
Abstract
Ex vivo gene therapy procedures targeting hematopoietic stem and progenitor cells (HSPCs) predominantly utilize lentivirus-based vectors for gene transfer. We provide the first pre-clinical evidence of the therapeutic utility of a foamy virus vector (FVV) for the genetic correction of human leukocyte adhesion deficiency type 1 (LAD-1), an inherited primary immunodeficiency resulting from mutation of the β2 integrin common chain, CD18. CD34+ HSPCs isolated from a severely affected LAD-1 patient were transduced under a current good manufacturing practice-compatible protocol with FVV harboring a therapeutic CD18 transgene. LAD-1-associated cellular chemotactic defects were ameliorated in transgene-positive, myeloid-differentiated LAD-1 cells assayed in response to a strong neutrophil chemoattractant in vitro. Xenotransplantation of vector-transduced LAD-1 HSPCs in immunodeficient (NSG) mice resulted in long-term (∼5 months) human cell engraftment within murine bone marrow. Moreover, engrafted LAD-1 myeloid cells displayed in vivo levels of transgene marking previously reported to ameliorate the LAD-1 phenotype in a large animal model of the disease. Vector insertion site analysis revealed a favorable vector integration profile with no overt evidence of genotoxicity. These results coupled with the unique biological features of wild-type foamy virus support the development of FVVs for ex vivo gene therapy of LAD-1.
INTRODUCTION
Leukocyte adhesion deficiency type 1 (LAD-1) is an inherited, primary immunodeficiency caused by a variety of autosomal recessive mutations of the ITGB2 gene. 1 ITGB2 encodes the β2 integrin common chain, CD18. Integrins comprise a large family of cell surface protein heterodimers primarily involved in receptor activity, cell-to-cell interactions, and binding to the extracellular matrix. 2 CD18 is common to a variety of integrins important for leukocyte activity, including lymphocyte function-associated antigen 1 (LFA-1; CD11a/CD18), macrophage-1 antigen (Mac-1; CD11b/CD18), complement receptor 4 (CR4; CD11c/CD18), and αDβ2 (CD11d/CD18). 3 CD18 mutation impairs binding of circulating leukocytes to activated vascular endothelium and subsequent leukocyte extravasation to sites of infection within tissues. Accordingly, individuals with LAD-1 are functionally immunocompromised and suffer from recurrent, life-threatening bouts of bacterial infection and high mortality rate in the first years of life.
Due to the genetic nature of the disease, current treatment regimens are limited to prophylactic antibiotics and ad hoc granulocyte infusions. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) can be curative, but is restricted by the availability of HLA-matched donors and is associated with significant risk, such as graft failure or graft-versus-host disease. More recently, ex vivo gene therapy, which utilizes a patient's autologous hematopoietic stem cells, has presented a promising alternative to allo-HSCT. This approach has been used in experimental treatments for various genetic disorders of the blood, including adenosine deaminase-deficient severe combined immunodeficiency (SCID), X-linked SCID (SCID-X1), Wiskott-Aldrich syndrome, chronic granulomatous disease, and sickle cell disease. 4 –6
The viral vectors of choice for these ex vivo gene therapy treatments, namely gammaretrovirus- or lentivirus-based vectors, have been derived exclusively from members of the Orthoretrovirinae subfamily of retroviruses. Although effective, gammaretrovirus-based vectors have been associated with adverse outcomes, 7,8 and both gammaretroviral vectors (GV) and lentiviral vectors (LV) demonstrate a propensity for integration near or within actively transcribed genes. 9 –11
In contrast to GV and LV, vectors derived from prototypic foamy virus, 12 a member of the Spumaretrovirinae subfamily of retroviruses, show a decreased preference for integration within genes or near proto-oncogenes. 13,14 In addition, wild-type foamy viruses, although prevalent among mammalian species, have not been associated with disease. 15 An interesting biological feature of foamy viruses that distinguishes them from other Retroviridae is the occurrence of reverse transcription within the maturing foamy virus capsid, most likely before egress. 16 –18 As a result of this replicative feature, a portion of packaged foamy virus vector (FVV) genomes is competent for DNA integration upon transduction, thus shortening the requisite exposure time of target cells to vector, as well as the duration of ex vivo culture.
This feature, therefore, allows transduction of freshly thawed CD34+ hematopoietic stem and progenitor cells (HSPCs) in the absence of pre-stimulation or prolonged culture in the presence of vector, cellular manipulations that can decrease the total number of repopulating HSPCs. 19 Moreover, foamy virus long terminal repeats have been shown to contain insulator sequences (i.e., cis-acting DNA sequences that establish partitions among transcriptional domains) 20 that allow the incorporation of strong heterologous promoters, while maintaining a reduced propensity for activation of nearby genes. 21 These features, combined with the broad tissue tropism of nonpseudotyped foamy viruses, have led to increasing interest in FVV as an alternative to GV and LV. 22 –24 Based upon the practical advantages and enhanced safety profile of FVVs, we have evaluated their use as gene transfer vehicles in CD34+ HSPCs isolated from an individual with a severe form of LAD-1.
MATERIALS AND METHODS
Materials and methods are detailed in the Supplementary Material section.
