
Abstract
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-based genome editing system exhibits marked potential for both gene editing and gene therapy, and its continuous improvement contributes to its great clinical potential. However, the largest hindrance to its application in clinical practice is the presence of off-target effects (OTEs). Thus, in addition to continuous optimization of the CRISPR system to reduce and eventually eliminate OTEs, further development of unbiased genome-wide detection of OTEs is key for its successful clinical application. This article summarizes detection strategies for OTEs of different CRISPR systems, to provide detailed guidance for the detection of OTEs in CRISPR-based genome editing.
INTRODUCTION
Effective genome editing in cells and organisms is necessary for both basic research and applied science. Early genome editing tools, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), relied heavily on protein–DNA interactions. 1,2 Since its introduction, CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas9 has been favored due to its simplicity and convenience, and its use quickly surpassed that of the previous generation of ZFN and TALEN technologies. 3 Thus, the emergence of this artificial endonuclease, CRISPR/Cas9, ushered in a new era of genome editing driving several major advances in medicine. Three types of CRISPR-based genome editing systems have been developed, CRISPR/Cas nucleases, 4 base editors (BEs), 5 and prime editors (PEs). 6
Natural CRISPR/Cas9 is an adaptive immune defense mechanism developed by bacteria and archaea during evolution 7 and is composed of two main parts: CRISPR sequence and CAS (CRISPR associate system). CRISPR/Cas nucleases work by generating site-specific double-strand breaks (DSBs) in the genome guided by the Cas9 protein and single-guide RNA (sgRNA). DSBs can be repaired by nonhomologous end joining (NHEJ), homology-directed repair (HDR), or microhomology-mediated end joining (MMEJ). 8 BEs are gene editing tools based on the CRISPR/Cas9 system 5 that contain a catalytically inactivated Cas nuclease fused to a single-stranded DNA-specific deaminase enzyme. Without the need for DSBs or donor DNA templates, and independent of HDR, BEs can precisely insert target-site mutations. 9 –11 Cytosine base editors (CBEs) 12 and adenine base editors (ABEs) 5 are classified at the DNA level according to the different bases to be replaced. A-to-I RNA BEs and C-to-U RNA BEs 13 are classified at the RNA level.
Another genome editing tool, PEs, can precisely and specifically introduce all 12 possible point mutations (all 6 possible base pair transitions), small insertions and small deletions, with a minimal editing INDEL ratio and without causing DSBs. 6 PEs consist of a fusion protein between the Cas9 nickase domain (inactivated HNH [His-Asn-His] nuclease) and the engineered reverse transcriptase domain. Although the number of clinical trials using the CRISPR gene-editing system to treat diseases has increased over the past 3 years, off-target effects (OTEs) pose serious risks in clinical practice. For example, CRISPR-Cas9 gene editing can cause unpredictable and heritable structural mutations in DNA. 14 CRISPR/Cas nuclease, BEs, and PEs all produce OTEs. Therefore, it is necessary to develop sensitive off-target detection methods to optimize genome editing technology. However, detecting off-target activity associated with CRISPR systems is extremely challenging. There are both in vitro and in vivo off-target detection methods.
In vivo detection is the preferred method for detecting OTEs, as it is a more realistic reflection of the actual off-target situation. This article reviews the strategies and considerations for choosing the most appropriate techniques for detection of OTEs of the CRISPR-based genome editing system (Table 1). In addition, recent developments aimed at improving the applicability and effectiveness of this approach are discussed.
Comparison of off-target detection methods
The quantity of *represents the extent of corresponding strategy's cost. ***: Lower cost; ****: High cost; *****: Higher cost.
