
Abstract
The phenomenon of no-reflow seriously limits the therapeutic value of coronary recanalization and leads to poor prognosis. Recent studies have demonstrated the potential role of pigment epithelium-derived factor (PEDF) in stabilizing endothelial cell junction, reducing vascular permeability and maintaining a quiescent vasculature. In this study, intramyocardial gene delivery was performed 5 days before the acute myocardial infarction/recanalization experiment in male rats. Positron emission tomography perfusion imaging with 13N-NH3 indicated PEDF to promote microvascular reperfusion significantly 4 h postcoronary occlusion. PEDF was observed to maintain the stability of endothelial adherens junctions (AJs), thus preventing the occurrence of no-reflow. PEDF reduced the hypoxia-induced vascular endothelial (VE)-cadherin endocytosis through PEDF/LR/Src/VE-cadherin S665 axis in vitro, which was remarkably observed to maintain endothelial AJs. Generally, PEDF might function as a relevant target for therapeutic vasculoprotection by way of regulating the phosphorylation level of VE-cadherin according to our data, thus being crucial for preventing no-reflow.
INTRODUCTION
Prompt mechanical reperfusion through primary percutaneous coronary intervention (PPCI) has been the fundamental countermeasure for managing acute myocardial infarction (AMI). 1 No-reflow has been reported to limit the therapeutic value of coronary artery recanalization and cause poor prognosis. Although no-reflow does not lead to myocardial cell death directly, it impedes the healing process by restraining blood flow into and out of the infarct area and delays the process of ventricular remodeling, eventually leading to heart failure. 2 –4 Therefore, the no-reflow phenomenon needs to be studied further, therapies need to be developed specifically targeting no-reflow, and the long-term consequences of such therapies should be determined.
Treatments aiming at minimizing microvascular damage are prioritized to protect the ischemic myocardium as recommended by the American Heart Association. 5,6 Capillaries in the no-reflow zone are dysfunctional, structurally disorganized, and highly permeable because of endothelial swelling, compression by interstitial fluid, myocyte edema, and neutrophil infiltration. 7 –9 Consequently, even if epicardial coronary artery reperfusion is achieved, microvascular recanalization and myocardial reperfusion fail. Adherens junctions (AJs) and their molecular components have been identified as major types of junction in endothelial cells. 10,11 Vascular endothelial (VE)-cadherin is a specific and exclusive vascular endothelial adhesion molecule linked to specific intracellular partners, which mediates anchorage to the actin cytoskeleton, and consequently stabilizes AJs. 12,13 Previous studies have shown that the amino acid residues of VE-cadherin S665, Y685, and Y731 (serine 665/tyrosine 685/tyrosine 731) can be modified, primarily phosphorylated, to control the permeability of vascular endothelium and leukocyte exudation. 14 However, whether the VE-cadherin endocytosis caused by hypoxia injury is related to phosphorylation of residues and it can be alleviated by regulating the phosphorylation level of the amino acid residues of VE-cadherin are yet to be discerned.
Pigment epithelium-derived factor (PEDF), a noninhibitory member of the serine protease inhibitor superfamily, has recently emerged as an endogenous antiangiogenic molecule. 15 Our previous study showed that PEDF significantly enhanced the binding of VE-cadherin with β-catenin, resulting in stabilized endothelial junctions and reduced vascular permeability in AMI rat models. 16 Laminin receptor (LR/67LR), known as PEDF receptor, is involved in cell translation, proliferation, adhesion, and movement. 17,18 Moreover, LR phosphorylation increases endothelial cell filopodia formation and motility stimulation. Therefore, it would be interesting to investigate whether there exists an association between LR phosphorylation and the disruption of the endothelial barrier function and to explore its possible therapeutic properties against vascular injury.
In this study, we specifically sought to address the following: (1) whether PEDF plays a role in no-reflow and prognosis of AMI and (2) mechanisms associated with the PEDF's effects on VE-cadherin endocytosis and its functions.
MATERIALS AND METHODS
Animals
Sprague-Dawley (SD) male rats (weighting about 250 ± 10 g, at 8–10 weeks of age) were obtained from the Experimental Animal Center of Xuzhou Medical University and housed in a controlled environment. All the experiments confirm to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication, 8th Edition, 2011). Animal experiments were approved by the Experimental Animal Center of Xuzhou Medical University (201802W010) and performed according to National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Preparation of lentivirus and plasmids
PEDF overexpression plasmids and the RNAi vector PEDF-RNAi-left ventricular (LV) of the PEDF gene that produce PEDF shRNA were successfully constructed and were then successfully packaged by 293T cells. 19 The concentrated titer of virus suspension was 2 × 1012 Tu/mL. Lentiviruses containing Src sgRNA, LR siRNA, PEDF-R siRNA, Human VE-cadherin (hVE-cad) CRISPR/Cas9 sgRNA, SrcYF (active from), SrcYF/KM (inactive form), and the wild-type (WT) hVE-cad; hVE-cadS665D and hVE-cadY685D (active from); hVE-cadS665V and hVE-cadY685F (inactive form); LRS43D, LRY47D, LRS75,78,79D, and LRT125D (active from); LRS43A, LRY47F, LRS75,78,79A, and LRT125A (inactive form) mutants were generated from Genechem (Shanghai, China). Plasmid carrying pCMV6-XL5-RPSA (LR) human cDNA clone was purchased from OriGene.
Rat AMI/recanalization model and intramyocardial gene delivery
AMI model was established surgically by ligation of the left anterior descending (LAD) coronary artery as described previously. 20 Briefly, SD rats were anesthetized with sodium pentobarbital (60 mg/kg) intraperitoneally and maintained under anesthesia using isoflurane (1.5–2.0%) mixed with air. After adequate anesthesia, the animals were intubated with a 14-gauge polyethylene catheter and ventilated with room air using a small animal ventilator (Model683; Harvard Apparatus, Boston, MA). 6-0 prolene monofilament polypropylene sutures were set up for myocardial infarction (MI) model building. Intramyocardial gene delivery was performed 5 days before the MI experiment in the rats. PEDF-LV or PEDF-RNAi-LV (2 × 107 TU) prepared in 20 μL enhanced infection solution (GeneChem, catalog no. REVG0002) was delivered with a 20‑μL syringe and 25‑gauge needle into the myocardium along the LAD. We tightened the reserved line to establish the AMI model, and released the knot to achieve coronary recanalization. We confirmed the occlusion and recanalization with ST segment elevation on the electrocardiogram. Sham-operated animals underwent an identical surgical procedure without artery ligation. All surgical procedures were performed under aseptic conditions. Mortality rate within 2 weeks following surgery was about 6%.
Myocardial positron emission tomography (PET) perfusion study and immunofluorescence analysis in vivo were performed immediately following the reperfusion by release of the ligature after 4 h of occlusion. Cardiac function evaluation, 2,3,5-triphenyltetrazolium (TTC) staining, and magnetic resonance imaging (MRI) were performed at 4 weeks after ischemia and reperfusion for long-term prognosis.
