
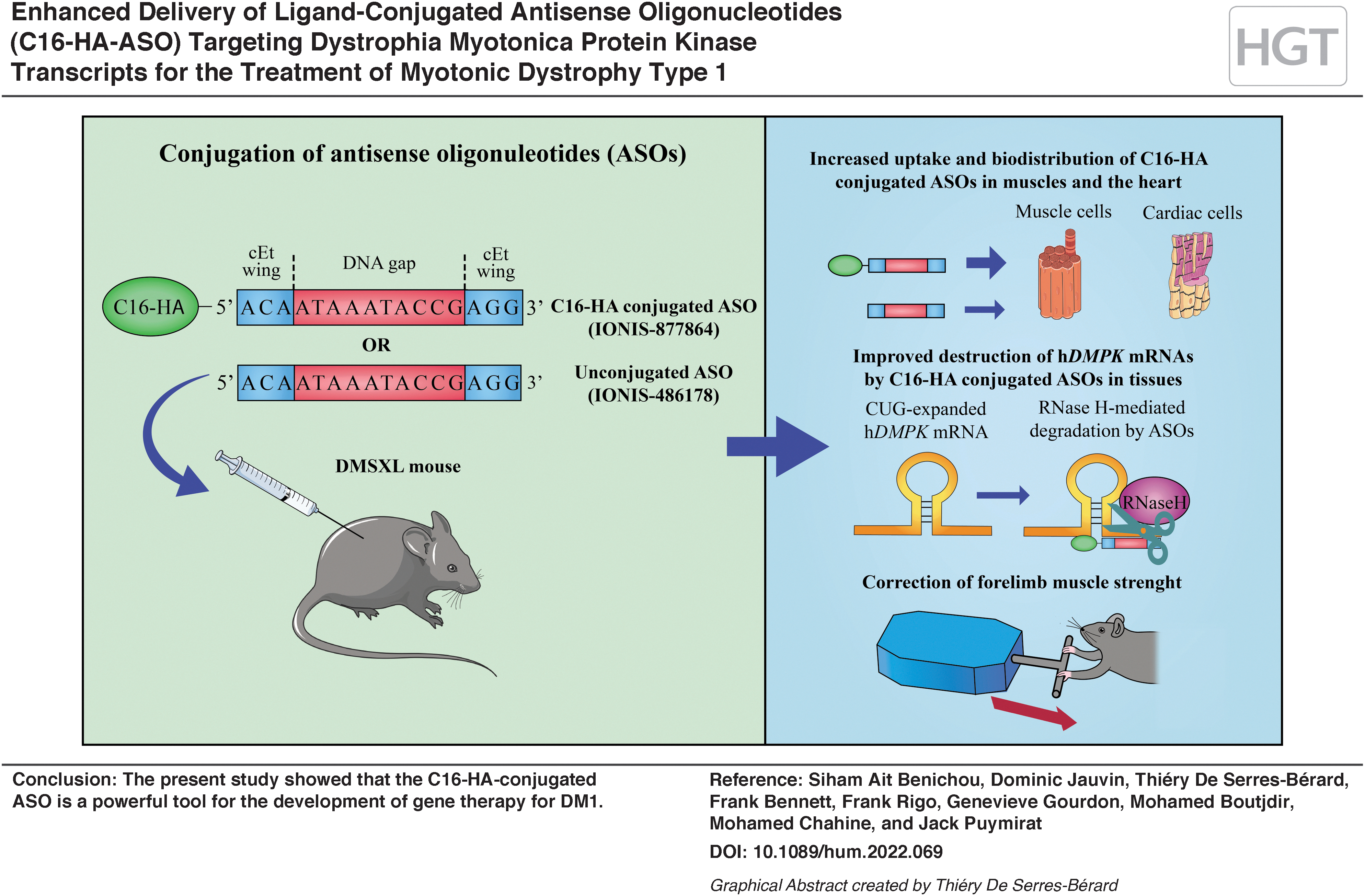
Abstract
Myotonic dystrophy type 1 (DM1) is a neuromuscular disorder that affects many organs. It is caused by the expansion of a cytosine–thymine–guanine triplet repeat in the 3′ untranslated region of the human dystrophia myotonica protein kinase (hDMPK) gene, which results in a toxic gain of function of mutant hDMPK RNA transcripts. Antisense oligonucleotides (ASOs) have emerged in recent years as a potential gene therapy to treat DM1. However, the clinical efficacy of the systemic administration of ASOs is limited by a combination of insufficient potency and poor tissue distribution. In the present study, we assessed the potential of a new ligand-conjugated ASO (IONIS-877864; C16-HA-ASO) to target mutant hDMPK mRNA transcripts in the DMSXL mouse model of DM1 carrying over 1000 CTG pathogenic repeats. DMSXL mice were treated subcutaneously for 9 weeks with either IONIS-877864 (12.5 or 25 mg/kg) or IONIS-486178 (12.5 or 25 mg/kg), an unconjugated ASO with the same sequence. At 25 mg/kg, IONIS-877864 significantly enhanced ASO delivery into the striated muscles of DMSXL mice following systemic administration compared with the unconjugated control. IONIS-877864 was also more efficacious than IONIS-486178, reducing mutant hDMPK transcripts by up to 92% in the skeletal muscles and 78% in the hearts of DMSXL mice. The decrease in mutant hDMPK transcripts in skeletal muscles caused by IONIS-877864 was associated with a significant improvement in muscle strength. IONIS-877864 was nontoxic in the DMSXL mouse model. The present study showed that the C16-HA-conjugated ASO is a powerful tool for the development of gene therapy for DM1.