RESULTS
Current good manufacturing practices-compatible FVV transduction of CD34+ LAD-1 HSPCs
Mobilized peripheral blood CD34+ HSPCs were isolated from a 19-year old, severely affected LAD-1 patient for evaluation of (i) FVV-mediated HSPC transduction, (ii) amelioration of LAD-1 myeloid cell chemotactic defects in vitro, (iii) long-term engraftment of gene-corrected LAD-1 cells in immunodeficient NSG mice, and (iv) in vivo transgene marking. The experimental design is shown in Fig. 1A. This patient has a homozygous deletion that spans from intron 11 to +4 of the splice donor site of intron 13 of the ITGB2 gene (c.1412 + 424_c.2080 + 4del).
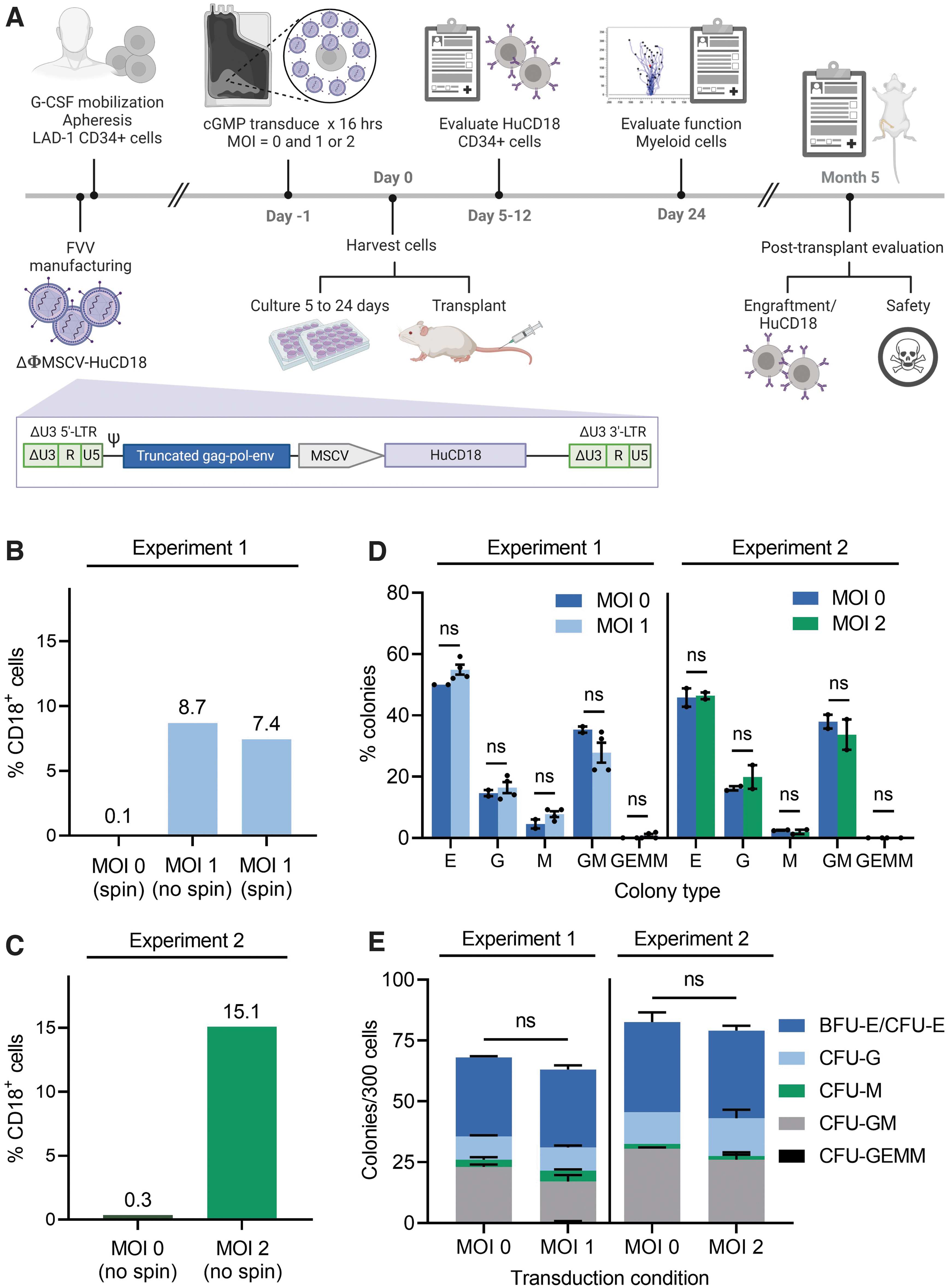
Experimental design and in vitro characterization of FVV transduction in CD34+ LAD-1 HSPCs.
Flow cytometric analysis confirmed a remarkable deficit of cell surface CD18 expression in HSPCs isolated from the LAD-1 patient compared to that of HSPCs isolated from a healthy donor (HD) (Supplementary Fig. S1). To facilitate translation of FVV-based ex vivo gene therapy for LAD-1 to a clinically relevant current good manufacturing practices (cGMP)-compliant protocol, we performed ex vivo transduction of LAD-1 HSPCs in scalable, single-use fluoroethyl polymer (FEP) cell culture bags under cGMP-compatible procedures.
