
Abstract
Sepsis is a common life-threatening pathology. This study investigated the role of transcription factor sex-determining region Y (SRY)-box 9 (SOX9) in sepsis-induced cardiomyocyte pyroptosis. A murine model of sepsis was established, followed by detection of cardiac functions and myocardial injury. HL-1 mouse cardiomyocytes were induced by lipopolysaccharides (LPS). The levels of interleukin (IL)-18, IL-1β, tumor necrosis factor-α (TNF-α), IL-6, malondialdehyde (MDA), and superoxide dismutase (SOD) in myocardial tissues and HL-1 mouse cardiomyocytes were determined. SOX9 ubiquitination level was measured. The binding relationships between SOX9-miR-96-5p and miR-96-5p-NLR pyrin domain containing 3 (NLRP3) were analyzed, and the interaction between ubiquitin-specific peptidase 7 (USP7) and SOX9 was measured. SOX9 was highly expressed in septic mice and LPS-induced HL-1 mouse cardiomyocytes. SOX9 silencing improved cardiac function, alleviated myocardial injury, reduced the levels of IL-1β, IL-18, cleaved caspase-1, gasdermin D N-terminal domain, TNF-α, IL-6, and MDA in myocardial tissues and HL-1 mouse cardiomyocytes, increased the level of SOD, and alleviated cardiomyocyte pyroptosis. USP7 upregulated SOX9 expression through deubiquitination. SOX9 inhibited miR-96-5p expression and miR-96-5p targeted NLRP3. miR-96-5p silencing or USP7 overexpression reversed the inhibitory effect of SOX9 silencing on cardiomyocyte pyroptosis. Collectively, USP7 upregulated SOX9 expression through deubiquitination, and SOX9 suppressed miR-96-5p expression by binding to the miR-96-5p promoter region, thereby promoting NLRP3 expression and then exacerbating sepsis-induced myocardial injury and cardiomyocyte pyroptosis.
INTRODUCTION
Sepsis has been regarded as systemic dissemination of infection and is deteriorated by inappropriate immune responses, leading to multiple organ failure and shock. 1 The pathogenesis of sepsis is exceedingly intricate, including imbalance of inflammatory responses, immune dysfunction, mitochondrial injury, coagulation disorder, and neuroendocrine immune network abnormality. 2 Despite the continual improvement of surgical and pharmacological treatments, sepsis and subsequent multiple organ failure remain the primary causes of mortality in intensive care units. 3 Myocardial injury is frequent in patients with severe sepsis syndrome, commonly manifested as global ventricular dysfunction, massive cardiomyocyte death, and interstitial edema. 4 Accumulating clinical and animal experiments have demonstrated the existence of cardiomyocyte inhibition in sepsis, which constitutes one of the factors leading to cardiac dysfunction and myocardial injury. 5
Pyroptosis, a pro-inflammatory form of programmed cell death, is triggered by various organism insults. 6 During the course of sepsis, pyroptosis destroys the integrity of the cell membrane, thus causing the secretion of inflammatory cytokines and the enhancement of inflammatory responses. 7 The elucidation of the pathophysiological processes of sepsis-induced cardiomyocyte pyroptosis may confer novel targets for sepsis therapy.
Transcription factors are a group of DNA-binding proteins and their gene regulatory capacity is crucial for the determination of the molecular state of cells. 8 Sex-determining region Y (SRY)-box 9 (SOX9) is a transcription factor that participates in mammalian development, which crucially modulates chondrogenesis and sex differentiation. 9 As a vital regulator of extracellular matrix genes, SOX9 is implicated in the pathogenesis of diverse fibrotic diseases, such as hepatic fibrosis and cardiac valve calcification. 9 Cardiomyocyte-specific SOX9 mediates cardiac injury and remodeling, and studies on mammalian hearts have confirmed that SOX9 expression is upregulated in fibroblasts after ischemic injury and associated with augmented fibrosis. 10,11 However, the role and mechanism of SOX9 in sepsis-induced myocardial injury remain unclear. Furthermore, the stability, expression, and localization of SOX9 have been demonstrated to be modulated by post-translational modifications, including ubiquitylation. 12
Ubiquitination has been acknowledged as a critical determiner of protein fate by labeling proteins for proteasomal degradation. 13 Ubiquitination thus affects a wide range of cellular processes and participates in multiple pathophysiological states, ranging from tumors to infectious diseases. 14 Ubiquitin-specific peptidase 7 (USP7) is a ubiquitin hydrolyzing enzyme that modulates the stability, function, or expression of its substrates through its deubiquitination activity. 15 The role of USP7 in sepsis has not been reported yet, but emerging evidence has indicated that USP7 overexpression exacerbates myocardial ischemic injury by enhancing inflammation and apoptosis of cardiomyocytes. 16 Suppression of the USP7-associated catabolic/apoptotic pathway can protect against doxorubicin-induced cardiotoxicity. 17 USP7 accelerates ferroptosis and exacerbates myocardial injury through activation of the p53/TfR1 pathway in ischemia/reperfusion-induced rat hearts. 18 Notably, a prior study has indicated the interaction between USP7 and SOX9 in the proliferation and differentiation of ATDC5 mouse chondrocytes. 19 Based on the above findings, we speculated whether SOX9 can be modified by USP7 to participate in sepsis-induced myocardial injury.
Transcription factors and microRNAs (miRNAs) exercise similar regulatory logic and exert synergistic effects in gene regulatory networks, and the combinatorial action of miRNAs and transcription factors triggers a regulatory cascade necessary for the proper execution of diverse biological events. 20 miRNAs are small noncoding RNAs (about 22 nucleotides) that mediate the post-transcriptional regulation of messenger RNAs (mRNAs) by binding to the 3′-untranslated regions (3′-UTRs) in multiple eukaryotic lineages. 21 miRNA profile is altered under various pathological conditions, and numerous deregulated miRNAs have been identified in the context of sepsis. 22 miR-96-5p is poorly expressed in the serum of sepsis patients, and miR-96-5p overexpression mitigates inflammatory responses in neonatal sepsis. 23 miR-96-5p overexpression can also alleviate myocardial infarction-triggered myocardial injury and cardiac remodeling. 24 Accordingly, we speculated that the transcription factor SOX9 is implicated in the regulation of sepsis by mediating miR-96-5p expression.
This study aims to investigate the mechanism of SOX9 in sepsis-induced myocardial injury and cardiomyocyte pyroptosis, hoping to confer a novel theoretical basis for SOX9 as a molecular target in the treatment of sepsis myocardial injury.
MATERIALS AND METHODS
Ethics statement
This study was performed following the approval of the Ethics Committee of Sichuan Provincial People's Hospital. All the animal experiments were implemented based on the Guide for the Care and Use of Laboratory Animals. 25
Experimental animals
C57BL/6 mice (8 weeks old; 25 ± 2 g) were purchased from Animal Experiment Center of Wuhan University [SCXK (Hubei) 2019-0004] and fed with food and water according to the standard diet at room temperature (22 ± 2°C), and the circadian rhythm was artificially adjusted for 12 h.
Establishment of a murine model of sepsis
By referring to the method mentioned in the previous literature, 26 a murine model of sepsis was established through cecal ligation and puncture (CLP). In short, the mice were anesthetized with 0.1% pentobarbital sodium. A small incision was made in the anterior abdomen to expose the cecum. Then, the cecum was ligated with 4–0 suture and punctured with No. 18 needle, and some feces were pressed out to ensure smooth puncture. Next, the cecum was placed back, and the incision was sutured. The mice were injected subcutaneously with 1 mL 0.9% normal saline. Except CLP, the mice in the sham operation group received the same surgical treatment as those in the CLP group. Twenty-four hours before CLP operation, the mice were injected with 100 μL lentivirus packaged sh-SOX9 or lentivirus packaged oe-USP7 through tail vein (1 × 108 plaque-forming units). 27,28 Lentivirus packaged sh-SOX9 and oe-USP7 and their negative control (NC) were provided by GenePharma (Shanghai, China).
Echocardiography
By referring to the method mentioned in the previous literature, 27 24 h after CLP operation, the mice were anesthetized with isoflurane inhalation. The cardiac function of mice was measured using a 30 MHz linear array transducer and preclinical ultrasound system (Vevo 1100; Fujifilm VisualSonics, Canada). Left ventricular ejection fraction (LVEF) and left ventricular fraction shortening (LVFS) were analyzed.
Blood collection and tissue treatment
After echocardiography, 1 mL whole blood was collected from the left ventricle of mice. The whole blood was centrifuged at 3,000 g for 15 min to obtain serum samples. After blood collection, the mice were injected with 200 mg/kg sodium pentobarbital for euthanasia. Afterward, the chest was opened immediately, and the heart was taken out. The hearts of 6 mice in each group were randomly selected and fixed in 4% paraformaldehyde solution for 24 h. After dehydration, the hearts were paraffin embedded and sectioned with a thickness of 4 μm. The remaining myocardial tissues were used for tissue homogenate experiment. In short, radio-immunoprecipitation assay (RIPA) lysis buffer was added to myocardial tissues and centrifuged at 12,000 g for 10 min to obtain the supernatant.