INTRODUCTION
Myotonic dystrophy type 1
Patients with more than 1,000 CTG repeats often develop a severe congenital form characterized by hypotonia and lower motor performance, including ambulation and grip strength. In addition, such patients have a neonatal mortality rate of around 20%. Mutant DMPK transcripts containing cytosine–uracil–guanine (CUG-expanded) repeats accumulate in nuclear aggregates (foci) and generate mis-splicing events of specific transcripts in affected tissues. 7 –10 They also disrupt RNA localization, polyadenylation and transcription, protein translation, and miRNA processing. 11 –14
There are currently no curative treatments available for DM1 patients. However, several therapeutic approaches have been investigated for restoring muscle function in DM1. Notably, metformin, a drug used to treat insulin resistance in patients with DM1 and promote alternative splicing correction through AMPK-dependent mechanisms, provides a significant functional benefit with respect to the grip test performance of DMSXL mice model carrying over 1000 CTG repeats and motor function in adult patients with DM1. 15
Subcutaneous administration of Arthex-01 (antagomiR-23b), an antagonist of the endogenous translational repressor of the MBNL1 gene, upregulates MBNL1 protein levels and improves grip strength and myotonia, with long-lasting effects in HSALR mice. 16 Other studies have reported improvement in the muscle force of HSALR mice following administration of rapamycin, which targets mTOR signaling 17 and GSK3 inhibitors such as TDZD-8 and lithium. 18
Other preclinical studies have indicated that the destruction of mutant transcripts may be a viable therapeutic strategy. 19 –22 Antisense oligonucleotides (ASOs), which have demonstrated promising results in degrading toxic CUG-expanded mRNA transcripts through the RNase-H mechanism, restore the normal activity of muscleblind-like protein (MBNL), reversing mis-splicing events and correcting myotonia in DM1 mouse models. 20 –24
Various chemical modifications of ASOs have been developed with the aim of decreasing their toxicity and degradation by nucleases while increasing their stability and binding efficiency. Multiple studies have investigated the potential of a constrained ethyl-modified antisense compound (cEt) that decreases mutant DMPK transcript levels by 70% in many skeletal muscle tissues 21,22,25 with no toxicity.
IONIS-DMPKRx, containing both 2′-MOE and cEt modifications, was the first ASO to be investigated in a clinical trial in adults with DM1 (NCT02312011). No significant biological changes were observed in skeletal muscle biopsies of patients, most likely due to the insufficient tissue delivery of ASO doses of up to 600 mg/wk over a 6-week treatment period. 26 Improving ASO delivery in tissues is thus a major requirement for the development of an effective ASO gene therapy for DM1.
Several therapeutic approaches have been developed to enhance skeletal muscle ASO delivery, including peptide-conjugated ASOs, 27 monoclonal antibody-conjugated ASOs, 28 and chemically modified ASOs. Ligand-conjugated antisense (LICA) technology involves combining an ASO with a ligand that has high affinity for specific cell surface receptors, making it possible to target specific tissues and increase the efficacy of drug uptake.
Several antisense drugs based on LICA technology have entered stage 2 and 3 clinical trials, including IONIS-GHR-LRX (metabolic disease, NCT03967249), IONIS-ANGPTL3-LRX (familial partial lipodystrophy disease, NCT03514420), IONIS-FB-LRX (kidney disease, NCT04014335), IONIS-AGT-LRX (chronic heart failure and hypertension, NCT04714320 and NCT04836182), IONIS-TMPRSS6-LRX (blood disease, NCT04059406), and IONIS-APO(a)-LRX (hyperlipoproteinemia (a), NCT03070782). Although the first LICA drugs were designed to target the liver, remarkable progress has been made in the development of LICA technology to improve the potency of ASOs in other organs. 26,29
In DMSXL mice, we evaluated the pharmacodynamic activity of a recently designed LICA oligonucleotide (IONIS-877864) that contains a palmitate hexylamine phosphodiester (C16-HA) conjugated to the IONIS-486178 ASO. This DM1 transgenic model carries a patient-derived genomic fragment that expresses a hDMPK transcript containing >1,000 CTG pathogenic repeats. 30 The present study showed that IONIS-877864 improves the uptake and efficacy of ASOs in DMSXL mice, making this ligand potentially very valuable for treating DM1.
MATERIALS AND METHODS
Animals
All animal experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee of CRCHU de Québec. The 2- to 4-month-old male and female homozygous DMSXL and wild-type (WT) mice used in the present study were generated in our animal facility. A stratified randomization of animals was performed between experimental groups with regard to their sex and age.
Subcutaneous injection of ASOs and grip test
Ionis Pharmaceuticals synthesized the ASOs, as previously described. 31,32 See Table 1 for the chemically modified oligonucleotide structures. IONIS-877864, IONIS-486178, and IONIS-885417 were dissolved and diluted in sterile phosphate-buffered saline to produce 12.5 and 25 mg/kg doses of IONIS-877864 and IONIS-486178. The IONIS-885417 ASO control was used at 25 mg/kg. The maximum dosage was chosen based on previous studies with IONIS-486178 showing efficient hDMPK knockdown with no toxicity when administrated at a concentration of 25 mg/kg in DMSXL mice. 21,22
Antisense oligonucleotide sequences
Phosphorothioate (
The ASOs were administered by subcutaneous (s.c.) injection in the interscapular region biweekly for the first 4 weeks and, subsequently, once a week for an additional 5 weeks, as previously described. 21 Mouse grip strength was assessed with a published protocol 21 using a Chatillon grip strength digital dynamometer (Columbus Instruments).
