiency virus 1
Acquired immunodeficiency syndrome (AIDS) is a life-threatening condition caused by infection with the human immunodeficiency virus 1 (HIV-1). HIV works by damaging the immune system, leaving the body defenseless against organisms that cause disease. The efficient replication of HIV-1 virus relies on the integration of protein cyclophilin A (CyPA) into HIV-1 virions. 42 A double-target U7 Sm OPT snRNA system was used by Liu et al. to target the 3′ and 5′ splice sites of CyPA exons 3 or 4 to promote their exclusion in the final transcript of CyPA. 42 Exclusion of these exons would result in a truncated protein unable to integrate into viral particles; hence delaying HIV-1 infection.
Efficient skipping of these exons and a reduction in CyPA protein was observed after lentiviral transduction of a human T cell line alongside delayed HIV-1 replication. 42 While this approach showed high rates of reduction of HIV-1 expression, it was significantly less efficient than small interfering RNA (siRNA) targeting the same regions, although there are still some questions regarding the clinical applications of siRNA given their reliance on the saturable RNA-induced silencing complex and Drosha complexes. 45 –47 Moreover, siRNAs and miRNAs can only reduce mRNA or protein levels produced and are not suited for changing of mRNA structure (exon inclusion/exclusion). Thus, using a modified U7 snRNA to induce gene knockdown by generating an out-of-frame transcript may prove superior as it has never been shown to saturate any cellular processes. To increase efficacy of the U7 snRNA approach, several U7 Sm OPT snRNA cassettes can be added into the same viral vector. 48
Spinal muscular atrophy
Spinal muscular atrophy (SMA) is a severe neuromuscular disorder that affects 1:6,000–10,000 patients and causes progressive muscle loss and premature death (reviewed in Kolb and Kissel 73 ). SMA is caused by homozygous mutations in the survival motor neuron 1 (SMN1) gene leading to a loss of expression of the SMN1 protein. In addition to SMN1, another similar gene (99% homology) named survival motor neuron 2 (SMN2) exists. This gene encodes the SMN2 protein, which in theory can compensate for the lack of SMN1. However, SMN2 contains a single-nucleotide polymorphism (C > T transition) in exon 7 compared to SMN1. This nucleotide variation disrupts an ESE site and creates an ESS site. In addition, exon 7 contains an inhibitory terminal stem loop and ISSs upstream and downstream of its vicinity, which causes it to be poorly recognized by the spliceosome machinery. Furthermore, SMN2 contains a weak 3′ splice site that competes with the strong 3′ splice site found in exon 8 for recognition by the spliceosome (reviewed in Nlend Nlend et al. 74 ). Altogether, this leads to an aberrantly spliced transcript that lacks exon 7.
Complete absence of the SMN protein is embryonic lethal and all SMA patients produce reduced levels of the protein from the SMN2 gene. The number of SMN2 genes and other factors influencing the efficiency of correct SMN2 mRNA splicing affect disease severity. Hence, increasing the correct splicing of the SMN2 pre-mRNA is an attractive therapeutic approach for SMA patients. Among the drugs investigated, Nusinersen, an AON, was the first approved drug used in treating SMA. 75 –77
Single-target U7 Sm OPT snRNA constructs with complementary sequences to the strong 3′ splice site found in exon 8 allowed the spliceosome to recognize the weak 3′ splice site found in exon 7, leading to its incorporation into the final mRNA and producing a full length SMN2 protein in vitro. 78 Alternatively, another group has used bifunctional U7 Sm OPT targeting the 3′ end of exon 7 with an ESE tail that attracts splicing enhancer proteins to force exon 7 inclusion. This strategy allowed generation of 90% full-length SMN2 transcript in a HeLa cell minigene screening system and in fibroblasts from SMA patients, leading to a 50% increase in SMN protein. 79
Treatment of a mouse model of SMA using this construct demonstrated extended lifespan, improved muscle performance, and extended motoneuron survival. When compared in the same mouse model to the gene replacement strategy, in which the SMN coding sequence is delivered via AAV, the splicing correction approach was slightly less efficacious. 80 However, this result may be attributable to the use of a strong ubiquitous promoter for the expression of the SMN cDNA. Due to their varied mechanisms of action, a combination of the two approaches might warrant further investigation and could lead to improved patient outcomes. The transgene replacement gene therapy (Zolgensma) was recently approved by the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and in Japan for treatment of SMA patients. 43
Duchenne muscular dystrophy
The potential of the U7 snRNA system has been best demonstrated in DMD (reviewed in Benchaouir and Goyenvalle 38 ), the most common form of muscular dystrophy. DMD is a lethal, X-linked, genetic disorder that affects roughly 1:5,000 boys born every year. 81,82 DMD is characterized by a loss of ambulation before 12 years of age. DMD can be caused by a wide variety of mutations in the DMD gene, which usually lead to an absence of the dystrophin protein. Exon skipping approaches for DMD seek to restore the DMD reading frame. Skipping of exons surrounding a deleteriously removed exon or direct skipping of an exon containing a nonsense mutation results in a partially functional but truncated dystrophin protein. Such proteins are characteristic of Becker muscular dystrophy (BMD), in which patients have a significantly milder phenotype (loss of ambulation past the age of 12 and increased life expectancy). In essence, exon skipping can convert a DMD patient's phenotype to that of BMD.