Hematoxylin and eosin staining
The obtained mouse myocardial tissue sections were dried. The dried tissue sections were immersed in xylene I and xylene II for 10 min for dewaxing, then immersed in absolute ethanol I, absolute ethanol II, 95% ethanol, 80% ethanol, and 70% ethanol for 2 min, and washed twice with phosphate-buffered saline (5 min/time). Afterward, the tissue sections were stained with hematoxylin for 5 min, eosin for 3 min, then immersed in 95% ethanol, absolute ethanol I, and absolute ethanol II for 3 min, and immersed in xylene I and xylene II for 5 min. Finally, the pathological changes of myocardial tissues were observed under a microscope.
Cell culture and treatment
HL-1 mouse cardiomyocytes were purchased from American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) containing 10% fetal bovine serum (Gibco, Life Technologies, Gaithersburg, MD) and 1% (v/v) Penicillin/Streptomycin-Glutamine (100 × ; Gibco, Life Technologies) at 37°C with 5% CO2. The cardiomyocyte model of sepsis was induced by lipopolysaccharides (LPS) treatment. Briefly, HL-1 mouse cardiomyocytes were seeded into 6-well plates (1 × 106 cells/mL) and cultured for 24 h till 60–70% confluence. Then, HL-1 mouse cardiomyocytes were cultured with 0.5 μg/mL LPS or normal saline for 12 h. Three small interfering (si)-SOX9 (si-SOX9#1, si-SOX9#2, and si-SOX9#3), three si-USP7 (si-USP7#1, si-USP7#2, and si-USP7#3), oe-USP7, miR-96-5p inhibitor, and their corresponding NC were provided by GenePharma. The cells were transfected in line with the instructions of Lipofectamine 2000 (Life Technologies). After 48 h, the cells were collected for LPS treatment.
Detection of cell viability
The viability of HL-1 mouse cardiomyocytes was measured using Cell Counting Kit 8 (CCK-8) (ab228554; Abcam, Cambridge, UK). Briefly, the cells were seeded into 96-well plates and treated with LPS for 1 h. The cells in each well were added with 10 μL CCK-8 reagent and cultured for 2 h at 37°C. The absorbance at 450 nm was measured.
Hoechst 33342/propidium iodide fluorescent staining
Hoechst 33342/propidium iodide (PI) fluorescent staining was performed to measure HL-1 mouse cardiomyocyte pyroptosis. Briefly, HL-1 mouse cardiomyocytes were seeded into 24-well plates, and the medium was added with Hoechst 33342 and PI. The cells were cultured at 37°C for 30 min, and the stained cells were observed under a laser confocal scanning microscope (Olympus, Tokyo, Japan).
Reverse transcription quantitative PCR
The total RNA was extracted from mouse myocardial tissues and HL-1 mouse cardiomyocytes using TRIzol reagent (Invitrogen Inc., Carlsbad, CA) and then reverse transcribed into complementary DNA using PrimeScript RT Kit (Takara, Otsu, Shiga, Japan). Reverse transcription quantitative PCR (RT-qPCR) was performed using SYBR green I Master Mix Kit (Invitrogen) on the 7,500 real-time PCR system (Applied Biosystems, Inc., Carlsbad, CA) to detect the expression of genes, with U6 as the internal reference of miR-96-5p 29,30 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal reference of other genes. The relative expression of genes was quantified using the 2−ΔΔCt method. The experiment was repeated thrice independently. The primers are shown in Table 1.
Quantitative PCR primers
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; miR-96-5p, microRNA-96-5p; NLRP3, NLR pyrin domain containing 3; SOX9, sex-determining region Y (SRY)-box 9; USP7, ubiquitin-specific peptidase 7.
Western blot
The total protein was extracted from mouse myocardial tissues and HL-1 mouse cardiomyocytes using RIPA buffer (Beyotime Biotechnology, Shanghai, China) and quantified using Bicinchoninic Acid Kits (Beyotime). The protein was separated on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The membrane was then blocked with 5% skim milk in 0.05% Tween 20/TBS (TBST) for 2 h at room temperature.
After TBST washing three times, the membrane was incubated with the following primary antibodies at 4°C overnight: SOX9 (ab185966, 1:1,000; Abcam), USP7 (ab108931, 1:1,000; Abcam), NLR pyrin domain containing 3 (NLRP3) (ab214185, 1:100; Abcam), cleaved-caspase-1 (PA5-38099, 1:500; Thermo Fisher Scientific Inc., Waltham, MA), gasdermin D N-terminal domain (GSDMD-N) (ab219800, 1:1,000; Abcam), and GAPDH (ab8245, 1:1,000; Abcam). Afterward, the membranes were incubated with the secondary antibody immunoglobulin G (IgG) (ab6721, 1:2,000; Abcam) for 2 h and developed using enhanced chemiluminescence solution (Thermo Fisher Scientific). The band density was measured using Quantity One 4.6 software (Bio-Rad Laboratory, Inc., Hercules, CA).
Enzyme-linked immunosorbent assay
The myocardial injury of mice was evaluated by detecting the levels of cardiac troponin T (c-TnT) and creatine kinase MB (CK-MB) in serum; the pyroptosis level was evaluated by detecting the changes of interleukin (IL)-1β and IL-18 in myocardial tissue and HL-1 mouse cardiomyocyte supernatant, and the inflammatory level was evaluated by detecting the levels of tumor necrosis factor-α (TNF-α) and IL-6 in tissue and cell supernatant. The contents of c-TnT and CK-MB in serum were determined using c-TnT Enzyme-Linked Immunosorbent Assay (ELISA) Kit (ab223860; Abcam) and CK-MB ELISA Kit (EHCKMBX10; Thermo Fisher Scientific). The contents of IL-18, IL-1β, TNF-α, and IL-6 in tissues or cells were measured using IL-18 ELISA Kit (ab216165; Abcam), IL-1β ELISA Kit (ab197742; Abcam), TNF-α ELISA Kit (ab208348; Abcam), and IL-6 ELISA Kit (ab222503; Abcam), respectively.
Detection of oxidative stress level
The level of oxidative stress was evaluated by detecting the levels of malondialdehyde (MDA) and superoxide dismutase (SOD) in tissue or cell supernatant. In short, MDA Kit (A003-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China) and SOD Kit (A001-3; Nanjing Jiancheng Bioengineering Institute) were used to detect the levels of MDA and SOD in myocardial tissues or HL-1 mouse cardiomyocytes.
In vivo ubiquitination assay
The transfected HL-1 mouse cardiomyocytes were treated with MG132 (10 mM) for 8 h. The cell lysates were incubated in RIPA buffer (Beyotime), collected, and boiled for 10 min. Then, the cell lysates were incubated with diluted buffer at 4°C for 30 min. The protein fraction was collected by centrifugation and then incubated with anti-ubiquitin (ab134953; Abcam) and anti-SOX9 (ab185230; Abcam) at 4°C overnight. The mixture and protein A/G agarose beads were incubated at room temperature for another 1 h. The beads were rinsed with washing buffer thrice, and then, the immunoprecipitation was analyzed by Western blot to detect the protein expressions of ubiquitinated protein and SOX9.
Co-immunoprecipitation
The interaction between SOX9 and USP7 was analyzed by co-immunoprecipitation (Co-IP). In short, HL-1 mouse cardiomyocytes were lysed using lysis buffer, and then, the cell lysates were collected and centrifuged at 15,000 g for 20 min. The supernatant was prewashed with A/G protein magnetic beads (Millipore, Billerica, MA) for 2 h, and the antibody was incubated with A/G protein magnetic beads overnight at 4°C. The magnetic beads were washed, boiled in the sample buffer containing mercaptoethanol for 10 min, and immunoblotted with anti-SOX9 (ab185230; Abcam).
Bioinformatics
The binding sites between SOX9 and miR-96-5p promoter were predicted through the Jaspar database (
Chromatin immunoprecipitation
To verify the binding of SOX9 to miR-96-5p promoter, we performed chromatin immunoprecipitation (ChIP) using EZ-ChIP Kit (Millipore). In short, HL-1 mouse cardiomyocytes were treated and chromatin crosslinked. DNA fragments were isolated by ultrasound. Then, anti-SOX9 antibody (ab185230; Abcam) or anti-IgG antibody (ab6789; Abcam) was used for immunoprecipitation. The DNA of immunoprecipitated DNA-protein complex was extracted and purified using fragment DNA Purification Kit (Intron Biotechnology), followed by RT-qPCR. GAPDH was used as NC. The primer sequence of miR-96-5p promoter region was as follows: forward primer: 5′-GGTGAGGTCTCTGTTTCGGG-3′ and reverse primer: 5′-TGTGAGAGGCAGGACCACTA-3′.