Ex vivo transduction experiments utilized a nonpseudotyped, self-inactivating FVV (designated ΔΦMSCV-HuCD18, Fig. 1A) bearing a 2.3-kb human CD18 open reading frame under the transcriptional control of a murine stem cell virus (MSCV)-derived promoter/enhancer. ΔΦMSCV-HuCD18 was packaged using previously described, cGMP-compatible production and purification procedures. 25 Two serial transduction/transplantation experiments were performed.
The first experiment utilized a multiplicity of infection (MOI) of 1 FVV transducing unit per cell (TU/cell) and the second experiment utilized an MOI of 2 TU/cell. In both instances, freshly thawed CD34+ LAD-1 HSPCs were transduced overnight (∼16 h) with ΔΦMSCV-HuCD18, and samples of transduced LAD-1 HSPCs were either transplanted into immunodeficient NSG mice or plated for colony-forming unit analysis immediately following vector removal. A portion of the LAD-1 HSPCs transduced at an MOI of 1 was cultured for ∼24 days under conditions supporting myeloid differentiation to assess correction of leukocyte chemotactic defects in genetically modified LAD-1 cells.
In the first experiment (Experiment 1; MOI = 1 cohort), the effect on transduction efficiency of brief centrifugation (“spinoculation”) of the cell- and inoculum-containing FEP culture bags prior to overnight transduction was evaluated (Fig. 1B). As similar transduction efficiencies were observed in the presence or absence of pre-incubation centrifugation (8.7% CD18+ LAD-1 HSPCs vs. 7.4% CD18+ LAD-1 HSPCs, respectively), the downstream experimental results were combined for analysis and the pre-incubation centrifugation step was eliminated in Experiment 2 (the MOI = 2 cohort), which yielded a transduction efficiency of 15.1% CD18+ LAD-1 cells (Fig. 1C). Compared to mock-treated samples, LAD-1 HSPCs transduced with FVV at MOIs of 1 or 2 demonstrated no significant difference in the relative frequency of myeloid and erythroid progenitors (Fig. 1D) or in overall clonogenic output (Fig. 1E).
FVV-mediated CD18 expression restores chemotactic response in myeloid-differentiated LAD-1 cells
Loss of CD18 expression in individuals with LAD-1 is associated with leukocyte motility defects, including diminished capacity for adhesion, chemotaxis, and extravasation. 26 –28 To characterize the ability of CD18 transgene expression to correct chemotactic defects in myeloid-differentiated cells derived from vector-transduced LAD-1 HSPCs, we performed image acquisition-based motility analysis using an EZ-TAXIScan instrument (Fig. 2A). Cellular motility in response to chemoattractant was characterized in terms of (i) average cellular velocity, (ii) average Euclidean migration distance (i.e., the average straight-line distance between cellular point of origin and migration endpoint), and (iii) average accumulated migration distance (i.e., the average total path length traversed by a given cell).
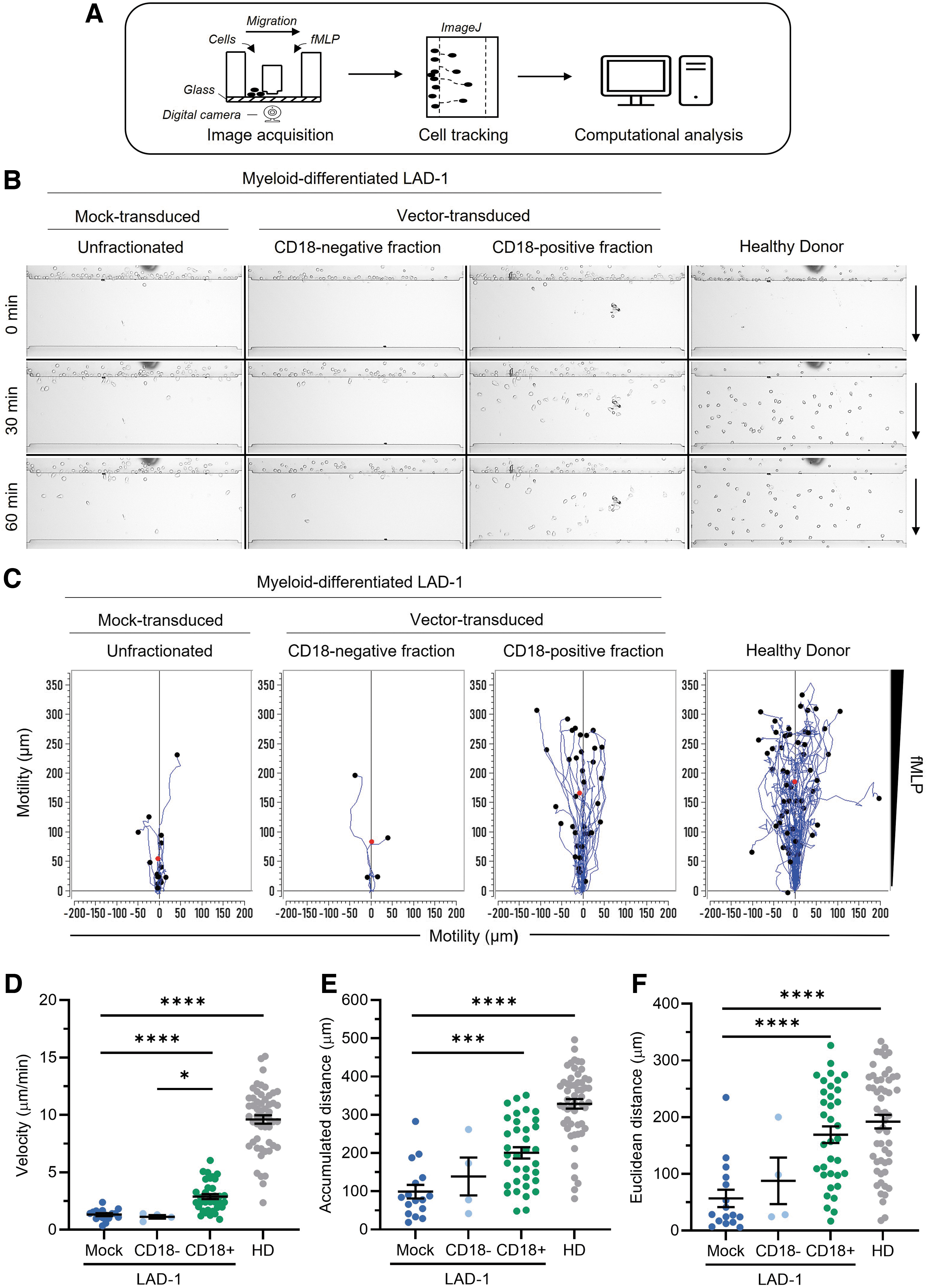
FVV-mediated CD18 expression partially rescues motility defects in myeloid-differentiated LAD-1 donor cells.