Exon skipping strategies in DMD are well-documented and AONs targeting various exons of DMD have been in preclinical and clinical trials (for reviews see Refs. 83 –88 ). However, as mentioned previously, a major challenge posed by AON-mediated exon skipping is the requirement to continuous delivery of the compound for long-term protein restoration, as such compounds get degraded by endogenous nucleases (for reviews see Refs. 89 –91 ). In addition, while such AONs can enter many muscles, their penetration in the heart and diaphragm is inefficient. 92 –94
To overcome these limitations, several groups have taken advantage of VES (reviewed in Benchaouir and Goyenvalle 38 ) to induce exon skipping and long-term dystrophin expression. They used a wide range of animal models of DMD such as the mdx mouse (nonsense mutation in exon 23 of DMD); the Dup2 mouse (out-of-frame duplication of DMD exon 2), and the golden retriever muscular dystrophy (GRMD) dog (out-of-frame natural skipping of DMD exon 7). In all these models, as described below, U7 snRNA demonstrated efficient exon skipping, long-term dystrophin expression, and functional muscle improvement.
One of the first in vivo proof of concepts of VES was performed by Goyenvalle et al., for DMD. This group demonstrated that VES induced persistent exon skipping and restored dystrophin expression in the mdx mouse. 44 Specifically, they used a double-target U7 Sm OPT snRNA targeting both the DMD exon 22 branch point and the DMD exon 23 splicing donor site. This engineered U7 Sm OPT snRNA was inserted into an AAV1 viral vector and injected into the tibialis anterior and extensor digitorum longus of the mice. Four weeks postinjection, dystrophin was being expressed in muscles along with improvements in muscle force generation. 44 Other studies using exon 51 cell lines showed that bifunctional U7 Sm OPT snRNAs carrying an antisense targeting exon 51 and a splicing silencer sequence induce complete skipping of exon 51 in vitro and allow restoration of near wild-type levels of dystrophin in DMD patient cells. 52
As all these studies were conducted in vitro or in mice, a looming question was whether VES would retain its functionality in larger animals. This was answered in a series of studies, in which VES proved both effective and safe in GRMD. This model carries a natural point mutation in intron 6 that leads to an out-of-frame deletion of exon 7. Skipping of exon 6 and 8 can restore the reading frame of the mRNA and leads to expression of a slightly truncated form of the protein. Thus, a U7 Sm OPT snRNA carrying two antisense sequences (U7 Sm OPT snRNA6/8) was used for these studies. 56 Percutaneous transendocardial injection of rAAV6–U7 snRNA6/8 resulted in corrected transcript and cardiac dystrophin expression measured up to 13 months postinjection in GRMD dogs. 56
Another group validated the longevity of these VES results using an rAAV1–U7 snRNA6/8 and measured protein expression up to 5 years postinjection. 95 Beyond establishing the efficacy of VES in a larger animal model, this cohort study concluded that VES was safe and the restored dystrophin did not induce an immune response, a promising sign for VES. While initial rates of dystrophin expression and exon skipping were high, a large decline in the treatment's effects over the course of the 5 years was observed, with an especially steep decline over years 1 and 2. These data were challenged by a later large-cohort study conducted in 18 GRMD dogs injected intramuscularly with VES. 61 Although this study did not look 1-year postinjection, they did not observe lower rates of dystrophin restoration in their 3.5- and 7-month follow-ups. They hypothesized that this difference could be attributed to their earlier injection age of GRMD dogs (3–4 months vs. 5–6 months). 61
Earlier injection may provide earlier dystrophin restoration, preventing muscle degradation and resulting in long-lasting effects by preventing leakage of AAV episomes encoding the therapeutic U7 snRNA. 96 Based on these data, it will likely be beneficial for patients to receive therapy as early as possible, supporting the effort to include DMD in new-born screening panels.