Dual-luciferase reporter gene assay
The 3′-UTR sequence of NLRP3 containing wild-type and mutant-type binding sites was cloned into pGL3 vector to construct wild-type luciferase reporter vector (NLRP3-WT) and mutant-type luciferase reporter vector (NLRP3-MUT). The constructed luciferase reporter plasmids were cotransfected with miR-96-5p mimic or mimic NC (GenePharma) into HL-1 mouse cardiomyocytes. miR-96-5p containing wild-type or mutant-type binding sites was cloned into the pGL3 vector, respectively, to construct wild-type luciferase reporter vector (miR-96-5p-WT) and mutant-type luciferase reporter vector (miR-96-5p-MUT). The constructed luciferase reporter plasmids were cotransfected into HL-1 mouse cardiomyocytes with oe-NC or oe-SOX9, respectively. After 48 h, the luciferase activity was measured using Firefly Luciferase Assay Kit.
Statistical analysis
SPSS 21.0 (IBM Corp., Armonk, NY) and GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA) were used for data analysis and map plotting. The measurement data are depicted as mean ± standard deviation. The t-test was used for comparisons between two groups. One-way or two-way analysis of variance was used for comparisons among multiple groups, followed by Tukey's multiple comparison test. A value of p < 0.05 indicated a significant difference.
RESULTS
SOX9 was highly expressed in myocardial injury in septic mice
Transcription factors play a regulatory role in sepsis-induced myocardial injury. 34,35 The transcription factor SOX9 is upregulated in myocardial injury. 9,36 To investigate the correlation between SOX9 and sepsis-induced myocardial injury, we established a murine model of sepsis by CLP operation. The cardiac function indexes LVEF and LVFS of CLP mice were decreased evidently (p < 0.05; Fig. 1A, B). Hematoxylin and eosin (H&E) staining exhibited that the cardiomyocytes of sham-operated mice were complete and arranged neatly, while the cardiomyocytes of CLP mice were disordered and showed obvious inflammatory infiltration (Fig. 1C). The contents of myocardial injury markers CK-MB and c-TnT were elevated significantly in the serum of CLP mice (p < 0.05; Fig. 1D, E). The mRNA level and protein expression of SOX9 in myocardial tissues of CLP mice were notably upregulated (p < 0.05; Fig. 1F, G). Briefly, SOX9 was highly expressed in sepsis-induced myocardial injury in mice.
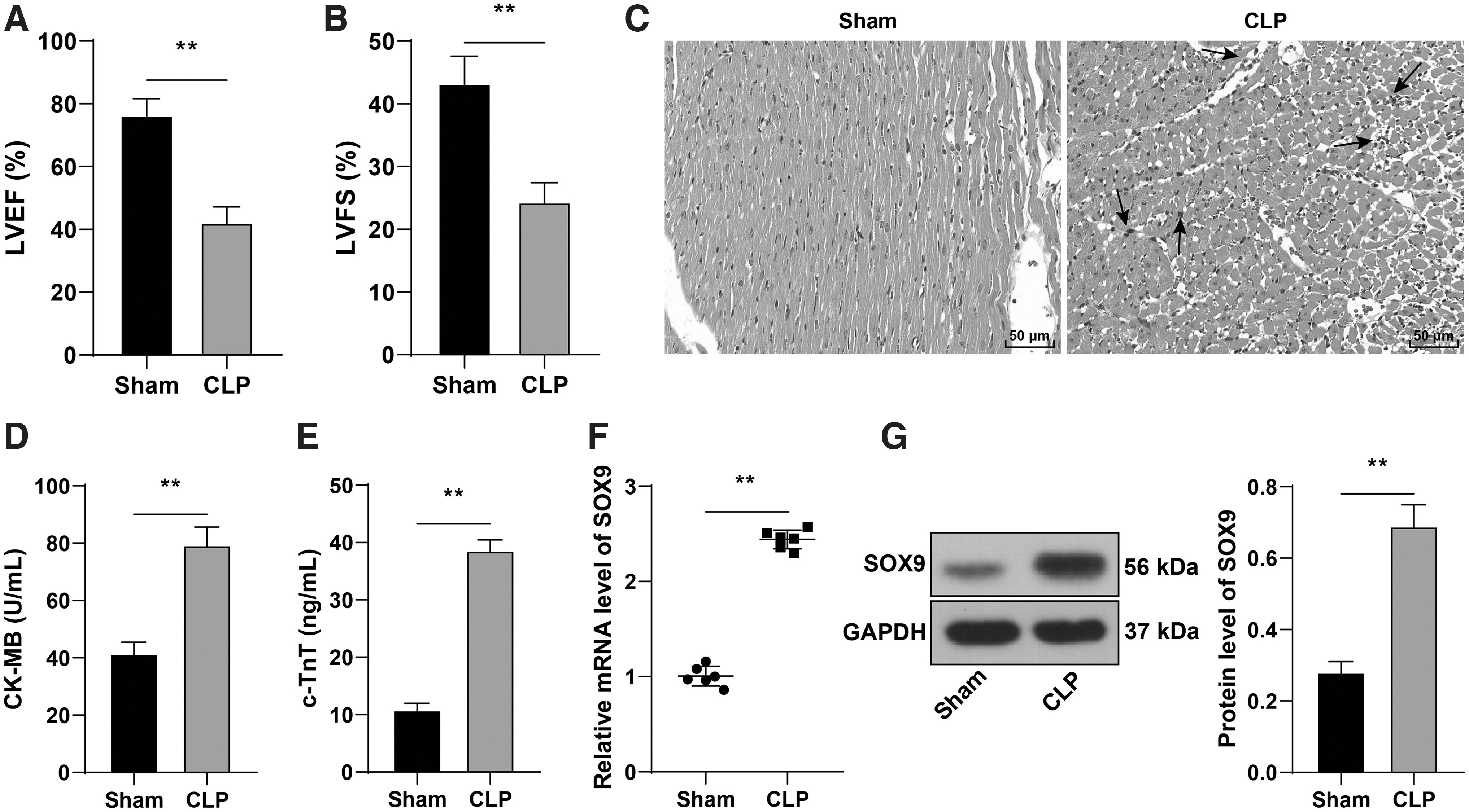
SOX9 was highly expressed in myocardial injury in septic mice. A murine model of sepsis was established by CLP.
Downregulation of SOX9 alleviated myocardial injury in septic mice
To further explore the regulatory effect of SOX9 on myocardial injury in septic mice, we injected lentivirus packaged sh-SOX9 into mice through the tail vein 24 h before CLP operation and found that SOX9 expression in myocardial tissues of septic mice was downregulated significantly (p < 0.05; Fig. 2A, B). Downregulation of SOX9 notably increased LVEF and LVFS of septic mice (p < 0.05; Fig. 2C, D), improved inflammatory infiltration of myocardial tissues (Fig. 2E), and reduced the contents of CK-MB and c-TnT (p < 0.05; Fig. 2F, G). Briefly, downregulation of SOX9 alleviated myocardial injury in septic mice.