RNA isolation and quantitative real-time PCR
RNA was extracted from frozen mouse tissues using a standard QIAzol protocol (Qiagen). The complementary DNA was synthesized using a QuantiTect Reverse Transcription kit (Qiagen), as previously described. 33 Real-time PCR assays were carried out using SYBR Green I Master hot start reaction mix on a LightCycler 480 Instrument II (Roche Diagnostics). Quantification of hDMPK mRNA was normalized using the ΔΔCT method, with Hprt1, Rpl13a, and Tbp as reference genes (see Table 2 for primers).
Real-time quantitative PCR primer details for human dystrophia myotonica protein kinase levels and reference genes
hDMPK, human dystrophia myotonica protein kinase.
High-performance liquid chromatography-UV
Triceps and quadriceps tissues from five mice per group were collected 24 h after the last ASO injection and stored at −80°C for high-performance liquid chromatography-UV (HPLC-UV) analysis. Ionis Pharmaceuticals assessed the ASO concentrations in frozen mouse tissues by HPLC-UV, as previously described. 34
Blood chemistry
Blood samples were drawn from the vena cava of the mice and analyzed as described previously 33 using Olympus reagents and an Olympus AU400e analyzer (Olympus) to determine the levels of alkaline phosphatase (ALP), alanine transaminase (ALT), aspartate transaminase (AST), creatine kinase (CK), and creatinine (CRE).
Histological analyses
Histological analyses were carried out as described previously. 33 Briefly, liver and kidney tissues from 10 mice per group were surgically removed and fixed in 10% buffered formalin for 24 h, embedded in paraffin, and stained with hematoxylin and eosin. An animal pathologist from IDEXX BioAnalytics examined the tissues under a microscope and rated the degree of histopathological damage.
Statistical analyses
Statistical analyses were carried out using PRISM 9. A one-way analysis of variance (ANOVA), followed by Tukey's post hoc analysis, was used for comparisons between multiple groups. An ordinary two-way ANOVA, followed by Tukey's post hoc analysis, was used for comparing the effect of two independent variables. Data are expressed as mean ± standard error of the mean, and p < 0.05 was considered significant.
RESULTS
Concentrations of IONIS-877864 and IONIS-486178 in the skeletal muscles of DMSXL mice
For treatments using ASOs, it has been reported that s.c. injections promote widespread oligonucleotide distribution and intracellular translocation. 35 In a first series of experiments, we compared the concentrations of IONIS-877864 with those of unconjugated IONIS-486178 in the triceps and quadriceps muscles. Five 2- to 4-month-old homozygous DMSXL mice were given s.c. injections of IONIS-877864 (12.5 or 25 mg/kg) twice a week for the first 4 weeks and once a week for the next 5 weeks, while another five mice were injected with IONIS-486178 (12.5 or 25 mg/kg).