Often overlooked is the role that dystrophin plays in nonmuscle tissues such as the brain. While the molecular mechanisms remain poorly understood, many DMD patients present with cognitive defects. 97 –99 Therefore, to create the greatest benefit for patients, it is important to consider targeting every tissue expressing dystrophin when designing treatments for DMD. Evidence of U7 Sm OPT snRNA's ability to function in the brain was demonstrated in the mdx mouse model. Groups found that a single intrahippocampal injection of rAAV1–U7 rescued dystrophin expression, restored gamma aminobutyric acid (GABA) receptor clusters in the hippocampus, and improved levels of long-term potentiation (LTP) measured at 2 months. 53,100 It has been shown that the lack of certain dystrophin isoforms in the brain reduces type A GABA receptor clusters and enhances LTP. 100 This demonstrated the potential of VES therapies to provide benefit to both muscular and neurological phenotypes. 101
The first clinical trial using VES recently began in the United States (NCT04240315). The therapy, developed by Wein et al., targets mutations in exon 2 of DMD. 48,102,103 Preclinical studies were conducted in a mouse model carrying an exon 2 duplication (Dup2 mouse) and using an AAV1-U7 to skip the duplicated exon 2. Treatment of the Dup2 mouse resulted either in single exon 2 skipping, which leads to the production of full-length dystrophin, or skipping of both copies of exon 2, which results in an out-of-frame transcript. However, this mRNA lacking both copies of exon 2 still produces a highly functional truncated protein, thanks to a downstream internal ribosome entry site. 48 This study further supported the ability of VES to induce efficient exon skipping, protein restoration, and force improvement. A follow-up dose escalation study was also performed in Dup2 mice using the same VES approach to determine the minimal efficacious dose (MED). 102 MED was found to be 3e13 vg/kg.
Finally, two additional studies were performed to assess potential off targets of this VES, one in the Dup2 mouse and one in wild-type nonhuman primates (NHPs). No off-target or other adverse events were found in NHPs. 103 Based on these results, a phase I/IIa clinical trial (NCT04240315) using this exon 2 VES approach began in 2020. Astellas Pharma, the sponsor of this trial, recently presented initial results of increased dystrophin expression and muscle strength in one patient. While these results are promising, the number of patients in the clinical trial is very small, and the data have not yet undergone peer review and thus must be interpreted with care (LBO3, Waldrop et al., 2020 WMS). 104
For DMD, several other gene therapy strategies are currently being explored in clinical trials. Since AAV packaging limits make delivery of the full-length dystrophin transcript impossible, microdystrophin (microdys) has emerged as a promising alternative. Various forms of microdys are currently being explored in three different clinical trials in the United States.
A major advantage to microdys is its potential to treat DMD patients regardless of their mutation. However, VES offers distinct advantages. Namely, exon skipping restores a near full-length dystrophin, while microdys only represents about 30% of the full-length protein. Thus, the VES induced protein is expected to have a higher functionality compared to microdys. Additionally, exon skipping utilizes the natural dystrophin promoter, ensuring that expression of the restored protein is properly regulated. Finally, the engineered microdys is more likely to be immunogenic as it contains several unnatural epitopes. Due to its several advantages, VES may be a superior choice when compared to microdys for patients with mutations correctable by VES approaches.
Beyond the described studies, U7 Sm OPT snRNA has also been explored as a possible treatment for Myotonic Dystrophy Type 1 (DM1), 72 Dysferlinopathies, 63 Pompe disease, 68 amyotrophic lateral sclerosis, 64 Hemophilia, and several other genetic disorders (Table 1).
Conclusion: Advantages/Disadvantages and Future Steps for U7 snRNA VES/VEI
U7 snRNA is a versatile platform for creating therapies that overcomes obstacles faced by AONs. However, important questions remain regarding U7 snRNAs. Since AONs have been more widely used than U7 snRNA for modulating splicing, the ability of AONs to compete with endogenous proteins for regulatory element binding sites is better characterized. 105 For example, results from DeepCLIP, 106 a tool which accelerates antisense design by providing predicted sites of splicing regulatory element binding to RNA, were validated using AONs. It will be of interest to perform similar studies that characterize U7 snRNA antisense's ability to compete with regulatory element binding sites. Such research will be valuable as an AON or U7 snRNA carrying the same antisense sequence may vary in their ability to bind to a particular RNA regulatory element.