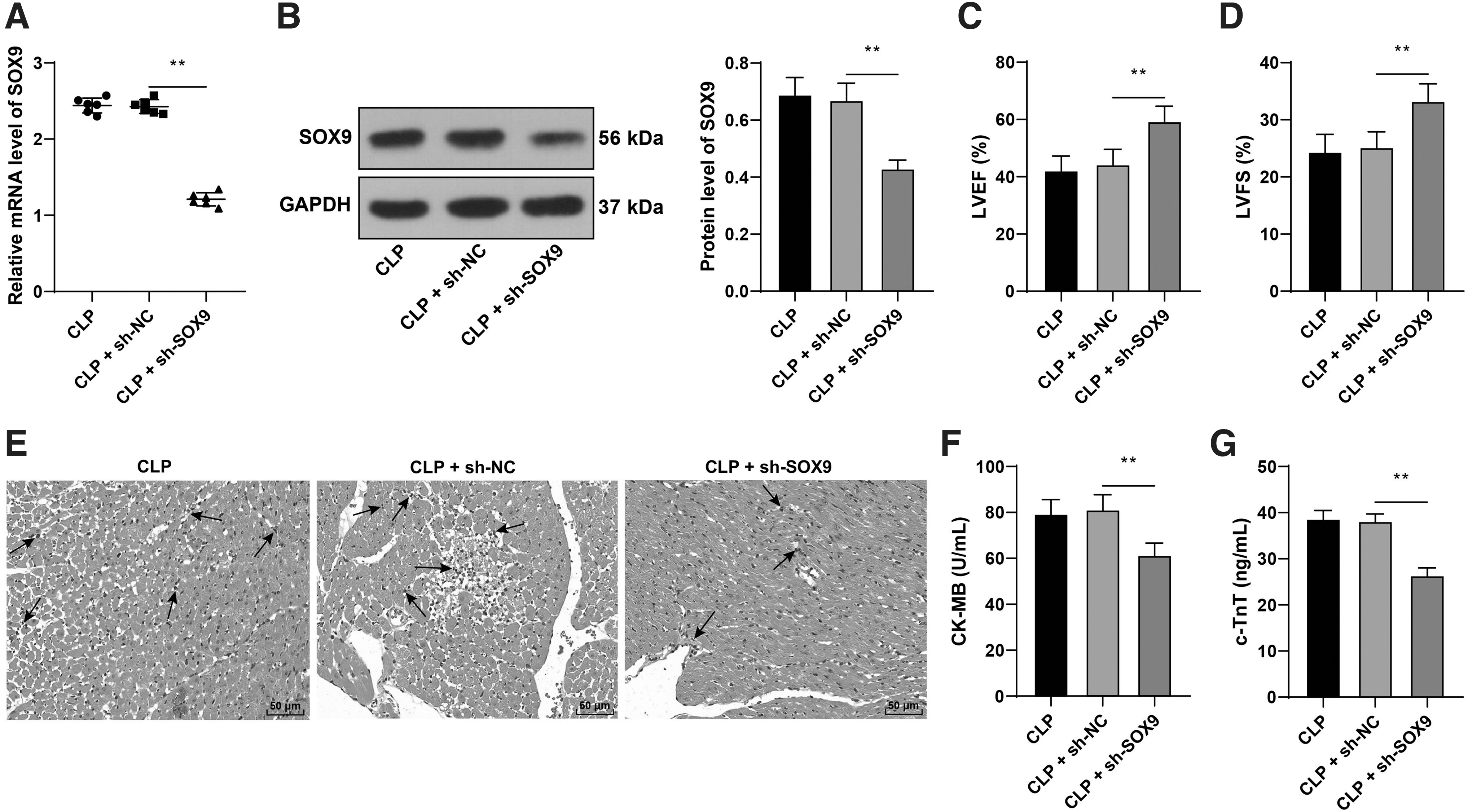
Downregulation of SOX9 alleviated myocardial injury in septic mice. Lentivirus packaged sh-SOX9 was injected into mice through tail vein 24 h before CLP operation, with sh-NC as negative control.
Downregulation of SOX9 suppressed cardiomyocyte pyroptosis in septic mice
Cardiomyocyte pyroptosis plays a vital role in myocardial injury. 37,38 To explore the regulatory effect of SOX9 on cardiomyocyte pyroptosis, we detected pyroptosis-related factors in myocardial tissues of mice. The contents of IL-1β and IL-18 and the protein expressions of cleaved caspase-1 and GSDMD-N in myocardial tissues of CLP mice were increased evidently, while downregulation of SOX9 inhibited the above increasing trends (p < 0.05; Fig. 3A, B).
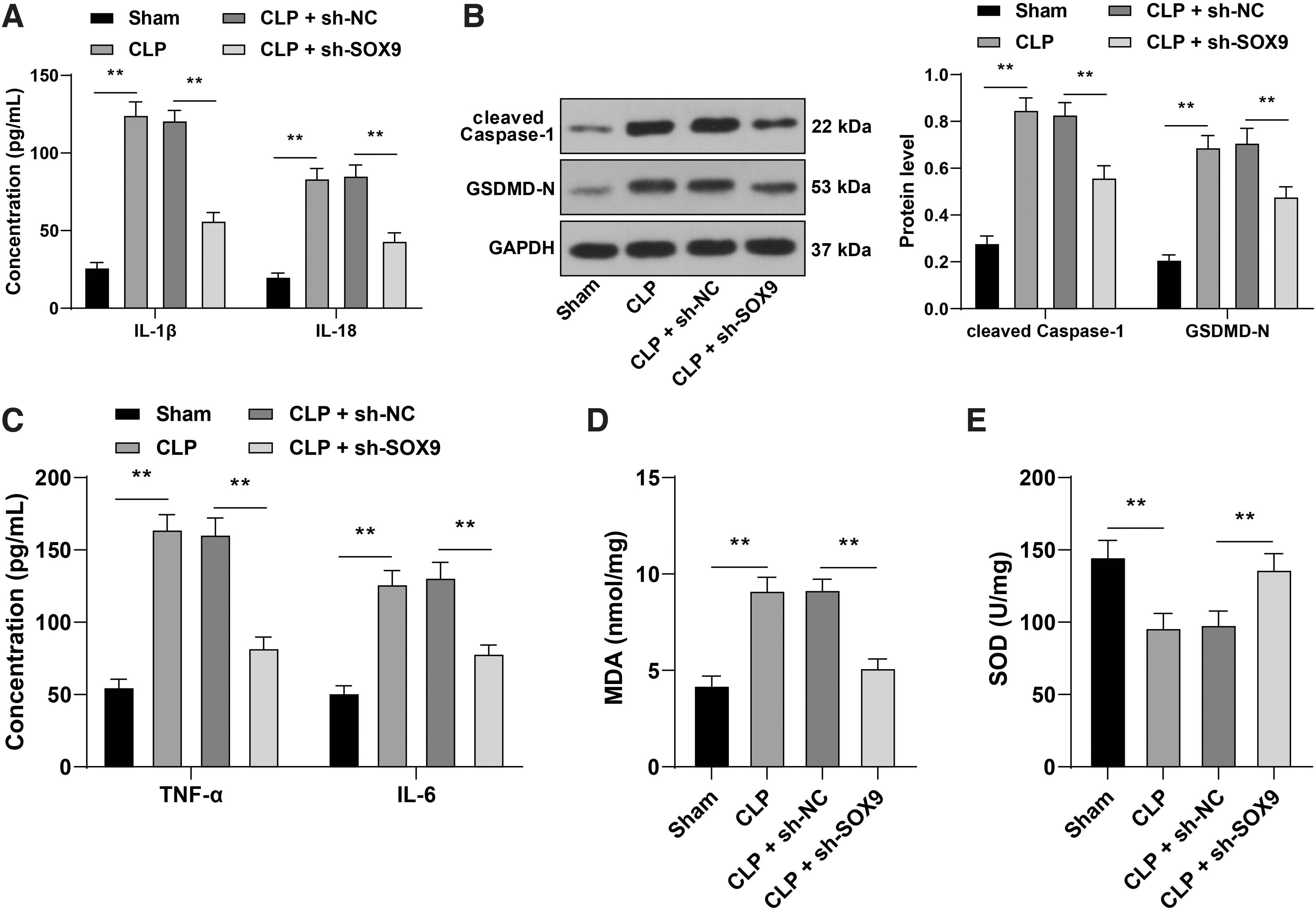
Downregulation of SOX9 suppressed cardiomyocyte pyroptosis in septic mice.
Inflammatory factors such as TNF-α and IL-6 39,40 and oxidative stress 41,42 contribute to pyroptosis. Our results exhibited that the contents of TNF-α and IL-6 in myocardial tissues of CLP mice were elevated significantly, while downregulation of SOX9 led to an opposite trend (p < 0.05; Fig. 3C). The contents of MDA and SOD in myocardial tissues of mice were detected to evaluate the oxidative stress level. The results demonstrated that the MDA level was increased and the SOD level was decreased in myocardial tissues of CLP mice, while downregulation of SOX9 reversed the trends of MDA and SOD (p < 0.05; Fig. 3D, E). Briefly, downregulation of SOX9 suppressed cardiomyocyte pyroptosis in septic mice.
Downregulation of SOX9 mitigated LPS-induced cardiomyocyte injury and pyroptosis in vitro
To further verify the regulatory role of SOX9 in sepsis-induced cardiomyocyte pyroptosis, we transfected si-SOX9#1, si-SOX9#2, or si-SOX9#3 into HL-1 mouse cardiomyocytes to downregulate SOX9 expression in cells and selected si-SOX9#1 with better transfection efficiency for subsequent experiments (p < 0.05; Fig. 4A). Then, HL-1 mouse cardiomyocytes were subjected to LPS induction to establish a cardiomyocyte model of sepsis. It was found that SOX9 expression in LPS-induced cells was enhanced significantly (p < 0.05; Fig. 4B, C). LPS treatment reduced the viability of HL-1 mouse cardiomyocytes and enhanced pyroptosis level, while the cell viability was enhanced and the pyroptosis level was reduced after downregulation of SOX9 (p < 0.05; Fig. 4D, E).