ASO concentrations measured in both tissues by HPLC-UV revealed a dose-dependent tissue distribution. A slight but nonsignificant difference in tissue concentrations (triceps) was observed at 12.5 mg/kg, with 5.24 ± 1.48 μg/g for IONIS-877864 and 4.19 ± 1.67 μg/g for IONIS-486178 (Fig. 1). At 25 mg/kg, the concentrations of IONIS-877864 were approximately twofold higher than those of IONIS-486178 (13.12 ± 3.26 μg/g vs. 7.41 ± 1.06 μg/g, p = 0.0059) in the triceps and quadriceps muscles.
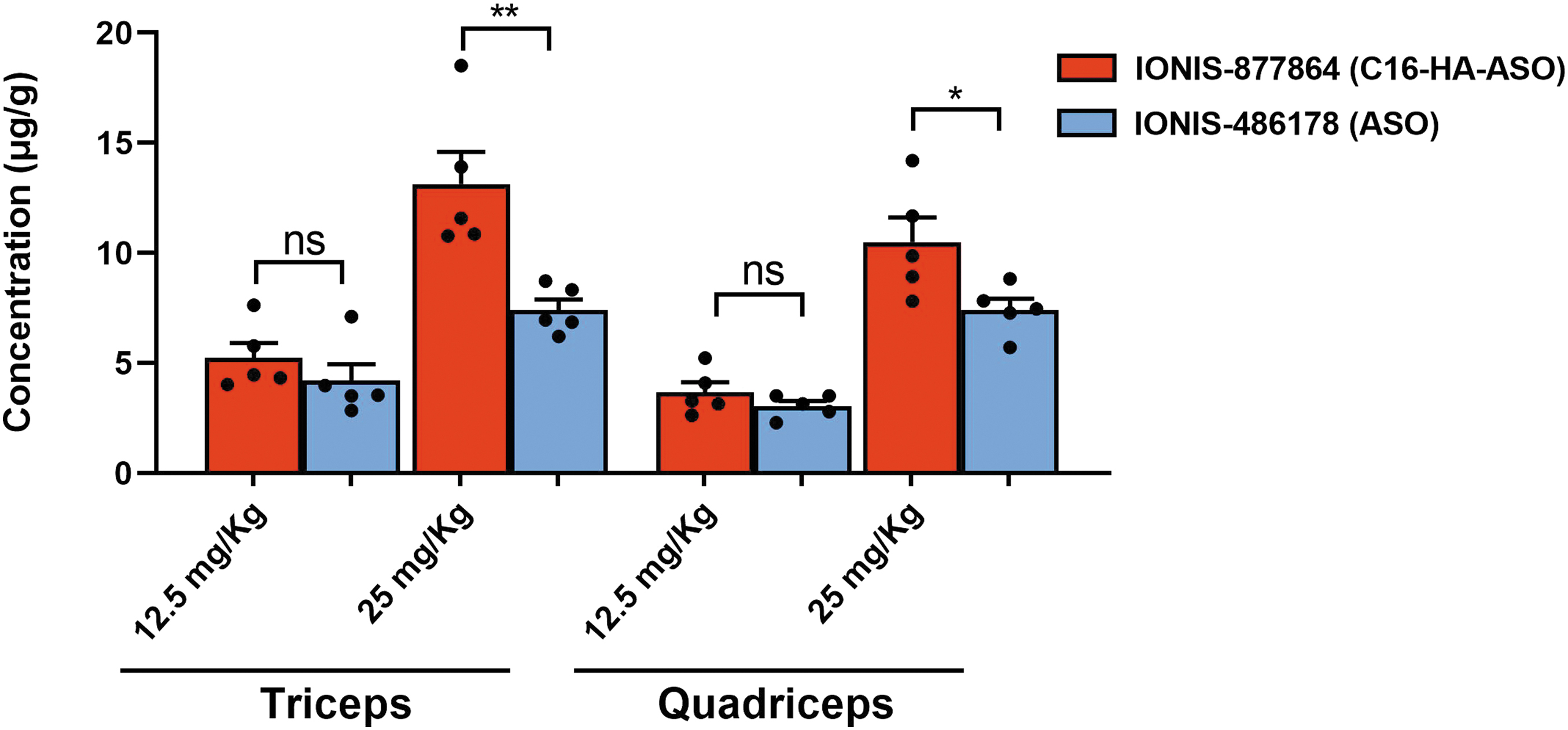
ASO concentrations in DMSXL mouse muscles after subcutaneous injection. Tissue content of IONIS-877864 and IONIS-486178 in the triceps and quadriceps muscles for each treatment dose. DMSXL mice were treated with subcutaneous injections of 12.5 or 25 mg/kg IONIS-486178 or 12.5 or 25 mg/kg IONIS-877864. n = 5, *p < 0.05, and **p < 0.001 using an ordinary two-way ANOVA, followed by Tukey's post hoc analysis, for comparing the effects of two independent variables. Data are expressed as means ± SEMs. ANOVA, analysis of variance; ASO, antisense oligonucleotide; DMSXL, mice model carrying over 1000 CTG repeats; ns, not significant; SEM, standard error of the mean.
It has been reported that ASOs with cEt chemistry are cleared from the blood within 2–5 h. 36 ASO concentrations measured in the present study thus likely result from ASO accumulation in tissues. Our results showed that C16-HA conjugation enhances ASO uptake by skeletal muscles.
Efficiency of IONIS-877864 in targeting mutant hDMPK transcripts in DMSXL mice
To investigate the in vivo potential of IONIS-877864 and IONIS-486178 to target mutant hDMPK transcripts, we evaluated their efficacy in DMSXL mouse skeletal muscles. DMSXL homozygote mice between 2- and 4-month-old express toxic transcripts in various tissues causing RNA foci and mild mis-splicing that perturb muscular, cardiac, brain, and respiratory functions. DMSXL mice display growth retardation, reduced muscle strength and lower motor performance, as well as impairments in cardiac, respiratory, and cerebral functions. 30
Ten 2- to 4-month-old homozygous DMSXL mice received s.c. injections of IONIS-877864 (12.5 or 25 mg/kg) and another 10 received IONIS-486178 (12.5 or 25 mg/kg) twice a week for a 4-week loading dose period and then once a week for the following 5 weeks to maintain high levels of ASOs. IONIS-885417 with the C16-HA chemistry was used as a control at 25 mg/kg. Nine weeks after the first injection, the mice were sacrificed and tissues of five muscles (triceps, tibialis anterior, soleus, quadriceps, and grand dorsal) were collected.
There was a dose-dependent decrease in mutant hDMPK transcripts in all five skeletal muscles with both ASOs (Fig. 2). However, IONIS-877864 was significantly more effective in knocking down hDMPK in muscles in vivo than the unconjugated ASO. With a 25 mg/kg dose, the IONIS-877864 treatment resulted in a 92 ± 2.5% reduction in mutant hDMPK transcripts compared with 63.8 ± 7.2% for the IONIS-486178 treatment (p = 1.8E-07), resulting in a 1.4-fold reduction in mutant hDMPK transcripts in the triceps muscle.