While AAV delivered splicing modulation is significantly longer lasting when compared to regular AONs, the precise longevity of the treatment remains unknown and is unlikely to be permanent. 96 This issue may be partially circumvented in some disorders by starting treatment early, but a form of readministration might eventually be necessary. Unfortunately, the potential immune response elicited by the AAV capsid complicates VES reinjection. However, in the future, immunosuppressants, alterations of the AAV capsid, or nonviral vectors may make readministration of U7 snRNA-based therapies possible.
Another alternative could also be to first treat with VES and later administer an AON treatment if necessary. This combined approach was recently tested and proved to be highly promising in a study where VES was used in combination with peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO). 107 Results showed significant improvements in skeletal muscle, diaphragm, and heart function that resulted in significant life extension beyond PPMO or VES alone. Alternatively, if long-term expression of VES/VEI is found to be toxic, strategies to remove the therapeutic AAV episomes will have to be developed. This may be done through designing an AAV that forms a less stable episome or introducing molecules to knockdown the introduced AAV episomes. A similar idea has been explored in the development of “self-deleting” AAV encoding CRISPR-Cas9. 108
An additional potential challenge for U7 Sm OPT snRNA approaches is that although they do not interfere with natural U7 snRNA pathways, the formation of U7 Sm OPT snRNPs and other U snRNPs could be saturated. When expressed at high levels, a fraction of U7 Sm OPT molecules are truncated at the 3′ end. It is expected that these misprocessed sequences are not assembled into snRNPs and lose therapeutic function. 109 Thus, reaching sufficiently high levels of intracellular antisense sequence may require a further optimized U7 Sm OPT that does not undergo such misprocessing. Although not detected in safety studies in mice, dogs, and nonhuman primates, it is possible that inducing high levels of snRNP formation poses a safety concern. This concern is somewhat mitigated by the finding that U7 Sm OPT misprocessing is a result of the Sm OPT sequence itself. 24 Nevertheless, additional study regarding the effects of expressing high levels of therapeutic U7 snRNPs on natural snRNP processing might be needed.
Recently, genome editing tools based on modified bacterial proteins have gained popularity for treating genetic disorders. While the advantages of modifying genomic DNA rather than RNA are clear and may one day prove superior, in vivo delivery of these DNA-editing technologies carries the risk of permanent off-target effects and immune responses triggered by the introduction of foreign proteins. Meanwhile, VES has been shown to have negligible off-target effects and its targeting of RNA makes for an inherently safer choice. 110
In some cases, DNA-editing may lead to undesired mutations inducing improper splicing. If predicted ahead of time, VES/VEI may be administered alongside the DNA-editing vector to correct the unintended alterations in splicing caused by DNA-editing off-targets or improper repairs. In addition, using DNA-editing and VES/VEI in tandem may be beneficial if a single gene harbors more than one deleterious mutation (e.g., an improperly included exon and downstream nonsense mutation). This may greatly reduce off-target risks when compared to using two DNA-editing vectors.
Altogether, VES/VEI have shown to be a highly adaptable and effective tools for modulating splicing in many genetic disorders and have the potential to play a prominent role in developing personalized treatments. Genetic disorders for which VES/VEI may be used as a treatment will typically fall into one of three categories: (1) diseases caused by the improper inclusion or exclusion of exons, (2) diseases in which the effects of a mutation in one gene can be alleviated by altering the splicing of a second gene, and (3) diseases caused by a nonsense mutation located within an exon whose removal is in-frame and does not affect overall protein function. For diseases that fall into these categories and more, VES/VEI is an emerging technology that has the potential to play a key role in treating patients with genetic disorders.
Footnotes
Authors' Contributions
D.L., Y.R., D.R., and N.W. wrote the article. Y.R. compiled and created the figures.
Author Disclosure
Nationwide Children's Hospital optioned a program to Astellas Pharma. N.W. received royalty payments from this option.
Funding Information
This review was written while D.L., Y.R., and N.W were supported by Nationwide Children's internal and D.R. and N.W. by a patient donation (Nicholoff Family).