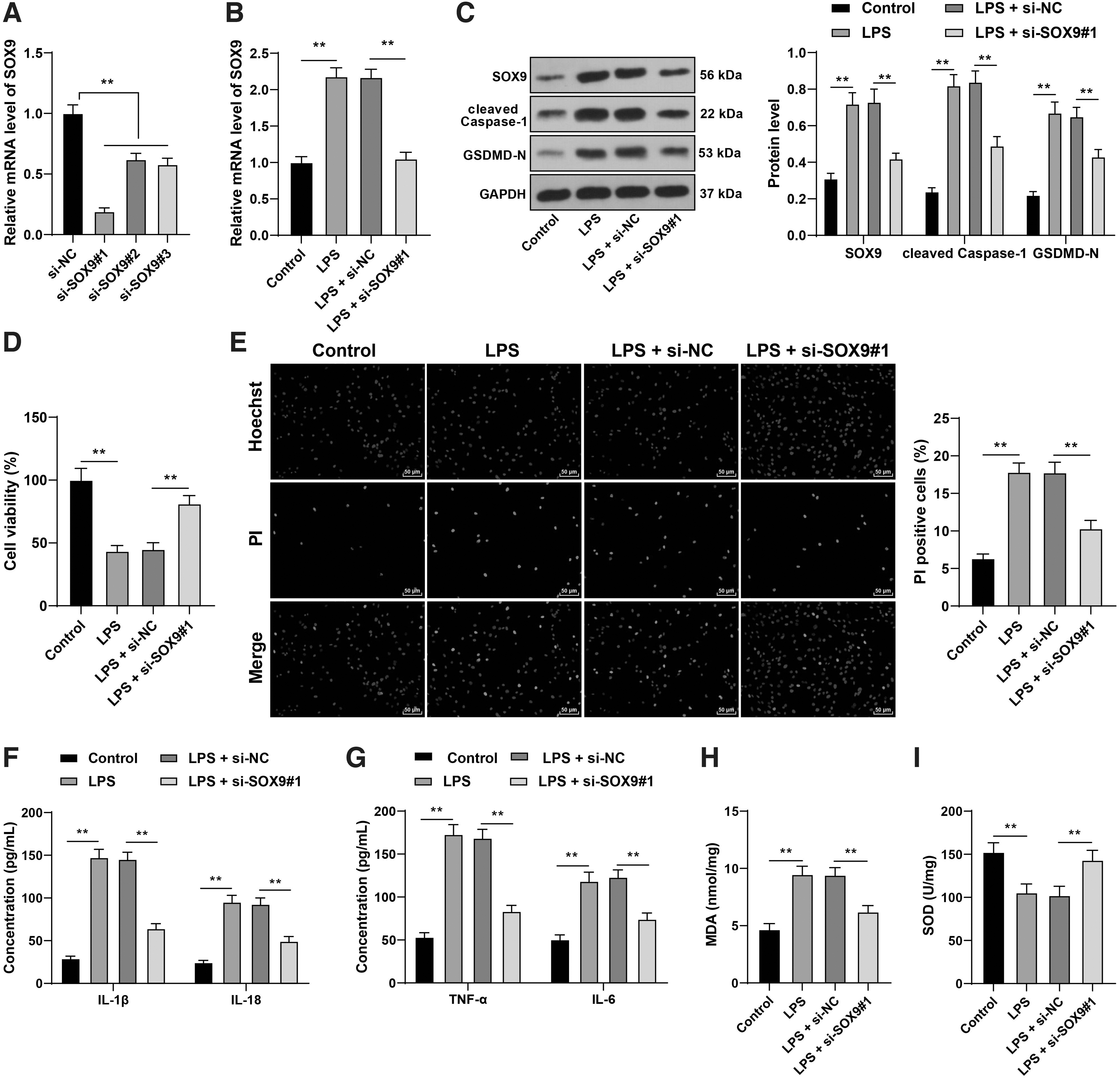
Downregulation of SOX9 mitigated LPS-induced cardiomyocyte injury and pyroptosis in vitro. A cell model of sepsis was established by treating HL-1 mouse cardiomyocytes with LPS. LPS-induced HL-1 mouse cardiomyocytes were transfected with si-SOX9#1, si-SOX9#2, or si-SOX9#3, with si-NC as negative control.
In addition, LPS treatment increased the protein expressions of cleaved caspase-1 and GSDMD-N in cells (p < 0.05; Fig. 4B), elevated the levels of IL-1β, IL-18, TNF-α, IL-6, and MDA, and decreased the level of SOD, while downregulation of SOX9 reversed the changes of the above factors (p < 0.05; Fig. 4F–I). Briefly, downregulation of SOX9 mitigated LPS-induced cardiomyocyte injury and pyroptosis in vitro.
USP7 upregulated SOX9 expression through deubiquitination modification
Studies have shown that SOX9 can be modified by ubiquitination. 12,43 USP7, as a deubiquitination enzyme, is upregulated in myocardial injury. 16 –18 Therefore, we detected the level of USP7 in mouse myocardial tissues and HL-1 mouse cardiomyocytes and found that USP7 was highly expressed after CLP or LPS treatment (p < 0.05; Fig. 5A–D). Co-IP verified the interaction between USP7 and SOX9 (Fig. 5E). Next, we transfected si-USP7#1 or si-USP7#2 or si-USP7#3 into HL-1 mouse cardiomyocytes to reduce USP7 expression in HL-1 mouse cardiomyocytes and selected si-USP7#1 with better transfection efficiency for subsequent experiments (p < 0.05; Fig. 5F, G). It was found that the protein expression of SOX9 in cells was significantly reduced after si-USP7#1 transfection (p < 0.05; Fig. 5G). The ubiquitination level of SOX9 was increased after downregulation of USP7, but decreased notably after the addition of MG132 (Fig. 5H). Briefly, USP7 upregulated SOX9 expression through deubiquitination modification.

USP7 upregulated SOX9 expression through deubiquitination modification.
USP7 overexpression reversed the inhibitory effect of SOX9 silencing on cardiomyocyte pyroptosis
To explore the role of USP7 in the regulation of cardiomyocyte pyroptosis by SOX9, we transfected oe-USP7 into HL-1 mouse cardiomyocytes to upregulate USP7 expression (p < 0.05; Fig. 6A, B) and conducted a combined experiment with si-SOX9#1. It was found that the protein expression of SOX9 was increased with the upregulation of USP7 (p < 0.05; Fig. 6B). After overexpression of USP7, the cell viability was reduced notably, and the pyroptosis level was enhanced significantly (p < 0.05; Fig. 6C, D). Overexpression of USP7 also elevated the protein expressions of cleaved caspase-1 and GSDMD-N (p < 0.05; Fig. 6B), increased the levels of IL-1β, IL-18, TNF-α, IL-6, and MDA, and decreased the level of SOD significantly (p < 0.05; Fig. 6E–H). Briefly, overexpression of USP7 reduced the alleviating effect of SOX9 silencing on cardiomyocyte pyroptosis.
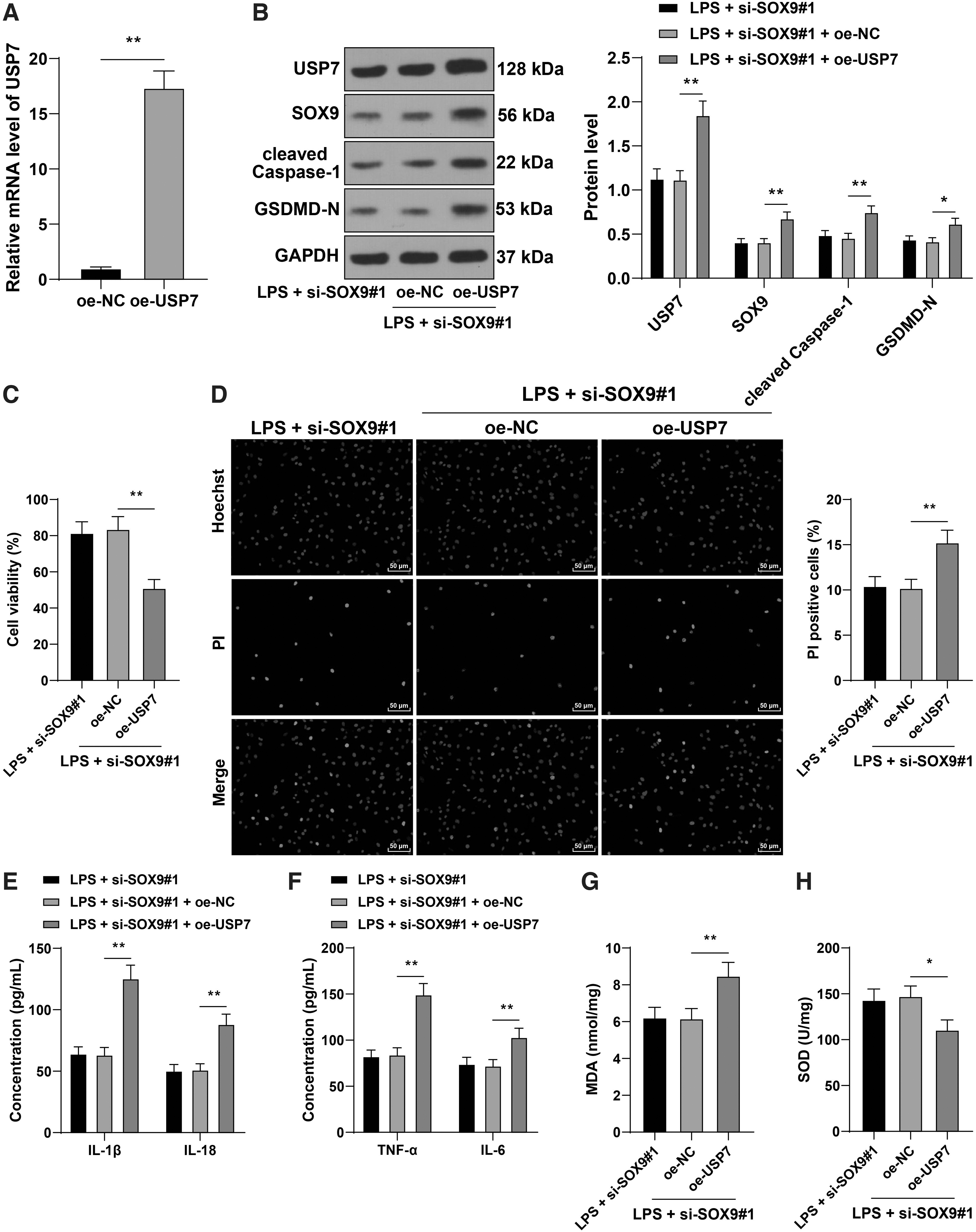
USP7 overexpression reversed the inhibitory effect of SOX9 silencing on cardiomyocyte pyroptosis. HL-1 mouse cardiomyocytes were transfected with oe-USP7, with oe-NC as negative control, followed by the combined treatment with si-SOX9#1.