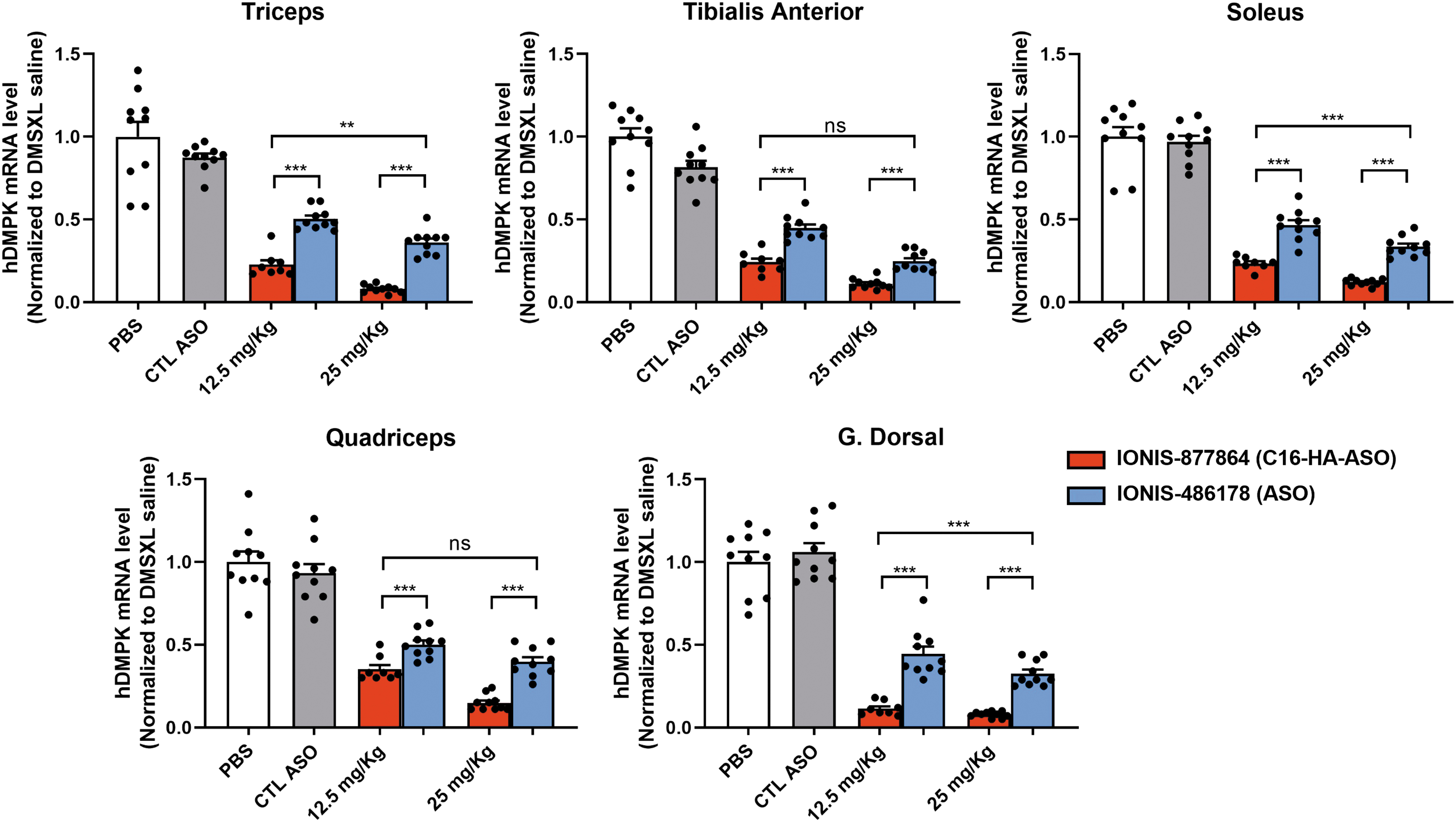
Mutant hDMPK transcript levels in DMSXL mouse skeletal muscles. Analysis of mutant hDMPK transcript levels by RT-qPCR of mice treated with 12.5 or 25 mg/kg IONIS-486178 or IONIS-877864. n = 9–10, **p < 0.001, and ***p < 0.0001 using a one-way ANOVA, followed by Tukey's post hoc analysis, for comparing multiple groups. Data are expressed as means ± SEMs. hDMPK, human dystrophia myotonica protein kinase; RT-qPCR, real-time quantitative PCR.
Similarly, with a 12.5 mg/kg dose, the IONIS-877864 treatment resulted in a 77.4 ± 7.7% reduction in mutant hDMPK transcripts in the triceps muscle compared with 49.8 ± 6.7% for the IONIS-486178 treatment. Interestingly, the 12.5 mg/kg IONIS-877864 treatment displayed an efficiency similar to that of the 25 mg/kg IONIS-486178 treatment. The superior efficacy of the IONIS-877864 treatment was observed in all five types of muscle tissues.
These results showed that the C16-HA-conjugated ASO reduces mutant hDMPK transcript levels to a greater extent in all skeletal muscle tissues examined than the unconjugated ASO.
Increased delivery of IONIS-877864 to the hearts of DMSXL mice
We then determined whether this advanced C16-HA conjugate enhances ASO biodistribution in DM1 mouse organs, including the heart, diaphragm, kidney, liver, and brain. Systemic delivery of IONIS-877864 produced a significantly higher dose-dependent decrease of the heart's mutant hDMPK transcripts than the unconjugated ASO. IONIS-877864 doses of 25 and 12.5 mg/kg reduced mutant hDMPK transcript levels by 78 ± 5.4% and 72.3 ± 6.5% compared with 64.9 ± 6.9% and 31.9 ± 5.7% for IONIS-486178 in the heart (p = 0.0001 and 1.28674E-09), respectively.
No significant differences in mutant hDMPK transcript levels were observed between the two ASOs in the diaphragm (reduced by 69% for both, p = 0.93) and the liver (reduced by 83.6 ± 3.2% for IONIS-877864 and 80.7 ± 6.4% for IONIS-486178, p = 0.25). No significant decrease in mutant hDMPK transcript levels was observed in the brain with either ASO, suggesting that the C16-HA modification did not allow the ASO to cross the blood–brain barrier.