Animal cardiac function evaluation
Echocardiography was conducted under sedation by sodium pentobarbital (30 mg/kg, i.p), as described previously. 21 Two-dimensional guided M-mode echocardiography was used to determine LV chamber volume at systole and diastole and contractile parameters, such as left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic volume, and left ventricular end-systolic volume. The left ventricular fractional shortening was calculated as follows: fractional shortening (FS) (%) = (LVEDD−LVESD)/LVEDD × 100. The ejection fraction (EF) was then derived as EF (%) = (EDV−ESV)/EDV × 100.
Myocardial PET perfusion study
Myocardial PET perfusion imaging with
13
N-NH3 was conducted as previously described.
22
In brief, PET was performed by MITRO Biotech Co., Ltd. (NanJing, China). Micro PET (Siemens, Erlangen, Germany) dynamic scan was performed after
13
N-NH3 (300 ± 150 μCi) injection, respectively. The standardized uptake volume (SUV) was calculated using the following equation:
The animal models were randomly divided into six groups as follows: (1) Sham group, surgical procedure without artery ligation; (2) AMI group, arterial ligation for 4 h; (3) AMI/recanalization (AMI/R), arterial ligation for 4 h+releasing the ligation immediately before PET scan; (4) AMI/R+Vector, arterial ligation for 4 h+releasing the ligation immediately before PET scan+Vector-LV injection; (5) AMI/R+shPEDF, arterial ligation for 4 h+releasing the ligation immediately before PET scan+shPEDF-LV injection; and (6) AMI/R+PEDF, arterial ligation for 4 h+releasing the ligation immediately before PET scan+PEDF-LV injection.
Histological determination of infarct size
The selected rats were euthanized, and then the hearts were removed for MI analyses by the method of Evans blue/2,3,5-triphenyltetrazolium staining as previously. 23 Briefly, 1 week after LAD arterial ligation, 1 mL 3% Evans blue dye (Sigma-Aldrich) was injected into the ascending aorta. Then hearts were removed for MI size analyses by TTC staining. The left ventricle was isolated and cut into 2-mm-thick sections perpendicular to the axis of the LAD. Then, slices were immediately immersed in 1% TTC (Sigma-Aldrich) in phosphate buffer (pH 7.4) at 37°C for 10 min to discriminate infarcted tissue from viable myocardium. All sections were scanned from both sides using a color charge-coupled device camera (FV-10; Fujifilm Holdings Corporation, Tokyo, Japan).
The animal models were randomly divided into five groups as follows: (1) Sham group, surgical procedure without artery ligation; (2) AMI/R, arterial ligation for 4 h+releasing the ligation for 4 weeks; (3) AMI/R+Vector, arterial ligation for 4 h+releasing the ligation for 4 weeks+Vector-LV injection; (4) AMI/R+shPEDF, arterial ligation for 4 h+releasing the ligation immediately for 4 weeks+shPEDF-LV injection; and (5) AMI/R+PEDF, arterial ligation for 4 h+releasing the ligation for 4 weeks+PEDF-LV injection.
MRI acquisition and analysis
Using a commercial 8-channel phased array knee coil, the rats were imaged at a 3.0T clinical MRI scanner (Trio; Siemens) with a maximum gradient capability of 45 mT/m. The cMRI was triggered by both endothelial cell growth and respiration using a small animal monitoring and gating system (SA Instruments, Inc., Stony Brook, NY). 24
All data were acquired during free breathing of the animal. Six to 10 short-axial slices of the LV were acquired with a slice thickness of 3.0 mm without gap for all the MRI sequences. Turbo spin echo T1WI/T2WI sequences were applied for cardiac morphology and infarct with the imaging parameters of time of repetition (TR): 621/750 ms; time of echo (TE): 15/74 ms; field of view (FOV): 240 × 195 mm2; flip angle: 180°; and in-plane resolution: 0.9 × 0.9 mm2. After intravenous injection of gadolinium-tetraazacyclododecanetetraacetic acid (Dotarem®, Guerbet, France) at 0.2 mmol/kg, three contiguous short-axis first-pass perfusion-weighted imaging (PWI) measurements were acquired using segmented turbo-FLASH sequence with 80 dynamic acquisitions with parameters of TR/TE: 241.96/1.95 ms; FOV: 240 × 180 mm2; inversion time: 150 ms; flip angle: 15°; matrix: 128 × 90 mm2; and in-plane resolution: 1.88 × 1.98 mm2. The MRIs in DICOM format were processed in ImageJ. Appropriate contrast enhancement of the images was done to maximize the signal from the hyperintense region and the null signal from the nonenhanced region. For each slice, the hyperenhanced region and total LV myocardial area were calculated. Slice hyperintense areas were then summated to generate infarct volume as a percentage of LV myocardial volume.
The animal models were randomly divided into five groups as follows: (1) Sham group; (2) AMI/R, arterial ligation for 4 h+releasing the ligation for 4 weeks; (3) AMI/R+Vector, arterial ligation for 4 h+releasing the ligation for 4 weeks+Vector-LV injection; (4) AMI/R+shPEDF, arterial ligation for 4 h+releasing the ligation immediately for 4 weeks+shPEDF-LV injection; and (5) AMI/R+PEDF, arterial ligation for 4 h+releasing the ligation for 4 weeks+PEDF-LV injection.
Lectin and dextran perfusion experiment
Rats were anesthetized by intraperitoneal injection of pentobarbital 90 mg·kg−1 and 0.2 mL heparin. Deep sedation was verified by the absence of reaction to pain. In this condition, rats were decapitated and an immediate thoracotomy was conducted. The heart was quickly excised and placed in ice-cold modified Tyrode's solution of composition (in mM) 93 NaCl, 20 NaHCO3, 1 Na2HPO4, 1 MgSO4, 5 KCl, 1.8 CaCl2, 20 Na-acetate, and 20 glucose. After release of the ligature, hearts were then mounted on a Langendorff system through the aorta onto a cannula and retrogradely perfused at 9 mL/min using Tyrode's solution containing 20 mg/mL Alexa Fluor™488 conjugate dextran (D22910; Thermo Fisher Scientific) or 50 μg/mL Alexa Fluor™594 conjugate lectin (L21416; Thermo Fisher Scientific). After 30 s, hearts were harvested immediately for making frozen sections. Next, sections were fixed for 15 min with 4% paraformaldehyde, and blocked with solution containing 5% bovine serum before applying primary antibody. Specimens were incubated with anti-CD31 (Abcam; cat no. ab24590, 1:200). Then, sections were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
Immunofluorescence analysis
For heart tissue staining, frozen myocardial tissue was horizontally sliced into 5 μm section and mounted on glass slide. Both sections and cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with Triton X-100 (0.1%), and blocked with solution containing 5% bovine serum before applying primary antibody. Specimens were incubated with anti-VE-cadherin (Abcam; cat no. ab33168, lot no. GR3248725-1, 1:200) overnight at 4°C, washed three times in phosphate-buffered saline (PBS), and incubated with the secondary antibody (Life Technologies; cat no. R37119, lot no. 2072295, 1:400) under light-protected conditions for 1 h at room temperature and counterstained using 4,6-diamino-2-phenyl indole (KeyGen Biotech; cat no. KGA215–10, lot no. 20180513). Then the sections were observed under a fluorescence microscope (Olympus) or confocal laser scanning microscope (Olympus).