SOX9 inhibited miR-96-5p expression by binding to miR-96-5p promoter region, thereby promoting NLRP3 expression
The transcription factor SOX9 binds to miRNA promoter in diseases to negatively regulate miRNA expression, 44 and miR-96-5p can be regulated by transcription factors. 45 Importantly, miR-96-5p expression is downregulated in myocardial injury and sepsis-induced inflammatory response. 23,24,46
We performed ChIP assay and confirmed that SOX9 could bind to miR-96-5p promoter in HL-1 mouse cardiomyocytes (p < 0.05; Fig. 7A). In addition, we predicted the binding of SOX9 to miR-96-5p promoter region through the Jaspar website (Fig. 7B) and conducted a dual-luciferase assay according to the binding site. The results showed that there was a binding relationship between SOX9 and miR-96-5p promoter region, and overexpression of SOX9 inhibited the expression of miR-96-5p (p < 0.05; Fig. 7C). Then, we detected miR-96-5p expression in mouse myocardial tissues and HL-1 mouse cardiomyocytes. It was found that miR-96-5p expression was decreased after CLP operation or LPS treatment, while miR-96-5p expression was increased to a certain extent after downregulation of SOX9 and overexpression of USP7 downregulated miR-96-5p expression (p < 0.05; Fig. 7D, E).
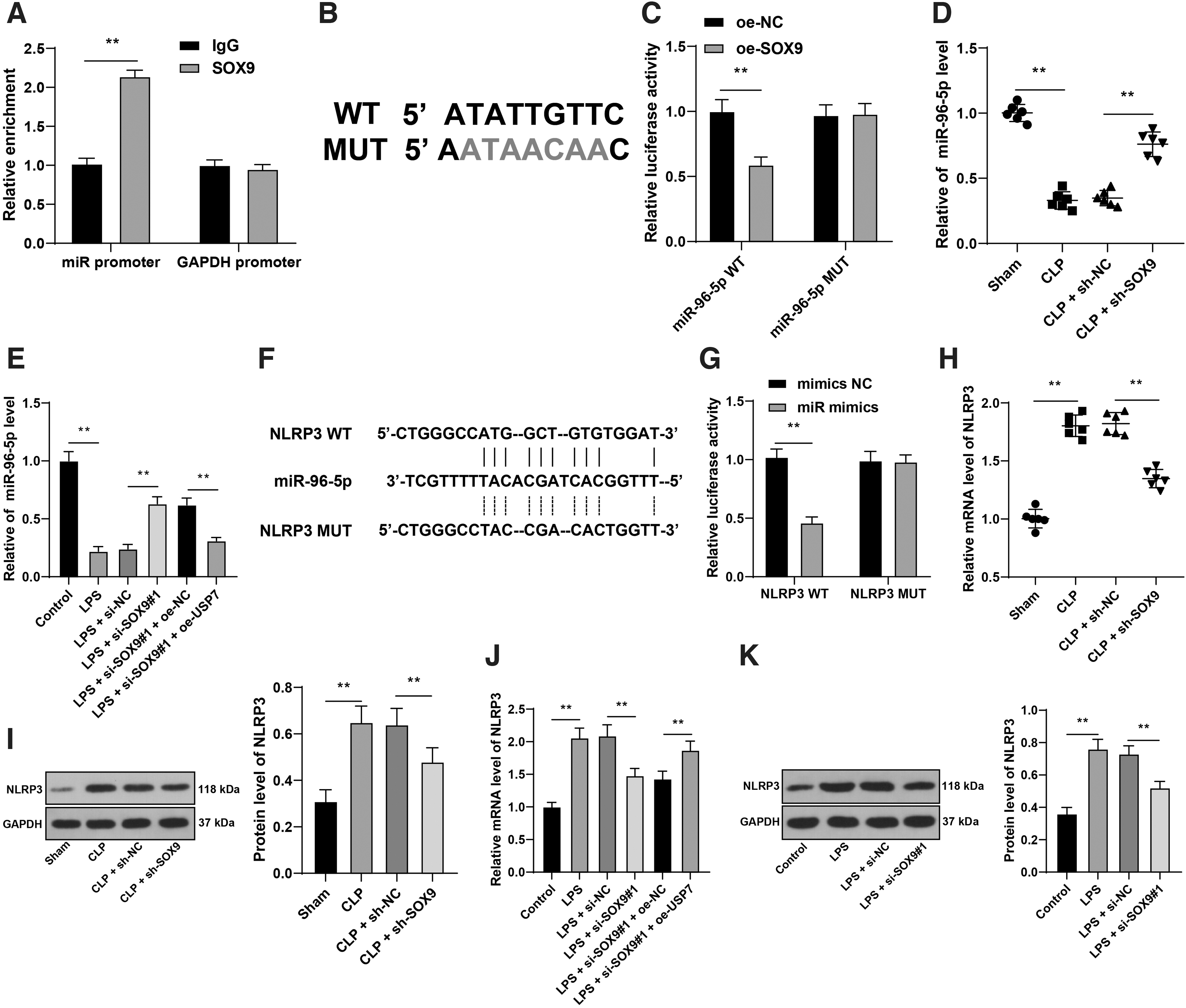
SOX9 inhibited miR-96-5p expression by binding to the miR-96-5p promoter region, thereby promoting NLRP3 expression.
Subsequently, the target genes of miR-96-5p were predicted through the miRWalk database, among which the activation of NLRP3 inflammasome has been reported to aggravate sepsis-induced cardiac dysfunction, inflammation, and pyroptosis. 47,48 The binding site between miR-96-5p and NLRP3 was predicted through the RNA22 v2 database (Fig. 7F). Dual-luciferase assay validated the binding relationship between miR-96-5p and NLRP3 (p < 0.05; Fig. 7G). Finally, NLRP3 expression in mouse myocardial tissues and HL-1 mouse cardiomyocytes was determined. CLP operation or LPS treatment notably elevated NLRP3 expression in mouse myocardial tissues and HL-1 mouse cardiomyocytes, while downregulation of SOX9 repressed the increase of NLRP3 and overexpression of USP7 upregulated NLRP3 expression (p < 0.05; Fig. 7H–K). Briefly, SOX9 inhibited miR-96-5p expression by binding to miR-96-5p promoter region, thereby promoting NLRP3 expression.
Downregulation of miR-96-5p reversed the inhibitory effect of SOX9 silencing on cardiomyocyte pyroptosis
To verify the role of the miR-96-5p/NLRP3 axis in SOX9 reducing cardiomyocyte pyroptosis, we transfected miR-96-5p inhibitor (miR inhibitor) into HL-1 mouse cardiomyocytes to downregulate miR-96-5p expression in cells (p < 0.05; Fig. 8A). Then, miR-96-5p inhibitor-transfected HL-1 mouse cardiomyocytes were treated with si-SOX9#1 for a combined experiment. Our results exhibited that downregulation of miR-96-5p notably elevated NLRP3 expression in HL-1 mouse cardiomyocytes (p < 0.05; Fig. 8B, C), reduced HL-1 mouse cardiomyocyte viability, and enhanced pyroptosis level (p < 0.05; Fig. 8D, E). Moreover, after downregulation of miR-96-5p, the protein expressions of cleaved caspase-1 and GSDMD-N in cells were increased evidently (p < 0.05; Fig. 8C); the levels of IL-1β, IL-18, TNF-α, IL-6, and MDA were elevated, and the level of SOD was reduced (p < 0.05; Fig. 8F–I). Briefly, downregulation of miR-96-5p upregulated NLRP3 expression and reversed the alleviating effect of SOX9 silencing on cardiomyocyte pyroptosis.