IONIS-877864 did not decrease hDMPK transcript levels in the kidneys compared with IONIS-486178, which resulted in 56% reduction in mutant hDMPK transcripts, suggesting that the C16-HA modification may lower distribution of the ASO to the kidneys (Fig. 3).

Mutant hDMPK transcript levels in DMSXL mouse organs. Analysis of mutant hDMPK transcript levels by RT-qPCR of mice treated with 12.5 or 25 mg/kg IONIS-486178 or IONIS-877864. n = 9–10, *p < 0.05, **p < 0.001, and ***p < 0.0001 using a one-way ANOVA, followed by Tukey's post hoc analysis, for comparing multiple groups. Data are expressed as means ± SEMs.
IONIS-877864 increases mouse forelimb strength
To determine whether the ASO treatments improve muscle weakness, we quantified the forelimb muscle strength of 2- to 4-month-old homozygous DMSXL mice using the grip test (Fig. 4). Muscle strength was analyzed before the start of the treatment (W0) and at the end of the experiment (W8) to determine the effect of the ASO treatments on the disease phenotype. DMSXL mice had significantly lower forelimb strength (19.64 g lower) than WT mice (70.8 ± 14.2 g and 90.44 ± 6.6 g, respectively, p = 0.0001), indicating that their muscular performance was impaired as previously described. 30
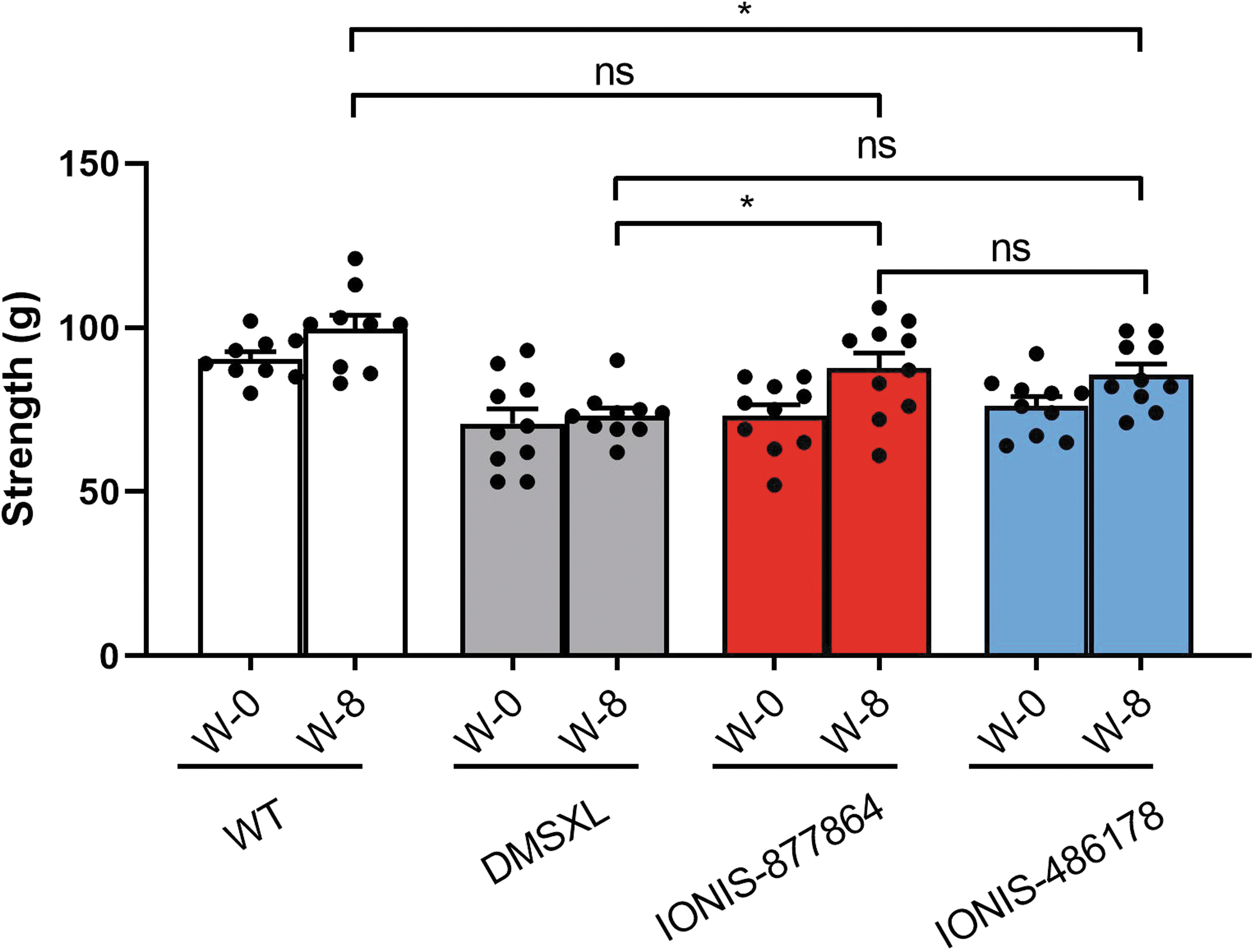
Maximum forelimb strength improvement in DMSXL mice after ASO treatments. Mouse forelimb strength measurements using the grip test after subcutaneous injections of 25 mg/kg IONIS-877864 or IONIS-486178 biweekly for the first 4 weeks and then once a week for the following 4 weeks (W-8). n = 9–10, *p < 0.05 using a two-way ANOVA. Data are expressed as means ± SEMs.