For this analysis, samples of LAD-1 HSPCs that were either mock-transduced or transduced with ΔΦMSCV-hCD18 at an MOI of 1 were cultured for ∼24 days in the presence of granulocyte colony-stimulating factor to promote myeloid lineage differentiation. Myeloid-differentiated, FVV-transduced LAD-1 cells were stained with a fluorochrome-labeled anti-CD18 monoclonal antibody and sorted by fluorescence-activated cell sorting into CD18-positive and CD18-negative fractions. Polymorphonuclear (PMN) cells isolated from a HD were included as a positive chemotactic control. Videos comparing the chemoattractant-stimulated and basal (i.e., unstimulated) migration activities of the CD18-positive versus the CD18-negative fraction of vector-transduced, myeloid-differentiated LAD-1 cells (Supplementary Videos S1 and S2, respectively) are presented in the Supplementary Material section, along with videos comparing the chemoattractant-stimulated and basal migration activities of HD PMN cells versus mock-transduced, myeloid-differentiated LAD-1 cells (Supplementary Videos S3 and S4).
Figure 2B shows still frame images of cellular migration patterns for each sample group at the 0-, 30-, and 60-minute time points in response to a gradient of the chemotactic tripeptide N-formylmethionine-leucyl-phenylalanine (fMLP; 10 nM gradient source), a strong neutrophil chemoattractant. 29 Figure 2C shows individual cellular migration tracks captured in each video plotted on a coordinate plane. A marked increase in the absolute number of chemoattractant-responsive cells within the CD18-positive, vector-transduced LAD-1 cell fraction compared to the CD18-negative, transduced-cell fraction or mock-transduced, bulk LAD-1 cells was observed (Fig. 2B, C).
Quantitative analysis revealed a statistically significant >2-fold increase in the average cellular velocity of CD18-positive, vector-transduced LAD-1 cells compared to the mock-transduced, bulk LAD-1 cell population or the CD18-negative fraction of the vector-transduced LAD-1 cell population, with average chemoattractant-stimulated velocities of 2.9 ± 0.2 μm/min (mean ± standard error of the mean [SEM]), 1.3 ± 0.1 μm/min, and 1.1 ± 0.2 μm/min, respectively (Fig. 2D).
In comparison, HD PMN cells demonstrated an average cellular velocity of 9.6 ± 0.4 μm/min in response to fMLP. The observed difference in chemoattractant-stimulated average velocities between CD18+ LAD-1 cells and HD PMN cells may be partly attributable to innate biological differences between in vitro- and in vivo-differentiated myeloid effector cells. Both the average accumulated distance migrated and the average Euclidean distance migrated were significantly improved within the CD18-positive, vector-transduced cell fraction relative to mock-transduced, bulk LAD-1 cells (Fig. 2E, F), consistent with CD18-mediated enhancement of chemotactic response.
In the absence of chemoattractant, mock-transduced LAD-1 cells and CD18-negative cells sorted from the vector-transduced LAD-1 cell population failed to demonstrate measurable motility during the duration of the image acquisition period, whereas the CD18-positive, vector-transduced LAD-1 cell fraction and HD PMN cells displayed low-level, random migration behavior that is typical of granulocytes lacking chemotactic cues (Supplementary Fig. S2 and Supplementary Videos S2 and S4). Basal velocities of motile CD18-positive LAD-1 cells and HD PMN cells in the absence of chemoattractant were 1.94 and 4.69 μm/min, respectively.