Cell culture and treatment
Human cardiac microvascular endothelial cells (HCMECs; ScienCell) were used between the third and fifth passage and cultured in endothelial cell medium (ScienCell) supplemented with 5% fetal bovine serum (ScienCell), 1% endothelial cell growth supplement (ScienCell), and 1% penicillin/streptomycin solution at 37°C in a humidified atmosphere containing 5% CO2. We replaced the medium every 3 days. Cells were subcultured or subjected to experimental procedures at 80–90% confluence. To establish the oxygen glucose deprivation (OGD) model, the culture medium was changed to glucose-free and serum-free medium (Gibco; Thermo Fisher Scientifc, Inc.) and placed into a tri-gas incubator (Heal Force Bio-meditech Holdings, Ltd., Shanghai, China) that was purged with 94% N2, 5% CO2, and 1% O2 for 4 h.
Establishment of stable cell lines
Approximately 0.8 × 106 HCMECs were seeded into 60-mm plastic dishes. After the cells reached about 30–35% confluence, lentiviruses containing Src siRNA or LR siRNA or hVE-cad CRISPR/Cas9 sgRNA were infected following the manufacturer's protocol at the desired multiplicity of infection (MOI = 20). After 8 h, the infection medium was removed and fresh medium was added. After an additional 64 h, GFP co-expression on the construct was used to determine efficiency of viral transduction.
Transmission electron microscopy imaging and analyses
Samples were fixed with 2.5% glutaraldehyde overnight and then incubated in 1% osmium tetroxide for 2 hours with lightproof. The final step in making the samples is embedment with fresh resin. Ultrathin sections were cut with an EM UC7 (Leica Microsystems GmbH, Wetzlar, Germany) and examined with a Tecnai G2 T12 (FEI, Hillsboro).
Protein extraction
For whole cell lysate, cells were lysed with Cell Total Protein Extraction Kit (C510003; Sangon Biotech) containing cocktail of phosphatase inhibitors and protease inhibitors according to the manufacturer's instructions. Membrane and cytoplasmic proteins were extracted using Membrane and Cytoplasmic Protein Extraction Kit (Beyotime; cat no. P0033) according to the manufacturer's instructions.
Rat tissue protein in the myocardium of the infarct zone was determined as previously described. 25 Protein was extracted with lysis buffer (pH 7.5) containing 50 mM Tris-HCl, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% Na-deoxycholate, 1% protease, and complete protease inhibitor cocktail (Sangon Biotech; catalog no. C510003). For the whole cell lysate, cells were lysed with a Cell Total Protein Extraction Kit (Sangon Biotech; catalog no. C510003) containing a cocktail of phosphatase inhibitors and protease inhibitors.
LR pulldown assays
Protein mixtures were incubated with 2 μg of anti-LR antibody at 4°C for 8 h on a mixing rotor. We added 50 μL agarose A/G protein (sc-2003; Santa) and incubated the samples overnight. After incubation, the beads were washed four times with 1 mL wash buffer (PBS containing 1 mg/mL BSA and 1% Nonidet P-40 [NP-40]). The beads were resuspended in 30 μL sodium dodecyl sulfate (SDS) sample buffer (62.5 mM Tris–HCl, 10% glycerol, 2% [w/v] SDS, 5% 2-mercaptoethanol, and 10 lg/mL bromophenol blue, pH 6.8), boiled for 5 min, and the supernatants were subjected to 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
Western blotting analysis
Immunoblotting was performed as previously described. 16 In brief, equivalent amount of protein was fractionated on a 10% or 12.5% SDS-PAGE and electrotransferred to 8 μm nitrocellulose membranes (SCWP04700; Millipore). Nonspecific binding was blocked by incubation with 5% non-fat milk in 0.1% Tween 20/Tris-buffered saline for 30 min, followed by overnight incubation at 4°C with the primary antibody(s). Primary antibodies for PEDF (Proteintech; cat no. 26045-1-AP, lot no. 00071340, 1:1,000), VE-cadherin (Abcam; cat no. ab33168, lot no. GR3248725-1, 1:1,000), pS665-VE-cad (Biosynergics; lot no. 2018078, 1:500), pY685-VE-cad (Biosynergics; lot no. 2018131, 1:500), pY731-VE-cad (Biosynergics; lot no. 2018061, 1:500), Src (Proteintech; cat no. 11097-1-AP, lot no. 00059200, 1:1,000), pY418-Src (Biosynergics; 1:500), LR (Proteintech; cat no. 14533-1-AP, 1:1,000), pT125-LR (Biosynergics; lot no. 2019021, 1:500), anti-phospho-Tyrosine (Cell Signaling; cat no. 8954, 1:400), anti-phospho-Serine (Cell Signaling; cat no. 9386, 1:400), anti-phospho-Tyrosine (Cell Signaling; cat no. 2351, 1:400), or β-Actin (Bioworld; cat no. AP0060, lot no. AA24142, 1:5,000) were followed by fluorescently labeled anti-mouse or rabbit antibodies (Li-Cor) and imaged using the Odyssey infrared imaging system (Li-Cor). Western blots were quantified using ImageJ software. Protein levels were calculated from the ratio of corresponding protein/β-Actin.
Identification of phosphorylation sites in LR by liquid chromatography/tandem mass spectrometry analysis
The solution containing LR-purified protein was reduced with 10 mM dithiothreitol followed by alkylation with 50 mM iodoacetamide (in the dark) and digested with TPCK Immobilized Magnetic Trypsin (Clontec; Saint-Germain-en-Laye, France) for 90 min. The digestions were stopped by removing the magnetic beads and addition of trifluoroacetic acid to a final concentration of 0.5%. Phosphopeptide enrichment was carried out using TiO2 and immobilized metal ion affinity chromatography. The tryptic peptides were Zip-Tip concentrated and reconstituted in 20 μL of 0.1% (v/v) formic acid. One μL of the sample was loaded onto a home-made reversed-phase analytical column packed with Reprosil-Pur Basic C18, 1.9 μm. The gradient comprised an increase from 2% to 22% solvent B (0.1% formic acid in 98% acetonitrile) over 40 min, 22% to 35% in 12 min, and climbing to 80% in 4 min and then holding at 80% for the last 4 min, all at a constant flow rate of 350 nL/min on an EASY-nLC 1000 UPLC system. The ion source was ElectroSpray ionization (ESI, nano-spray), fragmentation mode was collision-induced dissociation (y and b ions), mass spectrometry (MS) scan mode was FT-ICR/Orbitrap, and MS/MS scan in the range from 350 to 1,800 m/z was linear ion trap.