At 25 mg/kg, IONIS-877864, but not IONIS-486178, induced a significant increase in forelimb strength of 14.5 g (p = 0.023) after 8 weeks of treatment, which was not significant compared with the forelimb strength of WT mice. In contrast, the forelimb strength of DMSXL mice treated with IONIS-486178 remained significantly lower than that observed in WT mice (9.6 g, p = 0.0381). The gain of strength in DMSXL mice treated with IONIS-877864 was nonetheless small and not significant when directly compared with that of the group treated with IONIS-486178.
Although restricted to a specific muscle group, the increase in strength observed likely positively affected other parts of the muscular system given that hDMPK mRNAs are efficiently knocked down in most skeletal muscles.
IONIS-877864 is well tolerated by DMSXL mice
To assess the potential toxicity of both ASOs, we first quantified the blood biochemistry toxicity markers, ALP, ALT, AST, CK, and CRE. The normal values for mice range from 62 to 209 U/L for ALP; 28 to 200 U/L for ALT; 59 to 247 U/L for AST; 68 to 1,070 U/L for CK; and 0.15 to 0.7 mg/dL for CRE. Treatments of 10 homozygous DMSXL mice with 12.5 mg/kg and up to 25 mg/kg doses of the ASOs were well tolerated and the serum biochemistry levels remained in the normal range.
Although a significant increase in ALT levels was observed after the 25 mg/kg IONIS-877864 treatment and while CK levels appeared to decrease, although not significantly, the serum biochemistry levels of each mouse remained within the normal values for mice. Variability between mice was also observed in the homozygous control DMSXL mice treated with PBS (Fig. 5A). Histological preparations of liver and kidney tissues from 10 DMSXL mice per group were examined by an animal pathologist from IDEXX BioAnalytics.

IONIS-486178 and IONIS-877864 toxicity profiles in DMSXL mice.
After the IONIS-877864 ASO treatment, a small number of single cells, likely resident hepatic macrophages (Kupffer cells) with an amphiphilic finely vacuolated cytoplasm, were scattered throughout the hepatic parenchyma, which is consistent with drug accumulation in endolysosomal structures. Minimal infiltration of mixed inflammatory cells was observed in the gallbladder lamina propria of one or more animals after both treatments as well as in the control group.
Low levels of periportal mononuclear inflammatory cell infiltrates were also noted. Incidental and spurious findings included hepatic granulomatous foci and minimal renal pelvic inflammation. All tissues displayed mild postmortem artifacts (Fig. 5B). Overall, tissue damage and toxicity markers for the liver and kidney seemed to show no evidence of ASO-induced pharmacological toxicity.
DISCUSSION
The present study demonstrated that the systemic injection of IONIS-877864 improves the uptake and efficacy of ASOs for targeting mutant hDMPK transcripts in most striated muscles and hearts of DMSXL mice compared with the unconjugated ASO. These data agree with a previous study showing that the C16-HA-conjugated ASO was significantly better at engaging endogenous mouse Dmpk in skeletal and cardiac muscles. 32
However, we observed a similar hDMPK mRNA reduction by IONIS-877864 and the unconjugated ASO in the diaphragm of DMSXL mice, suggesting that the C16-HA conjugate may be less efficient at improving the targeting of CUG-expanded RNAs in this tissue. The C16-HA modification also improved the accumulation of ASO in the triceps and quadriceps muscles. Østergaard et al reported a slightly higher accumulation of conjugated ASO in the hearts, quadriceps, and livers of rats and monkeys. 32
IONIS-877864 had no effect on mutant hDMPK transcripts in the kidney, indicating that it has a lower potential toxic effect. It has previously been shown that ASOs are unable to efficiently cross the blood–brain barrier, likely because of their charge and size. Therefore, other delivery methods should be considered for treating DM1 brain deficits. 21,33
Despite similar ASO concentrations in the triceps and quadriceps muscles following the systemic injection of 12.5 mg/kg IONIS-877864 or IONIS-486178, 12.5 mg/kg C16-HA-conjugated ASO was more effective than 25 mg/kg unconjugated ASO at targeting mutant hDMPK transcripts in these skeletal muscles. It has recently been reported that subcellular localization significantly influences ASO therapeutic activities 37,38 and that cellular uptake pathways play an important role in the intracellular distribution of ASOs. 21,35,39
Our results are also in agreement with those reported by Hu et al, who showed that the C16-HA-conjugated ASO causes a dose-dependent therapeutic response in the gastrocnemius and lumbar paraspinal muscles within 7 days. 40 Interestingly, they also showed that C16-HA-conjugated ASO levels increased two-fold in skeletal muscles, suggesting a two-fold increase in targeting ACTA1-CUG-expanded transcripts. This effect persisted for several weeks, confirming the stability of the C16-HA-conjugated ASO.