Characterization of long-term engraftment, lineage reconstitution, and transgene marking
Five months post-transplantation of NSG mice with LAD-1 HSPCs, bone marrow was harvested and stained with a panel of human-specific monoclonal antibodies (Supplementary Table S1) for flow cytometric analyses of engraftment, lineage reconstitution, and transgene marking. In the first experiment, bone marrow isolated from mice that received LAD-1 HSPCs transduced with FVV at an MOI of 1 demonstrated an average engraftment level of 3.8% ± 1.7% (mean ± SEM) CD45+ human cells, comparable to the average level of human cell engraftment in mice transplanted with mock-transduced LAD-1 HSPCs (2.3% ± 0.5% CD45+ cells) (Fig. 3A, left). Mice in the MOI = 2 group (Experiment 2) demonstrated a similar engraftment outcome, with an average level of human CD45+ cells within the bone marrow of FVV-transduced, LAD-1 HSPC recipients and mock-transduced LAD-1 HSPC recipients of 3.3% ± 0.6% and 2.2% ± 0.7%, respectively (Fig. 3A, right).
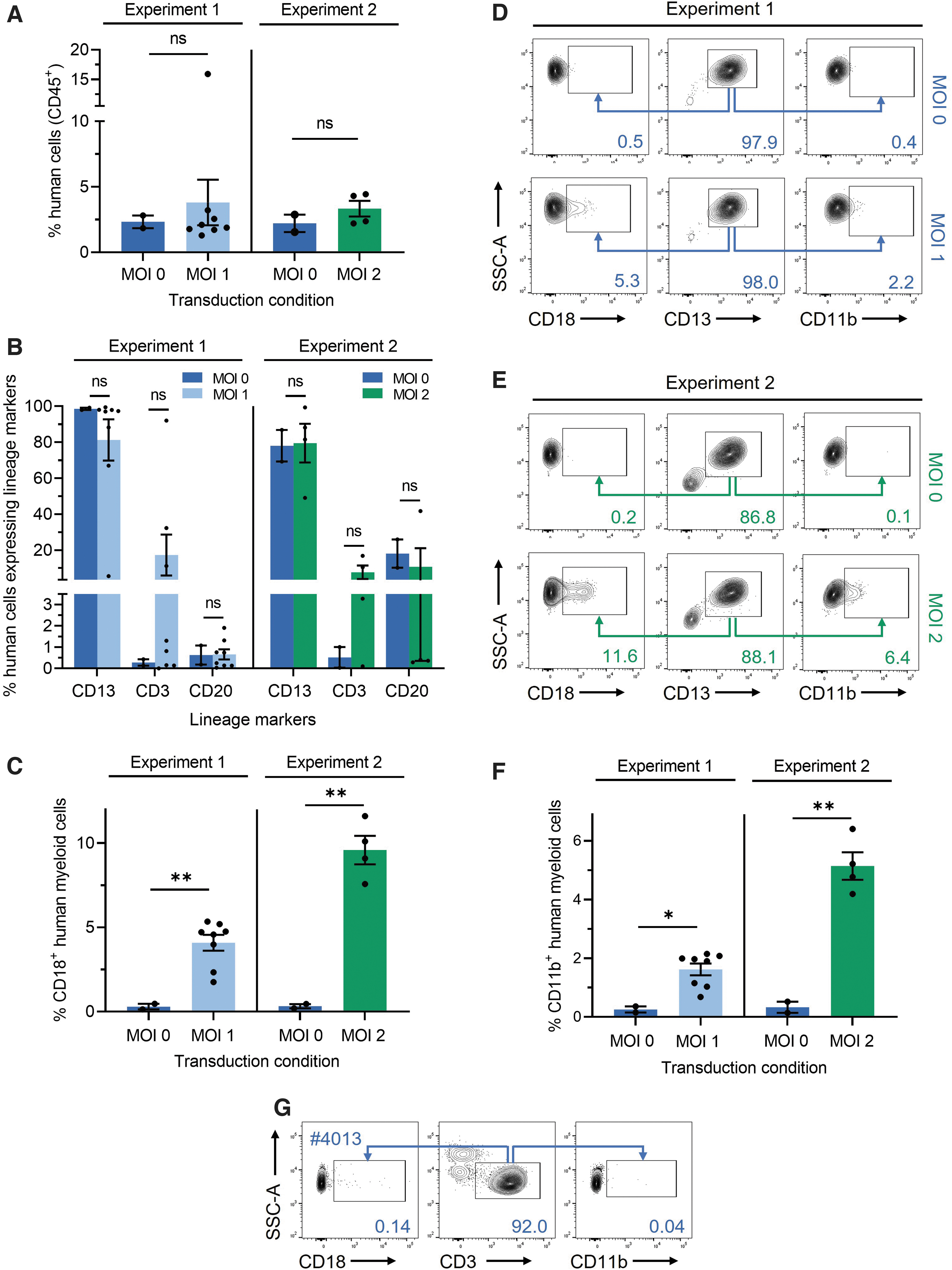
Characterization of LAD-1 HSPC engraftment, lineage reconstitution, and transgene marking in a mouse xenograft model.