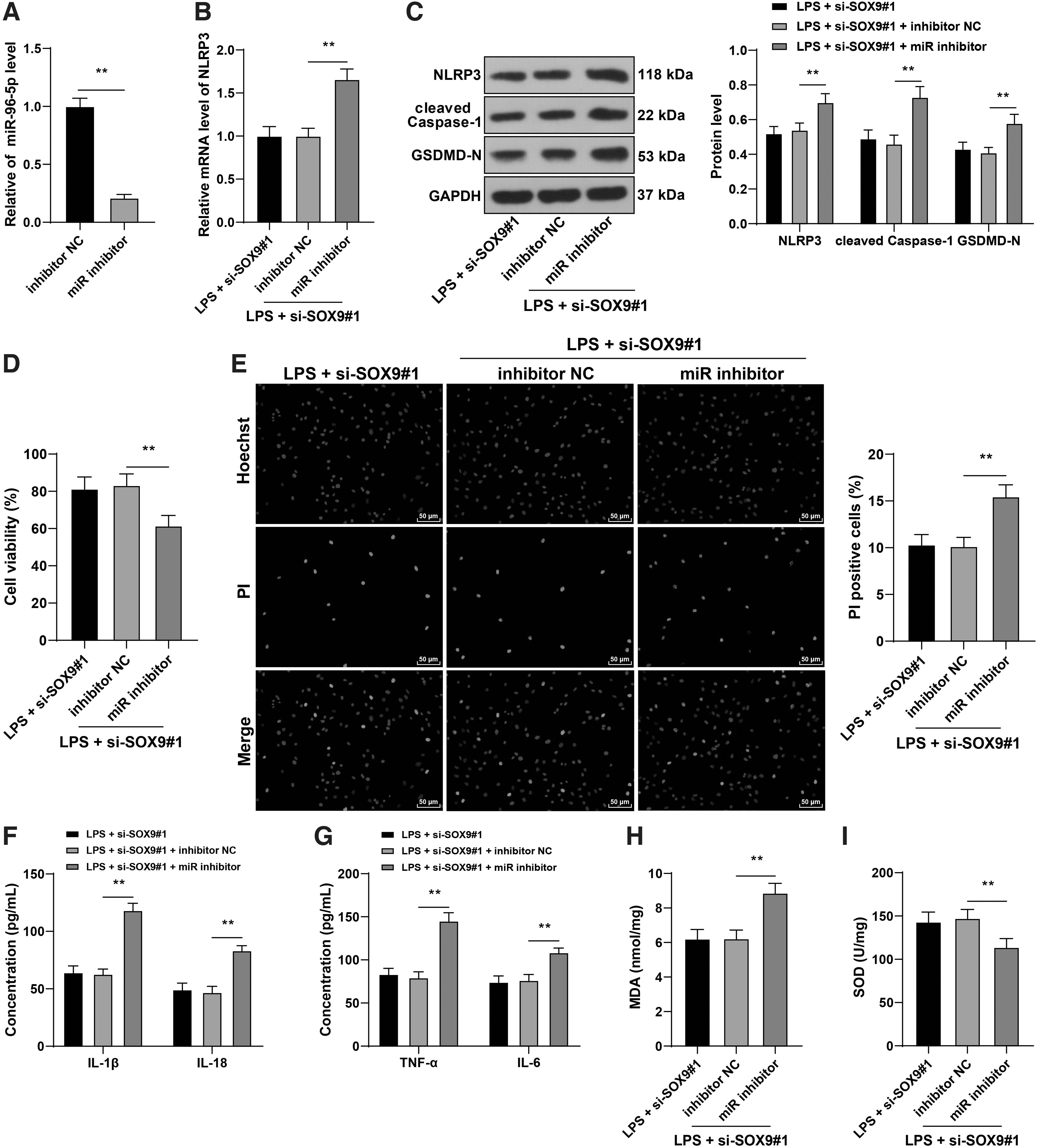
Downregulation of miR-96-5p reversed the inhibitory effect of SOX9 silencing on cardiomyocyte pyroptosis. HL-1 mouse cardiomyocytes were transfected with miR-96-5p inhibitor (miR inhibitor), with inhibitor NC as negative control. Then, miR-96-5p inhibitor-transfected HL-1 mouse cardiomyocytes were treated with si-SOX9#1 for a combined experiment.
Overexpression of USP7 inhibited the protective effect of SOX9 silencing on septic mice
To further verify the role of USP7 in septic mice, we upregulated USP7 expression in mouse myocardial tissues by injecting lentivirus packaged oe-USP7 into mice through tail vein (p < 0.05; Fig. 9A, B). Overexpression of USP7 increased the protein expression of SOX9 in myocardial tissues, decreased miR-96-5p expression, and elevated NLRP3 expression (p < 0.05; Fig. 9B–D).

Overexpression of USP7 inhibited the protective effect of SOX9 silencing on septic mice. Lentivirus packaged oe-USP7 was injected into mice through tail vein 24 h before CLP operation, with sh-NC as negative control, followed by combined treatment with sh-SOX9. USP7
In addition, LVEF and LVFS were reduced significantly after overexpression of USP7 (p < 0.05; Fig. 9E, F). H&E staining exhibited that there was obvious inflammatory infiltration in myocardial tissues after overexpression of USP7 (Fig. 9G). After overexpression of USP7, the contents of myocardial injury markers CK-MB and c-TnT were increased notably (p < 0.05; Fig. 9H, I); the contents of IL-1β and IL-18 were increased (p < 0.05; Fig. 9J); the expressions of cleaved caspase-1 and GSDMD-N were elevated (p < 0.05; Fig. 9B); the contents of TNF-α and IL-6 were increased (p < 0.05; Fig. 9K); the level of MDA was increased, and the level of SOD was decreased notably (p < 0.05; Fig. 9L, M). Briefly, overexpression of USP7 inhibited the protective effect of SOX9 silencing on septic mice.
DISCUSSION
Myocardial injury has been accepted as one of the manifestations of sepsis-induced multiple organ dysfunction. 49 Transcription factors are considered the main regulators of gene expression, and ∼10% of transcription factors in mammals are directly implicated in multiple human diseases. 50 Our study elucidated that the high expression of transcription factor SOX9 was related to deubiquitinase USP7, and SOX9 aggravated myocardial injury and cardiomyocyte pyroptosis in sepsis through the miR-96-5p/NLRP3 axis (Fig. 10).

Role and mechanism of SOX9 in septic cardiomyocytes. The high expression of transcription factor SOX9 in sepsis was related to deubiquitinase USP7. SOX9 inhibited miR-96-5p expression by binding to the miR-96-5p promoter region, thereby promoting NLRP3 expression, increasing cleaved caspase-1 and GSDMD-N expressions, elevating IL-1β and IL-18 contents, and eventually aggravating sepsis-induced cardiomyocyte pyroptosis.
Animal experiments and molecular studies have revealed the specific implication of SOX9 in mammalian development and diseases. 51 SOX9 downregulation mitigates myocardial pathological injury and attenuates oxidative stress and inflammatory responses in myocardial tissues of rats with myocardial ischemia–reperfusion injury. 6 However, the role of SOX9 in sepsis-induced myocardial injury remained unclear. In the current study, we established a murine model of sepsis by CLP to investigate the role of SOX9 in sepsis-induced myocardial injury. Myocardial injury and cardiac dysfunction are severe consequences of sepsis and substantially lead to high mortality. 52 Our results demonstrated that the cardiac function indexes LVEF and LVFS of CLP mice were decreased evidently. The cardiomyocytes of CLP mice were disordered and had obvious inflammatory infiltration. The contents of myocardial injury markers (CK-MB and c-TnT) in the serum of CLP mice were elevated. These results above were indicative of the successful establishment of a murine model of sepsis. Then, we detected SOX9 expression in myocardial tissues of septic mice and found that the mRNA level and protein expression of SOX9 were notably upregulated.
To explore the specific regulatory effect of SOX9 on sepsis-induced myocardial injury, we injected sh-SOX9 into mice through the tail vein 24 h before CLP operation. Our results exhibited that SOX9 silencing alleviated myocardial injury of septic mice, evidenced by elevated LVEF and LVFS, improved inflammatory infiltration in myocardial tissues, and reduced contents of CK-MB and c-TnT. To the best of our knowledge, our study is the first-of-its-kind to demonstrate that SOX9 was highly expressed in sepsis-induced myocardial injury, and SOX9 silencing alleviated myocardial injury of septic mice.