The resulting MS/MS data were processed using a Simple Modular Architecture Research Tool database with the PEAKS 7.0 (Matrix Science, UK) database search engine. Trypsin/P was specified as cleavage enzyme allowing up to three missing cleavages, three modifications per peptide and five charges. Monoisotopic peptide tolerance was set to 20 ppm, and fragment mass tolerance was set to 0.1 Da. Carbamidomethylation on Cys was specified as fixed modification and oxidation on Met; phosphorylation on serine, tyrosine, and threonine and acetylation on protein N-terminal were specified as variable modifications. Only matching proteins and peptides showing a −10 lgP value ≥30 and ≥15 were considered for identification purposes.
Statistical analysis
For all quantitative analyses, other than when specifically noted, data are expressed as mean ± standard error of the mean and repeated at least thrice in independent biological samples. Statistical significance was analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego). Two-tailed Student's t tests were performed for two experimental groups, and one-way analysis of variance (ANOVA) was performed for three or more experimental groups. p < 0.05 was considered significant difference.
RESULTS
PEDF prevents no-reflow and confers secondary cardioprotection in rat AMI/R model
To determine the detailed role of PEDF in the occurrence of no-reflow, intramyocardial gene delivery was performed 5 days before the AMI experiment. The overexpression and knockdown efficiency of the virus in rat hearts were examined by Western blot analysis (Supplementary Fig. S1A, B). To explore the myocardial reperfusion in rat AMI/R model, PET perfusion imaging with 13 N-NH3 was performed after temporary coronary occlusion for 4 h (Fig. 1A). For AMI/R rats, when the ligature was released, some portion of the myocardium did not receive sufficient blood perfusion, despite epicardial coronary artery reperfusion. In other words, recanalization of the LAD did not result in the reperfusion of the capillaries and no-reflow occurred (ischemic myocardial volume/total volume (%), 37.24 ± 1.79 [AMI] vs. 27.69 ± 1.44 [AMI/R], p < 0.01; SUV-mean, 2.50 ± 0.13 [AMI] vs. 3.14 ± 0.09 [AMI/R], p < 0.01). Compared with the AMI/R rats, overexpression of PEDF was found to significantly improve the perfusion level of ischemic myocardium and reduce the volume of ischemic myocardium after coronary recanalization (ischemic myocardial volume/total volume %], 27.69 ± 1.44 [AMI/R] vs. 22.49 ± 1.04 [AMI/R+PEDF], p < 0.05; SUV-mean, 3.14 ± 0.09 [AMI/R] vs. 3.48 ± 0.10 [AMI/R+PEDF], p < 0.05). Furthermore, knocking down of endogenous PEDF led to an increase in no-reflow area (Fig. 1B, C). To assess the effects of PEDF on the severity and prognosis of AMI/R rats, infarct size and cardiac function were determined by way of Evans blue/TTC staining (Supplementary Fig. S2), MRI analysis (Fig. 1D, E), and echocardiography. The results demonstrated that PEDF treatment could reduce the infarct size by 20–24%. Moreover, the EF and FS values of PEDF rats were both higher than those of the AMI/R rats (Fig. 1F, G).
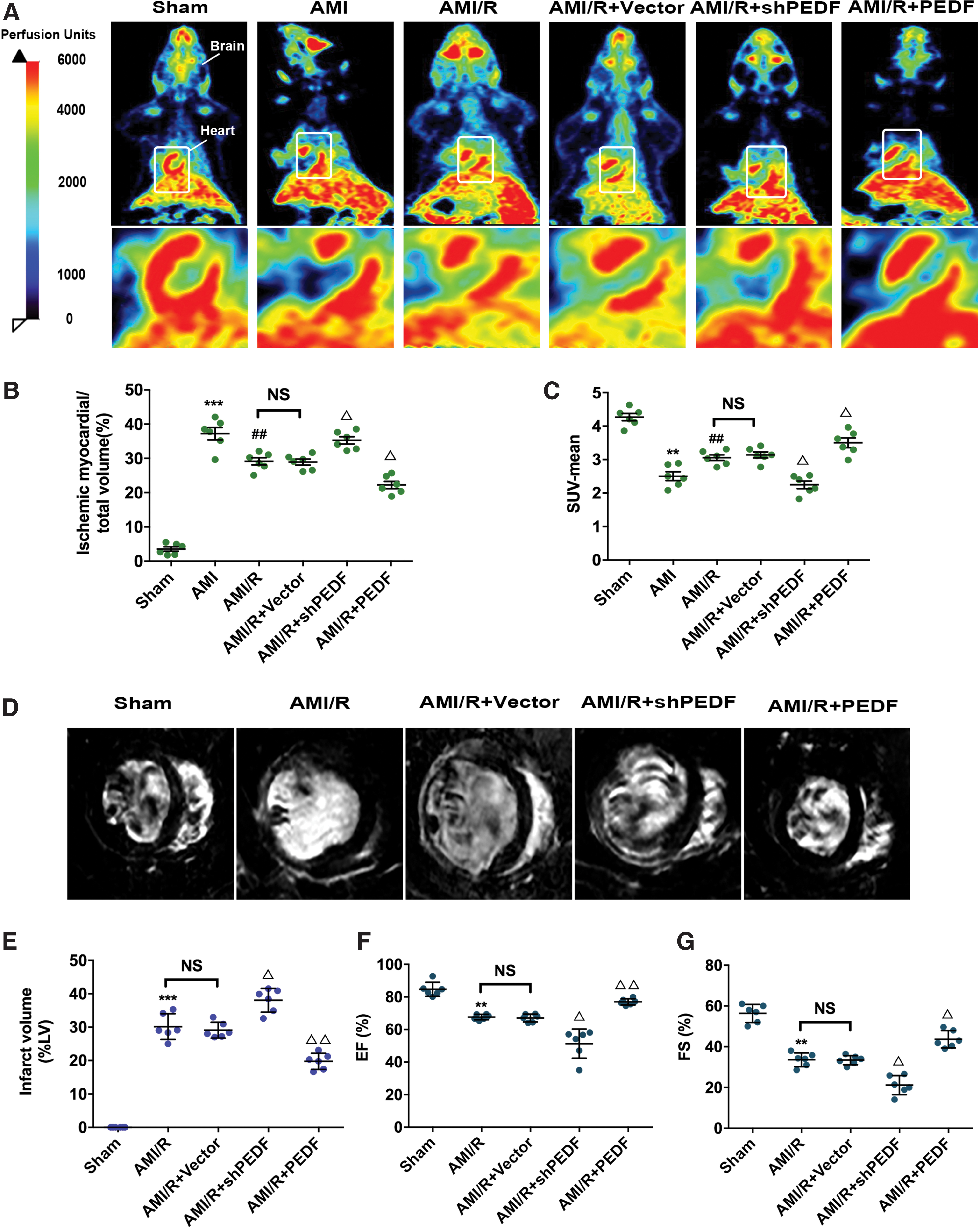
PEDF prevents no-reflow and confers secondary cardioprotection in rat AMI/R model.