Staining of bone marrow samples isolated from the MOI = 1 cohort for human hematopoietic lineage markers (CD13+, myeloid cells; CD3+, T cells; and CD20+, B cells) demonstrated that, in the majority of mice, CD13+ myeloid cells comprised the most abundant engrafted human cell compartment (Fig. 3B, left), with an average value of 81.3% ± 11.5% CD13+ cells in mice that received FVV-transduced HSPCs and an average of 98.6% ± 0.7% CD13+ cells in mice that received mock-transduced LAD-1 HSPCs. In comparison, the average CD3+ T cell and CD20+ B cell compartments within mice that received transduced or control LAD-1 HSPCs represented a much smaller fraction of total CD45+ bone marrow cells (Fig. 3B, left).
One mouse within the MOI = 1 cohort (mouse #4013) bearing the highest levels of human cell engraftment (15.9% CD45+ cells; Fig. 3A, left) displayed a CD3+ T cell subpopulation representing ∼92% of total CD45+ cells and is further characterized below. Lineage analysis of the MOI = 2 cohort (Fig. 3B, right) revealed similarly robust myeloid cell reconstitution, with an average of 78.0% ± 8.8% CD13+ human cells observed in recipients of mock-transduced LAD-1 HSPCs and 79.5% ± 10.8% CD13+ human cells observed in FVV-transduced HSPCs. Flow cytometric analysis of mouse peripheral blood collected at the time of bone marrow harvest (Supplementary Fig. S3A) revealed levels of circulating human CD45+ cells that were proportional to the levels of human lymphoid cells within the bone marrow of each animal.
Readily detectable levels of transgene marking were observed within engrafted, vector-transduced, human myeloid cells (i.e., CD45+CD13+ cells). At an MOI of 1, an average of 4.1% ± 0.5% of vector-transduced human myeloid cells expressed CD18, whereas recipients of mock-transduced cells showed background levels of CD18 staining (Fig. 3C, left). Representative flow plots are shown in Fig. 3D. Consistent with the twofold increase in vector MOI, the average percentage of CD18+ human myeloid cells within the MOI = 2 cohort increased to 9.6% ± 0.9% per mouse (Fig. 3C, right). Representative flow plots are shown in Fig. 3E. Staining for CD11b, a major CD18 binding partner in neutrophils and monocytes, revealed a lack of CD11b expression in nontransduced LAD-1 myeloid cells, which was rescued by ΔΦMSCV-HuCD18-mediated transduction in proportion to vector dosage (Fig. 3D–F).
Due to the predominance of myeloid cell engraftment in the majority of mice, accurate assessment of transgene marking within nonmyeloid subpopulations was untenable in most cases due to the paucity of flow cytometry events, with the exception of mouse #4013 in which the majority of bulk engrafted human cells were CD3+. Staining of bone marrow cells from mouse #4013 for vector-encoded transgene expression revealed only background levels of CD18 marking within the CD3+ subpopulation and a total lack of CD11b rescue (Fig. 3G), suggesting that this cell population is negative for ΔΦMSCV-HuCD18 transduction. The CD3+ compartment within this mouse may represent an autoreactive T cell expansion (and therefore is most likely clonal) or an immune response to the transgene. T cell proliferation in response to infection is also a possibility.
Preclinical biosafety evaluation
To assess safety and genotoxic potential of FVV-mediated gene transfer in LAD-1 HSPCs, transplanted mice were evaluated for health status, occurrence of tumors, and the genomic distribution of vector integration sites at the conclusion of the study. Pre-euthanasia, all mice were active and alert. Post-mortem gross examination revealed that the majority of animals were well hydrated, with signs of adequate nutritional intake and general health, including mouse #4013, which only displayed mild splenomegaly. No solid tumor or other lesion was observed within the heart, brain, liver, lungs, kidneys, spleen, reproductive tract, or gastrointestinal tract of any mouse examined. One mouse (#4018) that received LAD-1 HSPCs transduced with FVV at an MOI of 1 displayed multifocal lung congestion and a possible hemorrhage at the proximal end of the left tibia. Multifocal lung congestion was also noted in a cage mate (mouse #4021) that had received mock-transduced LAD-1 HSPCs.
For all mice, liver, lung, kidney, heart, and spleen were harvested for tissue sectioning, staining, and histopathological evaluation. Examination of hematoxylin and eosin-stained, formalin-fixed tissue sections of the harvested organs showed no overt sign of solid tumor formation (representative tissue sections are shown in Fig. 4A). Complete blood counts were conducted for the control and FVV-recipient mice within the MOI = 1 cohort. Median values associated with red blood cell (RBC) counts, white blood cell (WBC) counts, and hemoglobin, hematocrit, and platelet counts were similar between mock-transduced controls and FVV-recipient animals (Supplementary Fig. S3B). Mouse #4013 demonstrated an elevation in peripheral WBC count relative to other mice within the cohort (7,220 WBC/μL compared to a cohort median value of 1,020 WBC/μL).