The cEt chemistry in gapmers promotes ASO binding to albumin and histidine-rich glycoprotein (HRG) while the C16-HA conjugate improves binding to serum albumin, histidine-rich glycoprotein, highdensity lipoproteins (HDLs), low-density lipoproteins (LDLs) and also specifically targets cell surface receptors in muscle tissues. These attributes make the C16-HA particularly effective at enhancing ASO absorption in skeletal and cardiac muscles. 31,32
The palmitic acid conjugate has been shown to reduce ASO excretion in urine and highly increases plasma concentrations as well as in the interstitium of muscle tissues, although the improvement in intracellular delivery remained modest. 41 Another study showed that IONIS-877864 reduces RNA foci in the tibialis anterior muscle in a DM1 mouse model and that this reduction is associated with an increase in satellite cell numbers and fiber regeneration following tissue damage. 42 Our results suggest that decreasing mutant hDMPK transcript levels may be an appropriate approach for improving muscle strength.
Recent advances in targeted delivery of ASOs, including their conjugation to other vectors such as peptides and antibodies, have resulted in better uptake and accumulation leading to less frequent ASO dosing and enhanced intracellular targeting. Pip6a, an arginine-rich cell-penetrating peptide, conjugated to phosphoroamidate morpholino oligomer (PMO) has been used for systemic corrective therapy in DM1. 27
Administration of 12.5 mg/kg Pip6a-PMO decreased CUG-expanded RNA levels by 60%, which was sufficient to reverse splicing defects and correct myotonia in HSALR mice. However, no data on muscle strength and cellular biodistribution were provided. This ASO differs from our antisense strategy, in that it inhibits RNA-induced toxicity by steric hindrance to reduce pathogenic interactions with MBNL proteins rather than by RNase-H-mediated transcript degradation.
Avidity Biosciences recently initiated a clinical trial for the treatment of adult DM1 (NCT05027269) using an antibody–oligonucleotide conjugate (AOC 1001). AOC 1001 contains a monoclonal antibody that binds to the transferrin receptor 1 conjugated to a siRNA. It reduces the expression of DMPK and produces normal splicing variants. However, its potency cannot be compared with that of C16-HA conjugated ASOs as no preclinical data have been published. 28
Alternatively, the prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR/Cas9) system has also been investigated as an alternative therapeutic approach to target RNA toxicity in DM1. In this regard, systemic administration of an adeno-associated virus that induces the expression of Cas9 as well as single-guide RNAs that recognize expanded CUG repeats in RNAs resulted in prolonged correction of the molecular and motor phenotypes of DM1 mice. 43
New ASOs that incorporate LICA to increase their uptake in the liver and other tissues have been shown to have improved potency. 44,45 Conjugation of N-acetylgalactosamine (GalNAc) to ASOs enhances the uptake in hepatocytes by high-affinity ligand binding to the asialoglycoprotein receptor. This improves hepatocyte potency by 10- to 60-fold. 46,47 These studies support the translational potential of LICA for the development of therapeutics to treat several disorders.
The present study indicated that ASO conjugation is also a viable strategy for increasing target engagement in the heart and muscles for the treatment of muscular diseases. Conjugation of the IONIS-486178 ASO with C16-HA may facilitate clinical trials by reducing the dosage, thus limiting potential secondary effects while improving the efficacy of the treatment.
It is noteworthy that the toxicity associated with most ASOs is caused by their accumulation in the lysosomes of proximal tubules in the kidney. ASO accumulation has been associated with perturbation of tubular absorptive capacity and, at high doses, increased tubular proteinuria and chronic progressive nephropathy. 48 –50 IONIS-486178 decreased mutant hDMPK transcript levels in the kidney by 56%, whereas no significant decrease was observed with a C16-HA-conjugated ASO.
In addition, there was no significant increase in serum CRE levels in mice following ASO administration. No toxicity was observed in the liver with either ASO. These data suggest that C16-HA-conjugated ASOs are probably efficiently absorbed in tissues following systemic delivery, which likely results in a lower renal risk profile.
Østergaard et al reported very little free C16-HA-conjugated ASO in the plasma due to its high binding affinity to albumin, histidine-rich glycoprotein, HDL, and LDL, which would reduce the kidney's ASO exposure and explain the lack hDMPK mRNA knockdown. 31,32
CONCLUSIONS
Our results show that C16-HA conjugates can be effective in modulating antisense uptake, distribution, and efficacy at the cellular level, which results in higher internalization of ASOs and can improve muscle strength in DMSXL mice. Our findings strongly support the development of LICA-ASO for DM1 clinical trials and the treatment of other microsatellite expansion disorders.
Footnotes
ACKNOWLEDGMENTS
The art figures used in the graphical abstract were provided by Servier Medical Art (
AUTHORs' CONTRIBUTIONS
S.A.B. and J.P. were involved in conceptualization and supervision. S.A.B. was involved in writing the original draft and investigation. D.J. was involved in investigation. F.B. and F.R. were involved in resources and investigation. G.G. was involved in methodology. M.C. was involved in visualization. J.P., M.C., T.D.S-B., and M.B. were involved in writing, review, and editing.
All the authors revised and approved the article.
AUTHOR DISCLOSURE
F.B. and F.R. are employees of Ionis Pharmaceuticals. The remaining authors declare no competing interests.
FUNDING INFORMATION
This work was supported by Ionis Pharmaceuticals, Inc., the French Muscular Dystrophy Association (grant no. 21438 to J.P.), and the American Muscular Dystrophy Association (grant no. 417929 to J.P.). This work was also supported by a Canadian Institutes of Health Research grant (MOP-130373) to M.C., a U.S. Department of Defense grant (USAMRAA W81XWH-21-1-0426) to M.C., and a U.S. Department of Defense grant (W81XWH-21-1-0424) to M.B.