PEDF maintains the stability of endothelial AJs to preserve the perfusion function of microvessels during reperfusion post-AMI
The destructions of microvascular structure and function are the main pathogenesis of no-reflow. In vivo, lectin perfusion assay was performed to determine the effects of ischemia and PEDF overexpression on vascular permeability. Lectin functioned as a marker of perfused vessels under respective conditions. As shown in Fig. 2A and C, a decrease in microvascular perfusion efficiency was observed in control rats after LAD recanalization. Overexpression of PEDF was found to improve microvascular perfusion efficiency significantly compared with that in the control group. It was also found that PEDF shRNA administration aggravated the deterioration of microvascular perfusion. This demonstrated that PEDF exhibits protective effects on microvascular perfusion during reperfusion post-AMI. Dextran perfusion experiment to determine the effects of PEDF on vascular permeability (Fig. 2B) was performed thereafter. Ischemia for 4 h was observed to cause a significant increase in vascular permeability, and silent PEDF aggravated this process. Contrarily, the PEDF treatment showed lighter dextran leakage after reperfusion (Fig. 2D). Electron microscopy of no-reflow zones showed clear-cut evidence that PEDF reduces hypoxia-induced interstitial edema and focal swelling of the endothelium and myocardium. In addition, breaks in the endothelial continuum were observed, as were occasional microvascular hemorrhage and the infiltration of inflammatory cells after ischemia for 4 h (Supplementary Fig. S3A).

PEDF maintains the perfusion function of microvessels and reduces leakage during reperfusion post-AMI.
The stability of adherens junctions is critical for the maintenance of the endothelial permeability and integrity. Therefore, we further checked the gap at cell-cell junctions by TEM observation in in vitro cultured HCMECs. The expression level of PEDF in OGD-treated endothelial cells is shown in Supplementary Fig. S1C and D. After OGD for 4 h, gaps were observed between adjacent endothelial cells and the elevated distance could be clearly found with knocking down of endogenous PEDF. Overexpression of PEDF prominently reduced the size of endothelial gaps in HCMEC OGD models compared with OGD control group (Supplementary Fig. S3B).
PEDF inhibits hypoxia-induced VE-cadherin endocytosis mostly by regulating the phosphorylation of VE-cadherin at S665
Next, focused analyses were performed on the endothelial adherens junctions in HCMECs. The results of Western blot and PCR analysis demonstrated that significant decrease in the protein expression of VE-cadherin can be observed after OGD 8-h treatment, while OGD 4 h-induced internalization of VE-cadherin did not cause its significant degradation and PEDF treatment did not affect the RNA expression levels of VE-cadherin in both normoxic and hypoxic environments (Supplementary Fig. S4A and B). Immunofluorescence analysis indicated that OGD treatment for 4 h induced severe VE-cadherin endocytosis in endothelial cells. PEDF significantly inhibited the transfer of VE-cadherin from the membrane to the cytoplasm, whereas PEDF shRNA treatment aggravated this process (Fig. 3A). As shown in Western blot analysis, after OGD for 4 h, an increased expression of VE-cadherin was observed in cytoplasm. The expression of cytosolic VE-cadherin was decreased in the PEDF group compared with the control group, whereas in the shPEDF group, cytoplasm localization of VE-cadherin was substantially reduced (Fig. 3B). Further data indicated that the phosphorylation of VE-cadherin has no significant correlation with serum-free culture (Supplementary Fig. S4C, D).

PEDF inhibits hypoxia-induced VE-cadherin endocytosis by regulating the S665 phosphorylation of VE-cadherin in HCMECs.
To address whether VE-cadherin phosphorylation could participate in the regulation effect of PEDF on OGD-induced VE-cadherin endocytosis, the expression of phosphorylated S665, Y685, and Y731 was detected thereafter. The results indicated that OGD treatment for 4 h led to the phosphorylation of VE-cadherin at S665, Y685, and Y731. PEDF overexpression was observed to reduce the expression of pS665-VE-cad and pY685-VE-cad significantly (Fig. 3C).
Then we engineered VE-cadherin mutants that were nonphosphorylable (S665V and Y685F) or that mimicked the phosphorylated states (S665D and Y685D) to analyze whether PEDF inhibits VE-cadherin endocytosis by reducing S665 or Y685 phosphorylation. The transfection efficiency of VE-cadherin sgRNA and mutants has been shown in Supplementary Fig. S5. Immunofluorescence and Western blot indicated that, as a control, WT VE-cadherin was mainly distributed on the cell membrane, whereas hVE-cadS665D remained mostly in the cytoplasm spontaneously. VE-cadherin was found to be lightly internalized in the hVE-cadY685D group (Fig. 3D, E). OGD treatment for 4 h induced severe VE-cadherin endocytosis in endothelial cells with depletion of PEDF. Similar results were also found in WT VE-cadherin and PEDF shRNA group. Furthermore, we found it was hVE-cadS665V rather than hVE-cadY685F that stably remained at the cell borders and was almost unaffected by OGD treatment for 4 h in endothelial cells with depletion of PEDF (Fig. 3F, G).
Src kinase phosphorylation mediates the regulation of the pS665-VE-cad protein level by PEDF
We theorized that Src was involved in the transduction of phosphate cascade signals. Western blotting analysis indicated that OGD treatment for 4 h increased the expression of phospho-Src, whereas PEDF overexpression significantly decreases the protein level of phospho-Src. Moreover, the effects of various treatments on the protein levels of phospho-Src and pS665-VE-cad were exhibited to be similar on quantitative analysis (Fig. 4A). To investigate whether phosphorylation of Src is involved in the regulation of pS665-VE-cad and subsequent VE-cadherin endocytosis by PEDF, lentiviruses containing SrcYF (ASrc, active from) and SrcYF/KM (ISrc, inactive form) were engineered. The infection efficiency of Src mutant lentiviruses has been shown in the Supplementary Fig. S6. Immunofluorescence determination indicated that ISrc almost completely blocked the serious increase in VE-cadherin endocytosis caused by OGD treatment for 4 h in endothelial cells with depletion of PEDF. Moreover, we noted that ASrc has the activity to antagonize the attenuated effect of PEDF on VE-cadherin endocytosis induced by OGD for 4 h (Fig. 4B). Next, we examined the expression of pS665-VE-cad in endothelial cells with corresponding treatments. ISrc was found to effectively inhibit the increase of pS665-VE-cad protein level induced by OGD for 4 h in endothelial cells with depletion of PEDF. ASrc was observed to almost block the attenuated effect of PEDF on S665-VE-cad phosphorylation induced by OGD for 4 h (Fig. 4C).