Post-transplant histology and vector integration site analysis.
The elevated WBC count was associated with a decreased RBC count (3.89 × 106 RBC/μL vs. 7.39 × 106 RBC/μL median cohort value) and hematocrit (25.8% vs. 40.9% median cohort value) relative to other vector-recipient mice. Platelet counts and hemoglobin values in this animal were similar to the cohort medians of 1,205 × 103/μL and 10.9 g/dL, respectively. Microscopic examination of stained blood smears showed no cytological evidence of leukemia in any mice.
Integrated vector copy number (VCN) determination was performed using bone marrow samples isolated 5 months post-engraftment from all mice (Fig. 4B). VCN per diploid human genome was determined using TaqMan-based quantitative polymerase chain reaction (PCR) of FVV sequences and a single-copy human gene (albumin) as an internal reference. PCR primers are indicated in Supplementary Table S2. Control mice that received mock-transduced LAD-1 HSPCs were negative for vector sequences. The mean VCN of the MOI = 2 cohort (0.0753 vector copies/diploid human genome) was ∼2.7-fold greater compared with the MOI = 1 cohort (0.0281 vector copies/diploid human genome).
Moreover, the frequency of vector genome-containing human cells (∼3% at MOI of 1 and ∼8% at MOI of 2) correlated with the average levels of CD18 expression observed within engrafted human myeloid cells for each transduction cohort (i.e., 4.1% CD18+ human myeloid cells at an MOI of 1 and 9.6% CD18+ human myeloid cells at an MOI of 2), consistent with ∼1 vector copy per transduced cell.
Despite high engraftment levels, mouse #4013 demonstrated the lowest average VCN among mice receiving FVV-transduced LAD-1 HSPCs (0.0065 vector copies per diploid genome), indicating that the majority of human T cells engrafted within this animal were ΔΦMSCV-hCD18 negative. This conclusion was further supported by the observation that even though the MSCV promoter is highly active in human T cells, 30 the CD3+ T cell compartment within mouse #4013 was CD18 negative by flow cytometry (Fig. 3G).
Bone marrow samples from five mice (four from the MOI = 1 cohort and one from the MOI = 2 cohort) were used to prepare Illumina sequencing libraries enriched for vector integration junctions. Vector insertion sites (VIS) were enriched and amplified using an adaptation of the modified genomic sequencing PCR approach. 31 Primers utilized for vector insertion site analysis are indicated in Supplementary Table S3. A total of 2,641 VIS were mapped to the human reference genome. Analysis revealed that integration events were distributed among all human chromosomes, with a roughly equal occurrence of VIS within genes and outside of genes (Fig. 4C).
The frequency of integration events was approximately proportional to chromosome length, with VIS most frequently observed in chromosome 1. Integration events occurring within genes mapped predominantly to introns (91%), as opposed to exons (4%) or gene promoter sequences (5%) (Fig. 4C, inset). We assessed the proximity of VIS to annotated transcription start sites (TSS), as well as TSS associated with a list of 723 known cancer genes cataloged in COSMIC Cancer Gene Census database (Fig. 4D). 32
There was no preference for integration near TSS. Of insertion sites mapping near oncogenes, ∼90% were >5 kb from the TSS, with ∼36% being >50 kb away. To detect potential clonal dominance in transplanted mice, we quantified the relative capture frequency of FVV integration junctions in each mouse. Using an insertion site capture frequency of ≥20% of all reads per sample as a threshold for the determination of clonal dominance, 33 we observed that all five mice examined demonstrated at least one dominant clone, with three of the five mice demonstrating two dominant clones (Fig. 4E). However, despite the existence of dominant clones, overall quantification of the most represented VIS per mouse supported a polyclonal engraftment profile among vector-positive cellular compartments in all mice examined.
DISCUSSION
In this report, we examined the ability of an FVV bearing a human CD18 transgene under the transcriptional control of an MSCV-derived promoter/enhancer to successfully transduce CD34+ HSPCs isolated from an individual with a severe form of LAD-1. In a series of experiments, immunodeficient NSG mice that received either transduced or nontransduced LAD-1 HSPCs demonstrated long-term engraftment at 5 months post-transplantation, providing evidence that, in a mouse xenograft model, both unmanipulated human LAD-1 HSPCs and gene-corrected LAD-1 HSPCs maintain post-mobilization engraftment competency.
An interesting feature of LAD-1 HSPC engraftment within NSG mice was the preponderance of myeloid cell reconstitution over lymphoid lineages in the majority of recipient mice, a finding distinct from the lymphoid (B cell) engraftment generally favored in NSG mice engrafted with HD HSPCs. 34 The myeloid cell preponderance within LAD-1 HSPC-recipient animals was observed regardless of transduction status, suggesting that potential cell-intrinsic properties (e.g., myeloid-biased HSPCs in the context of an impaired granulocytic compartment in LAD-1), rather than vector-mediated effects, are involved.