Cardiomyocyte death is recognized as a crucial determinant of myocardial injury severity, and the involvement of cardiomyocyte pyroptosis has been well elucidated in sepsis-induced myocardial injury. 52,53 Pyroptosis is a caspase-1-dependent programmed cell death triggered by multiple cellular insults.
Activated caspase-1 protease can cleave and activate GSDMD, and the N-terminal fragment of GSDMD then forms plasma membrane pores that lead to cytosolic leakage and cell membrane rupture, eventually resulting in the release of pro-inflammatory factors, including IL-1β and IL-18. 48,54 We observed that the contents of IL-1β and IL-18, as well as the expressions of cleaved caspase-1 and GSDMD-N in myocardial tissues of CLP mice, were significantly enhanced, indicating that CLP induced cardiomyocyte pyroptosis in mice. After downregulation of SOX9, the contents of IL-1β and IL-18 and the expressions of cleaved caspase-1 and GSDMD-N were notably reduced.
The inflammatory cytokines TNF-α and IL-6 are essentially responsible for the characteristics of systemic inflammatory response syndrome and are commonly used as biomarkers of septic inflammatory responses. 55 During sepsis, biochemical alternations induce the imbalance of redox system and the formation of oxidative state, which seems to exacerbate the systemic inflammatory response syndrome and downstream adverse events. 56 Inflammation contributes to the production of reactive oxygen species (ROS), which in turn exacerbates tissue injury by activating oxidative stress. MDA, a product of lipid oxidation, is responsible for ROS generation and oxidative damage degree, and ROS in cells is mainly cleared by antioxidant enzymes, mainly SOD. 5 Our results demonstrated that the myocardial tissues of CLP mice had elevated contents of TNF-α and IL-6, increased MDA level, and decreased SOD level. SOX9 aggravates hepatic ischemia–reperfusion injury by enhancing inflammatory response, as supported by the elevated contents of TNF-α, IL-6, and IL-1β. 57 Downregulation of SOX9 can reduce the levels of inflammatory cytokines, including TNF-α, IL-8, and IL-18) in the serum of biliary atresia mice. 58 Inhibition of SOX9 also protects rat neurons from doxorubicin-induced toxic damage by ameliorating oxidative stress. 59 Similarly, we found that downregulation of SOX9 reduced the contents of TNF-α and IL-6, decreased MDA level, and increased SOD level.
To validate the effect of SOX9 on sepsis-induced cardiomyocyte pyroptosis in vitro, we treated HL-1 mouse cardiomyocytes with LPS to establish a cell model of sepsis. In line with our findings in vivo, SOX9 expression was notably elevated in HL-1 mouse cardiomyocytes after LPS treatment, accompanied by reduced cell viability and enhanced pyroptosis, while downregulation of SOX9 relieved LPS-induced cardiomyocyte injury and pyroptosis. SOX9 stability and expression have been demonstrated to be closely controlled by ubiquitination-dependent proteasome degradation. 12
Ubiquitination is essential for a plethora of physiological processes and thus plays a vital role in the pathogenesis and progression of autoimmune, inflammatory, and infectious diseases. 13 USP7, an important deubiquitinase, is involved in diverse cellular processes, including host–virus interactions, DNA damage and repair, epigenetic modulations, gene expression, and protein function regulation, and immune functions. 15 We were the first to report that USP7 was highly expressed in myocardial tissues of septic mice and LPS-treated HL-1 mouse cardiomyocytes. USP7 is reported to regulate the proliferation and differentiation of ATDC5 cells by interacting with SOX9. 19 We verified the interaction between USP7 and SOX9 in HL-1 mouse cardiomyocytes. USP7 silencing notably upregulated SOX9 protein expression and elevated the ubiquitination level of SOX9. Briefly, USP7 upregulated SOX9 expression through deubiquitination modification.
Augmentation of USP7 expression dramatically promotes hypoxia-triggered apoptosis of cardiomyocytes, accompanied by an increase in the secretion of IL-1β, TNF-α, and IL-6, as well as an elevation of myocardial injury markers such as c-TnT and CK-MB. 16 Inhibition of USP7 activates the p53 pathway by suppressing deubiquitination, which decreased ferroptosis and alleviated myocardial ischemia/reperfusion injury. 18 In the current study, USP7 overexpression in HL-1 mouse cardiomyocytes led to increased SOX9 protein expression, reduced cell viability, and enhanced pyroptosis level, indicating that USP7 overexpression inhibited the alleviating effect of SOX9 silencing on cardiomyocyte USP7 pyroptosis.
Subsequently, we determined the downstream pathways of SOX9 regulation in cardiomyocyte injury and pyroptosis, particularly its miRNA-mediated regulation. miR-96-5p has been identified as a critical regulator in myocardial infarction-induced myocardial injury and cardiomyocyte apoptosis. 24 miR-96-5p expression is reduced in neonatal sepsis and LPS-induced inflammatory responses. 23 ChIP results confirmed the binding relationship of SOX9 and miR-96-5p promoter in HL-1 mouse cardiomyocytes. After CLP or LPS treatment, miR-96-5p expression showed a downward trend, while downregulation of SOX9 elevated miR-96-5p expression and USP7 overexpression downregulated miR-96-5p expression.
Thereafter, the target genes of miR-96-5p were predicted through the database, in which we focused on NLRP3. NLRP3 is a member of the nucleotide-binding domain-like receptor family 60 that activates caspase-1 and promotes the maturation of IL-1β and IL-18, which plays a vital role in septic inflammatory responses. 54 NLRP3 inflammasome activation contributes to the progression of sepsis-induced myocardial injury and acute inflammation. 61,62 CLP or LPS treatment upregulated NLRP3 level in myocardial tissues or HL-1 mouse cardiomyocytes, while downregulation of SOX9 depressed the upregulation of NLRP3 and USP7 overexpression upregulated NLRP3 expression. Briefly, it was indicated that SOX9 suppressed miR-96-5p expression by binding to the miR-96-5p promoter, thereby promoting NLRP3 expression.
Functional rescue experiments were performed to verify the role of the miR-96-5p/NLRP3 axis in SOX9 reducing cardiomyocyte pyroptosis. HL-1 mouse cardiomyocytes were transfected with miR-96-5p inhibitor and then treated with si-SOX9#1 for a combined experiment. Our results exhibited that miR-96-5p inhibitor upregulated NLRP3 level in HL-1 mouse cardiomyocytes, reduced cell viability, and enhanced pyroptosis. miR-95-5p has been demonstrated to relieve LPS-induced inflammatory responses in neonatal sepsis by targeting nicotinamide phosphoribosyltransferase. 23 Activation of NLRP3 inflammasome triggered by exogenous stimuli and endogenous injury signaling promotes caspase-1-dependent cardiomyocyte pyroptosis. 54 All these confirmed that downregulation of miR-96-5p reversed the alleviating effect of SOX9 silencing on cardiomyocyte pyroptosis by upregulating NLRP3 expression.
Finally, to further verify the role of USP7 in septic mice, we injected lentivirus packaged oe-USP7 into mice and found that overexpression of USP7 resulted in a significant increase in the protein expression of SOX9, a decrease in the expression of miR-96-5p, and an increase in the expression of NLRP3. Overexpression of USP7 aggravates left ventricular remodeling and depresses left ventricular function of hypoxia-induced rats. 16 Similarly, our results exhibited that overexpression of USP7 inhibited the protective effect of SOX9 silencing on septic mice.
CONCLUSIONS
To sum up, the transcription factor SOX9 was regulated by deubiquitination of USP7, and SOX9 suppressed miR-96-5p expression by binding to the miR-96-5p promoter, thus facilitating NLRP3 expression and thus aggravating cardiomyocyte pyroptosis and myocardial injury in sepsis. However, whether other transcription factors also play a role in sepsis-induced cardiomyocyte pyroptosis remains to be studied. We only discussed the deubiquitination effect of USP7 on SOX9, and whether other ubiquitin ligases can regulate the ubiquitination of SOX9 remains unclear. Moreover, whether SOX9 can bind to other miRNA promoters and play a role in cardiomyocyte pyroptosis in sepsis remains unclear. We also have not verified the role of the miR-96-5p/NLRP3 axis in sepsis myocardial injury in animal experiments. From the above points, we will further improve the mechanism of the SOX9/miR-96-5p/NLRP3 axis in sepsis-induced myocardial injury and explore other ubiquitin ligases that can regulate SOX9 ubiquitination.
Footnotes
AUTHORs' CONTRIBUTIONS
X.G. and Y.L. validation, research, resources, Y.H. data reviewing and writing, F.Z. review and editing. All authors read and approved the final article.
DATA SHARING STATEMENT
The datasets and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
AUTHOR DISCLOSURE
The authors declare that they have no competing interests.
FUNDING INFORMATION
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.