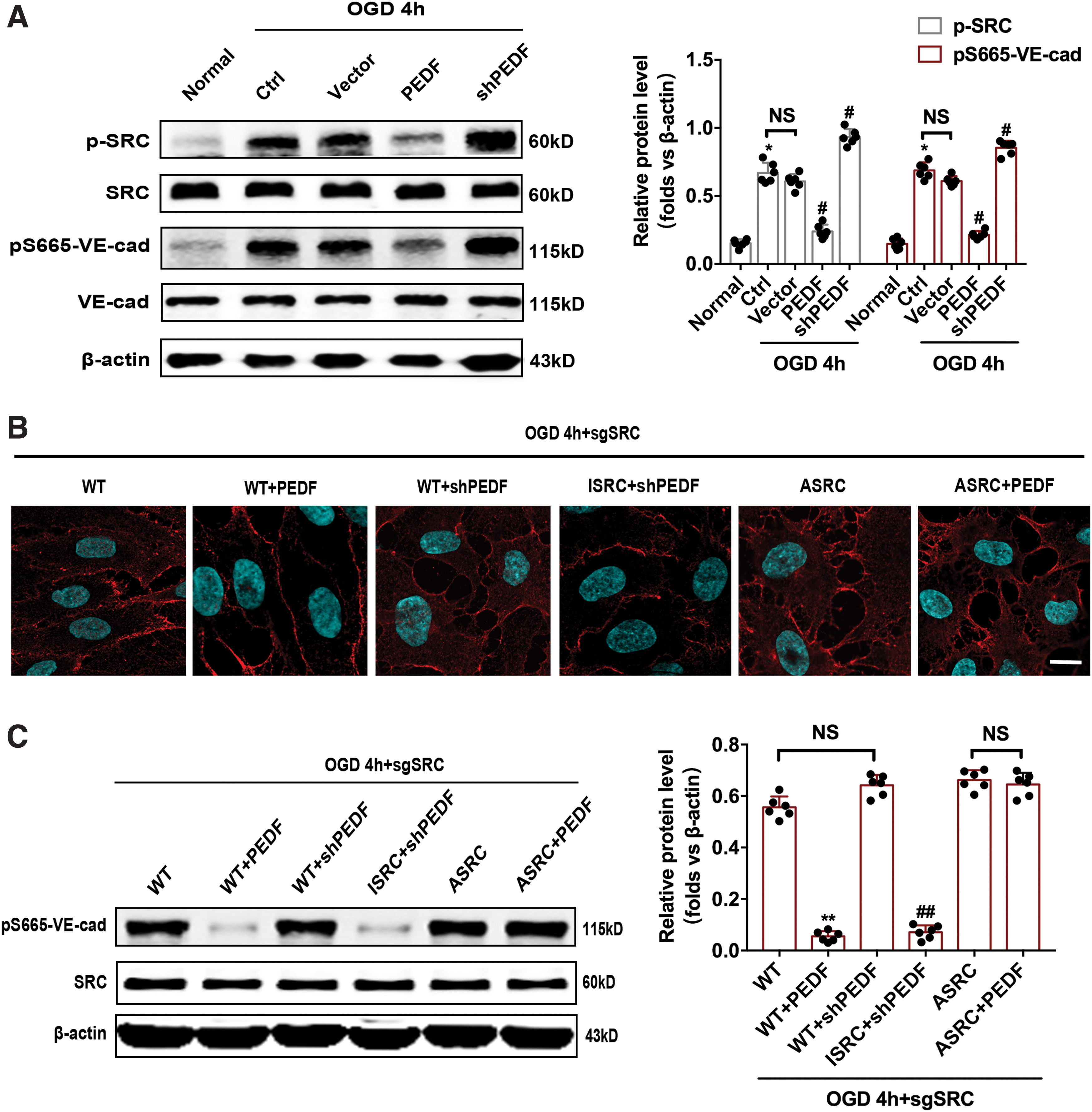
Src kinase phosphorylation mediates the regulation of the pS665-VE-cad protein level by PEDF.
PEDF regulates pS665-VE-cad protein levels by downregulating T125 phosphorylation of LR
To further determine the role of LR in this phosphorylation-related signaling pathway, the infection efficiency of siLR and siPEDFR lentiviruses was confirmed by Western blot analysis (Supplementary Fig. S7A). The results show that siLR abolished the PEDF effect on reducing the expression of pS665-VE-cad (Fig. 5A). It is clear that PEDF regulating pS665-VE-cad protein levels requires its membrane receptor, LR. We hypothesized that LR is also involved in the transduction of this phosphorylation signal and subsequent studies showed that OGD treatment for 4 h caused the serine/threonine/tyrosine phosphorylation of LR. PEDF overexpression significantly inhibited this process (Fig. 5B).
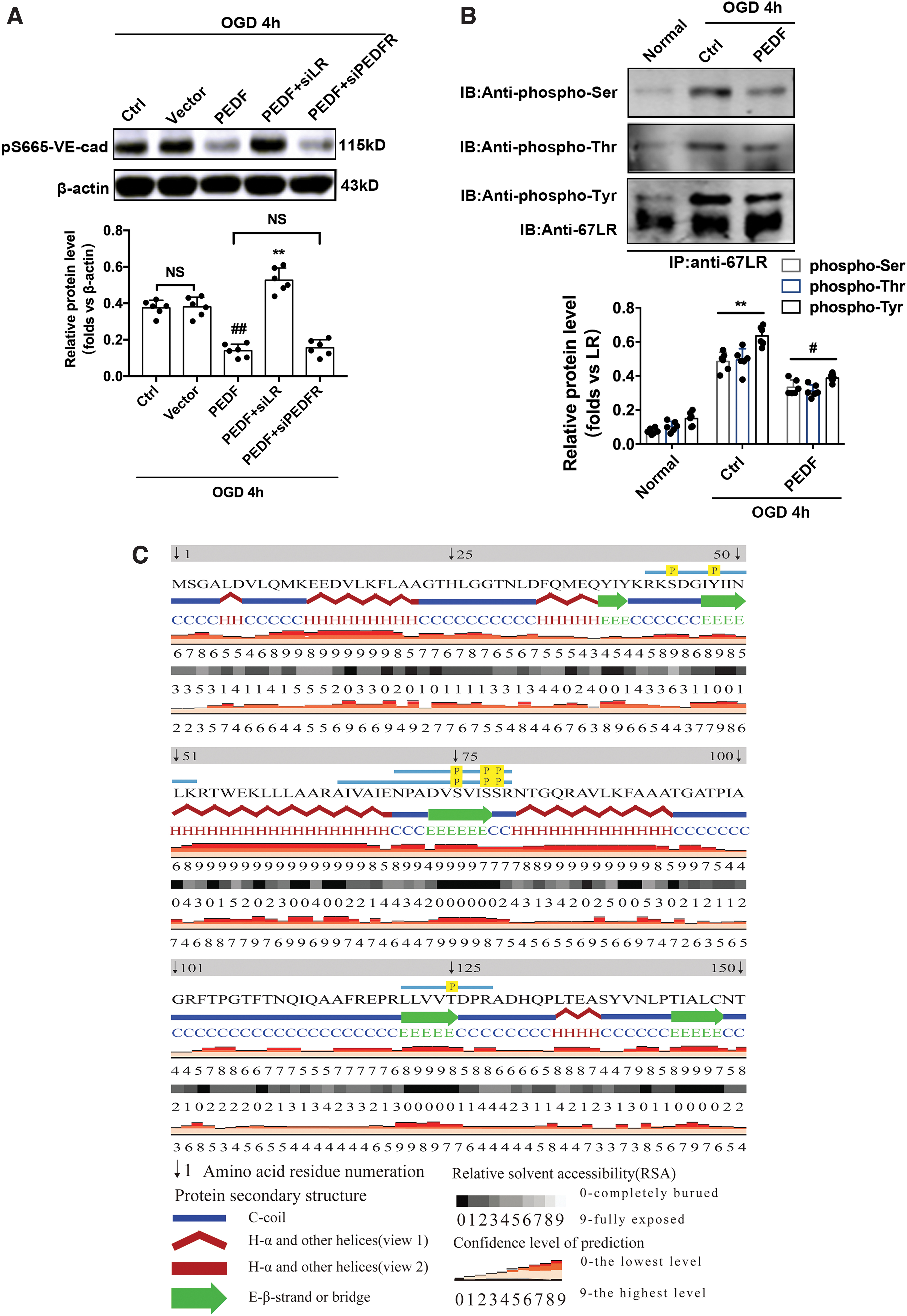
PEDF regulates pS665-VE-cad protein levels through cell surface receptor LR.