Although myeloid cell engraftment predominated over that of lymphoid cell engraftment, significant T or B cell compartments were observed in 5 of 16 LAD-1 HSPC-recipient animals (range 10–30% T or B cell engraftment), consistent with multilineage potential. Following LAD-1 HSPC transduction, the percentage of bone marrow-resident CD18+ myeloid cells at 5 months post-transplant ranged from an average of 4.1% per mouse at an MOI of 1 to an average of 9.6% per mouse at an MOI of 2. Importantly, peripheral blood leukocyte marking levels in this range (≥5%) have been shown to be sufficient to ameliorate disease in a large animal model of LAD-1. Notably, Bauer et al 35 reported FVV-based, long-term phenotypic correction in a canine model of LAD-1, known as canine leukocyte adhesion deficiency (CLAD).
Four CLAD dogs that underwent ex vivo FVV-mediated gene therapy displayed CD18+ peripheral blood leukocyte levels of ∼5–10% for the duration of the study reporting period (∼2 years). Nontreated littermate control animals succumbed to lethal infection within the first year of the study, whereas FVV-treated animals saw resolution of recurrent infectious episodes in the first year and long-term survival thereafter. A follow-up report showed that the dogs that received FVV-transduced autologous blood stem cells maintained clinical phenotypic correction for 4–7 years post-transplantation. 36 Our current study, utilizing human patient-derived LAD-1 HSPCs, also extends the findings of a previous murine LAD-1 gene therapy model based upon CD18-encoding retrovirus-mediated transduction of murine bone marrow cells and transplant into syngeneic recipients. 37
Alternative approaches to correct LAD-1 deficiency include LV 38 and, more recently, CRISPR/Cas9-mediated gene addition (for a proof-of-principle CRISPR/Cas9-mediated targeting of the ITGB2 gene, see Bloomer et al 39 ). CRISPR/Cas9-based strategies for the treatment of LAD-1, however, are still in the early stages of development. LV have been successfully utilized in the treatment of several monogenic diseases 40,41 and contract manufacturing of cGMP-grade drug product is widely available. Lack of cGMP-grade FVV manufacturing currently presents a disadvantage to the use of FV vectors in translational studies. Both LV and FVV can transduce nondividing cells; however, unlike FVV-mediated transduction, standard LV transduction protocols generally require pre-stimulation of target cells and the addition of transduction-enhancing adjuvants for efficient transduction. 42 –46
A limitation of our study was the inability to evaluate FVV-mediated gene transfer in multiple LAD-1 HSPC donor samples or to utilize large MOIs of vector due to the paucity of LAD-1 patients available to participate in this pre-clinical study and the limited supply of FVV particles produced under the pilot-scale, cGMP-compatible vector production and purification protocol.
Currently, a lack of dedicated bioproduction infrastructure and manufacturing organizations with expertise and validated standard operating procedures for large-scale, clinical-grade production of FVVs represents a primary challenge to translational studies and subsequent investigational new drug applications that require significant quantities of vector for pharmacology/toxicology studies in small animal models and, potentially, nonhuman primates. In addition, side-by-side comparison of FVV- versus LV-mediated gene transfer and xenotransplantation utilizing LAD-1 patient-derived blood stem cells would directly address the theoretical benefits of FVV-mediated ex vivo gene therapy for the treatment of LAD-1. The overall results of our study, however, demonstrate the utility of a human CD18-encoding FVV for the therapeutic correction of the LAD-1 genetic defect in human blood stem cells isolated from an affected individual and support further evaluation of foamy viral vectors as a potential alternative to LV for gene therapy-mediated treatment of LAD-1.
Footnotes
ACKNOWLEDGMENTS
The authors wish to thank Dr. David Stroncek and the NIH Department of Transfusion Medicine and Cell Processing Section staff for providing leukapheresis, isolation, and cryopreservation of CD34+ LAD-1 HSPCs; Dr. Yusheng Li and the NHLBI Sequencing Core for performing Illumina-based next-generation sequencing of vector insertion site-derived amplicons; Dr. Zu-Xi Yu and the NHLBI Pathology Core for histological preparation of tissue samples; and Dr. Michael A. Eckhaus and the NIH Division of Veterinary Resources for mouse pathology reports. Graphics in ![]() were created with BioRender.com
were created with BioRender.com
AUTHORs' CONTRIBUTIONS
R.H.S. and A.L. designed the experiments. R.H.S. and D.F. performed experiments. D.K. supervised cell motility analysis. H.B. performed motile cell tracking. R.H.S., R.D., and K.G.B. harvested and processed murine bone marrow for flow cytometry. T.R.B. and D.D.H. provided LAD-1 CD34+ cells and technical advice. D.W.R provided vector reagents. M.N., P.M., and J.C.M.L. produced and purified cGMP-compatible vector. S.L.H. performed cGMP-compatible vector transduction. F.S. performed CBC analysis. L.J.A. and M.P. analyzed vector insertion site data. R.H.S. and A.L. wrote the article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (Z99 HL999999 and ZIA HL006172), a Doris Duke Clinical Scientist Award to Punam Malik, and funded, in part, with federal funds from the National Cancer Institute, NIH, under contract number 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
SUPPLEMENTARY MATERIAL
Supplementary Material
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
Supplementary Video S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.