Next, LR-purified protein was analyzed by mass spectrometry. It was found that S43, Y47, S75,78,79, and T125 were phosphorylated induced by OGD for 4 h in endothelial cells (Fig. 5C and Supplementary Fig. S8).
Immunofluorescence and Western blot determination indicated that expression level S665 phosphorylation of VE-cadherin was increased in endothelial cells by transfection of LRT125D mutant virus (Fig. 6A, B). Endothelial cells transfected with LRT125D virus almost reverse the attenuated effect of PEDF on S665-VE-cad phosphorylation induced by OGD for 4 h. However, S665-VE-cad phosphorylation was still at a low level, despite knockdown of PEDF in endothelial cells transfected with LRT125A virus (Fig. 6C). These findings suggested that PEDF regulates S665 phosphorylation and internalization of VE-cadherin by regulating LR phosphorylation at T125. The results of Western blot indicated that OGD treatment for 4 h significantly increase the expression of phospho-T125-LR in endothelial cells. PEDF overexpression could substantially inhibit the phosphorylation of LR at T125, but due to knockdown of PEDF by short interfering RNA, a significant increase of phospho-T125-LR was observed compared with the control group (Fig. 6D).
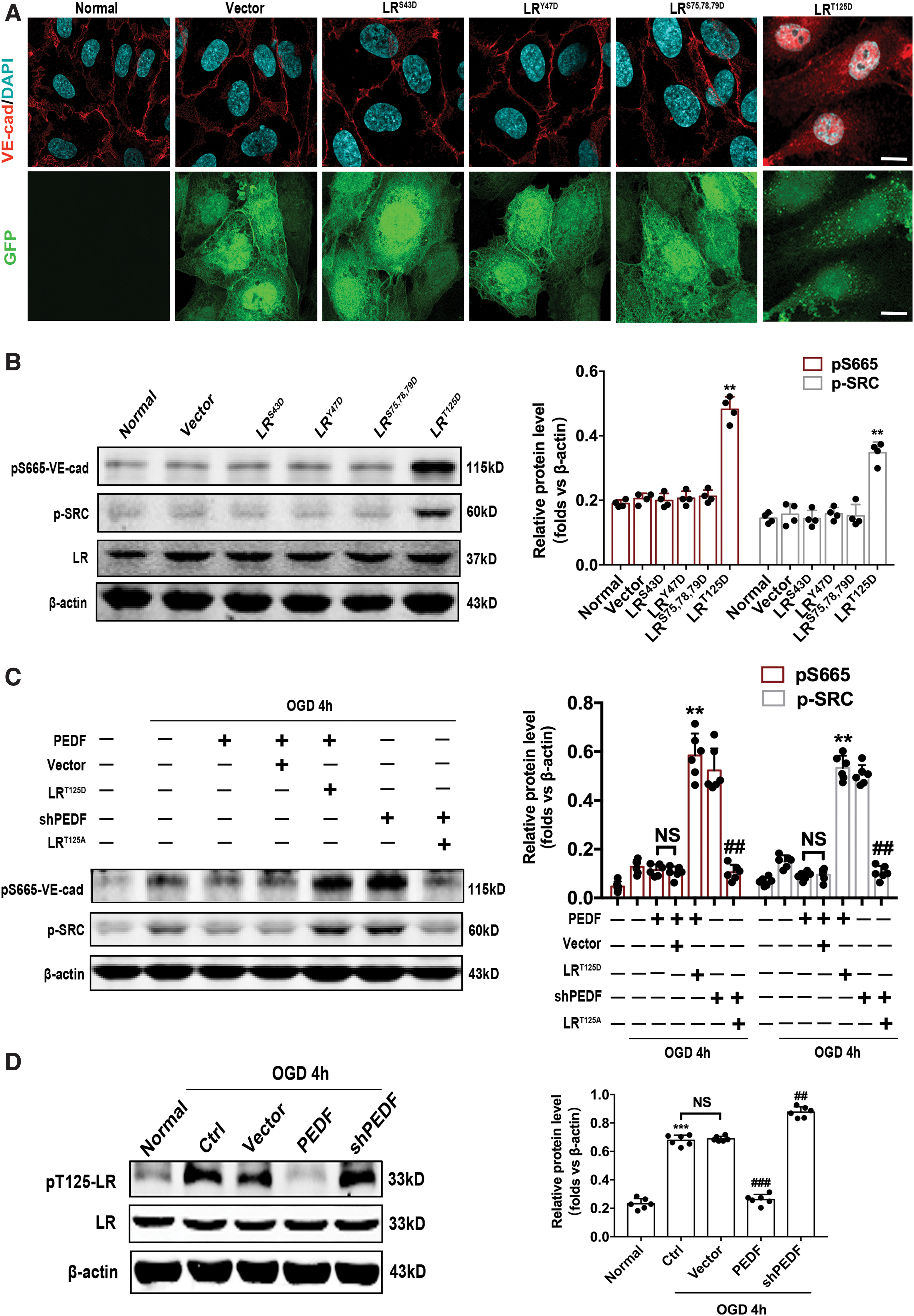
The T125 phosphorylation of 67LR mediates regulation of VE-cadherin endocytosis and the protein level of pS665-VE-cad by PEDF.
DISCUSSION
The current guideline-recommended treatment for AMI patients is mechanical revascularization by way of PPCI, which is effective at reducing mortality. However, although this procedure restores blood flow through the coronary artery, impaired myocardial perfusion, also known as no-reflow, is frequently observed. 23 Many drugs, such as Verapamil, Abciximab, and others, have been tried as adjunctive treatments to PPCI. Some of these have been proved to be effective, but not very significantly. 26,27 In this study, we introduced an efficient method to reduce the hypoxia-induced VE-cadherin endocytosis through PEDF/LR/Src/VE-cadherin S665 axis that markedly maintains endothelial AJs, reduces the incidence of no-reflow, and eventually, improves prognosis in rat AMI/R models.
To better understand no-reflow and explore effective strategies, we constructed an AMI/R model that can support intramyocardial gene delivery in the rat heart. Compared with open-chest AMI/R models, the advantage of this model is that it is more convenient and time-saving to achieve coronary artery occlusion and recanalization without the need for secondary anesthesia and secondary surgery. However, it should be noted that it is very important to bury the reserved line in the correct position, otherwise it may lead to incomplete LAD ligation or inconsistent ligation positions. Then we performed PET perfusion imaging with 13 N-NH3 in AMI/R rats to assess the degree of no-reflow and the results indicated that 22–28% of myocardium was still in ischemic state after LAD recanalization and PEDF treatment. PEDF treatment effectively reduces the area of no-reflow. In the long term, the overexpression of PEDF effectively limits the infarct size and ameliorates cardiac function 4 weeks after coronary artery recanalization. These findings highlight the important value of PEDF in preventing no-reflow and improving the prognosis of AMI. However, it is still unclear whether PEDF has a positive therapeutic effect on no-reflow that has occurred, which is one of the limitations of this study. We recognize the importance of this issue and try to address it in the future.
The pathogenetic components of no-reflow are very complicated. Indeed, coronary microvascular dysfunction caused by ischemic injury during AMI has been increasingly identified as a vital contributor to the occurrence of no-reflow. 28,29 It follows the guidelines set by the American Heart Association that treatments capable of minimizing microvascular damage should be prioritized to protect injured myocardium. In this study, AMI (4 h; in vivo) or OGD (4 h; in vitro) treatments were observed to induce VE-cadherin endocytosis, which resulted in the loss of AJ's function and the disassembly of the blood-vessel walls. We believe that it is the coronary microvascular dysfunction and its secondary severe interstitial edema and local swelling of the endothelium and myocardium that hinder the flow of red blood cells and ultimately lead to no-reflow. In this study, PEDF has showed particularly excellent biological activity in inhibiting hypoxia-induced endocytosis of VE-cadherin, maintaining the stability of AJs and protecting vessels' function. At the mechanistic level, further evidence indicated that this process was brought about by a series of complex phosphorylation cascade signals.
Several studies have reported a correlation between the phosphorylation of VE-cadherin amino acid residues and a decrease in the stability of AJs. 30 We found that OGD treatment can simultaneously trigger the phosphorylation of VE-cadherin S665/Y685/Y731. Phosphorylation of S665 was mainly found to be responsible for the internalization of VE-cadherin, which lead to the collapse of AJ. We further established hVE-cad mutants that were nonphosphorylable (S665V) or that mimicked the phosphorylated state (S665D). We observed that hVE-cadS665D spontaneously enters into the cytosol even in the absence of the OGD treatment. In other words, the phosphorylation level of S665 directly determines the distribution of VE-cadherin on the cell membrane or in the cytoplasm. Therefore, it is precisely because PEDF significantly downregulates the phosphorylation level of VE-cadherin S665 in the endothelial cells under OGD treatment, which in turn plays a role in maintaining the endothelial cell junction stability.
It is evident that after hypoxia stimulation, the phosphorylation-related signaling pathway is activated and transmitted downstream in endothelial cells, thus causing phosphorylation of VE-cadherin S665. It is a process wherein the activity of the Src is indeed required. In this study, the effects of OGD (4 h), PEDF, and shPEDF treatments on the phosphorylation of Src and VE-cadherin S665 were observed to be synchronous. PEDF significantly downregulated the phosphorylation of Src induced by OGD treatment. The application of ASrc form blocked the activity of PEDF to reduce the phosphorylation of VE-cadherin S665, while ISrc treatment significantly inhibited the phosphorylation of VE-cadherin S665 and endocytosis induced by OGD and shPEDF treatment. Thus, our findings are in agreement with the hypothesis that Src acts as an important intermediate signaling protein in this phosphorylation cascade pathway for signal transduction. In addition, it is easy to understand that the tyrosine kinase Src does not act directly on VE-cadherin and phosphorylates S665. However, how the signal is transmitted from SRC to VE-cadherin is still unknown. Gavard et al. 14 proposed that VEGF regulated the phosphorylation of VE-cadherin through the SRC/VV2/PAC/p21-activated kinase (PAK)/VE-cadherin axis. In brief, the activation of the small GTPase Rac by VEGFR-2 was through the Src-dependent phosphorylation of Vav2, a guanine nucleotide-exchange factor. Rac activation, in turn, promotes the PAK-mediated phosphorylation of a highly conserved motif within the intracellular tail of VE-cadherin. In this study, whether the hypoxia-induced signal transduction from SRC to VE-cadherin also depends on the above pathway or through other unknown signal pathways remains to be further verified. We recognize the importance of this issue and try to address it accordingly.
As shown in our earlier studies, LR is an important receptor found on the endothelial cell membrane and binds to its ligand (PEDF), which in turn mediates various biological activities of PEDF occurring on endothelial cells. LR shRNA was found to almost completely block the benefits of PEDF to AJs, suggesting that LR participates in the regulation of PEDF on hypoxia-induced endocytosis of VE-cadherin. Therefore, we theorized that LR may be the upstream link of this phosphorylation cascade signaling pathway. Results of MALDI-TOF-MS analysis indicated that LR amino acid residues (S43, Y47, S75,78,79, and T125) are phosphorylated in OGD conditions. Further evidence suggests that the phosphorylation of T125 stimulates the downstream phosphorylation of Src and VE-cadherin S665. PEDF may regulate S665 phosphorylation and internalization of VE-cadherin by downregulating the phosphorylation of LR at T125. The biological effects of other phosphorylation residues of LR call for further investigations.
To summarize, we demonstrated the remarkable efficacy of PEDF in maintaining microvascular AJ stability and vascular structural integrity. The application of PEDF to minimize microvascular damage during ischemia is a feasible strategy to prevent the occurrence of no-reflow (Fig. 7). These data provide a promising approach for optimizing AMI management strategies and improving the prognosis of AMI.

The mechanism of PEDF in maintaining microvascular AJ stability and preventing the occurrence of no-reflow. AJ, adherens junction. Color images are available online.
Footnotes
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (81870235, 81400227, and 81570242), the Social Development Projects of Key R&D Programs in Jiangsu Province (BE2019643), the National Natural Science Foundation of Jiangsu Province (BK20171178), the General Program of Jiangsu Commission of Health (H2017083), the Project of Invigorating Health Care through Science, Technology and Education, the Jiangsu Provincial Medical Youth Talent (QNRC2016778), and the Foundation of Jiangsu Province Six Talents Peak (2015-WSN-063).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.