BLESS, breaks labeling, enrichment on streptavidin, and NGS; CHANGE-seq, circularization for high-throughput analysis of nuclease genome-wide effects by sequencing; ChIP-seq, chromatin immunoprecipitation with NGS technology; CIRCLE-seq, circularization for in vitro reporting of cleavage effects by sequencing; Digenome-seq, digested genome sequencing; DISCOVER-seq, discovery of in situ Cas off-targets and VERification by sequencing; DSBs, double-strand breaks; GOTI, genome-wide off-target analysis by two-cell embryo injection; GUIDE-seq, genome-wide, unbiased identification of DSBs enabled by sequencing; HTGTS, high-throughput genome-wide translocation sequencing; IDLVs, integration-deficient lentiviral vectors; Indels, insertional deletions; NGS, next-generation sequencing; OTEs, off-target effects; SITE-seq, selective enrichment and identification of tagged genomic DNA ends by sequencing; SNVs, single nucleotide variants; VIVO, verification of in vivo off-target; WGS, whole-genome sequencing.
IN SILICO PREDICTION OF OTES
CasFinder and other online bioinformatics tools (Table 2) are used for in silico prediction of OTEs. 15 The process for this includes screening for sites with a high probability of off-target events, then enriching and identifying the fragments containing the predicted off-target sites through methods such as PCR amplification and sequencing. This is a biased way of detecting OTEs that generates large amounts of data and identifies off-target sites through different methods. These results can then be used to construct a more accurate algorithm with which to detect OTEs. 16 While these biased detection methods are simple, fast, and cost-effective, there are many factors that affect the specificity of the CRISPR/Cas system. Because all prediction tools are biased and it is impossible for one prediction tool to identify all off-target sites, online bioinformatics tools for use in prediction of off-target sites should be carefully designed and correctly evaluated to diminish bias. 17
Bioinformatics online off-target prediction tools
PAM, protospacer adjacent motif.
WHOLE-GENOME SEQUENCING
Whole-genome sequencing (WGS) is an unbiased method that can screen out all potential off-target sites and their corresponding frequencies, including detection of small indels and single nucleotide polymorphisms (SNPs). 18 This method involves sequencing the entire genome before and after editing and analyzing the mutations generated outside the desired loci. 16 WGS is suitable for analysis of single-cell clones or F1 gene-edited animals. 16 Currently, the most commonly used WGS platform is Illumina sequencing, which uses a sequencing-while-synthesizing method. However, WGS is both expensive and has low sensitivity, resulting in inaccurate identification of low-frequency OTEs. 19 Moreover, it is difficult to use for large-scale evaluation and screening, which greatly restricts development of clinical approaches.
David Liu's laboratory designed the r-loop experimental scheme in 2020, 20 which is fast, convenient, and more economical than WGS. In addition, PCR introduced by next-generation sequencing (NGS) can increase the error rate of sequencing to a certain extent, and has systematic bias. To overcome these shortcomings, Nguyen Tran et al developed a method for detection of PE off-target events using gel electrophoresis based on Sanger sequencing and NGS. This method can detect off-target sites of PEs in a rapid, scalable, and sensitive manner. 21 Although the PCR process introduced by Illumina sequencing can increase the error rate of sequencing to a certain extent and has systematic bias, it has played a huge role in genomics research, medical research, drug development, breeding, and other fields.
TAGGING DETECTION
In vivo detection
In cells
Chromatin immunoprecipitation with NGS technology
Chromatin immunoprecipitation with NGS technology (ChIP-seq) (Fig. 1) was originally developed by Johnson et al. 22 ChIP is an indirect method for detection of off-target events in vivo based on protein DNA binding. 23 The principle of this technology is to split cells after crosslinking DNA and protein in the physiological state. Chromosomes are then separated and randomly cut using ultrasound or enzyme treatment. DNA fragments bound to the target protein are precipitated through the specific recognition reaction of antigen by antibody. The DNA fragment of the binding protein is then released through reverse crosslinking. Finally, the sequence of the DNA fragment is obtained using second-generation sequencing technology. 23 In off-target detection of CRISPR/Cas9, dead Cas9 (dCas9) is used to bind to DNA sequences without cleavage, thereby identifying specific binding sites for Cas9, including on-target and off-target sites. 24
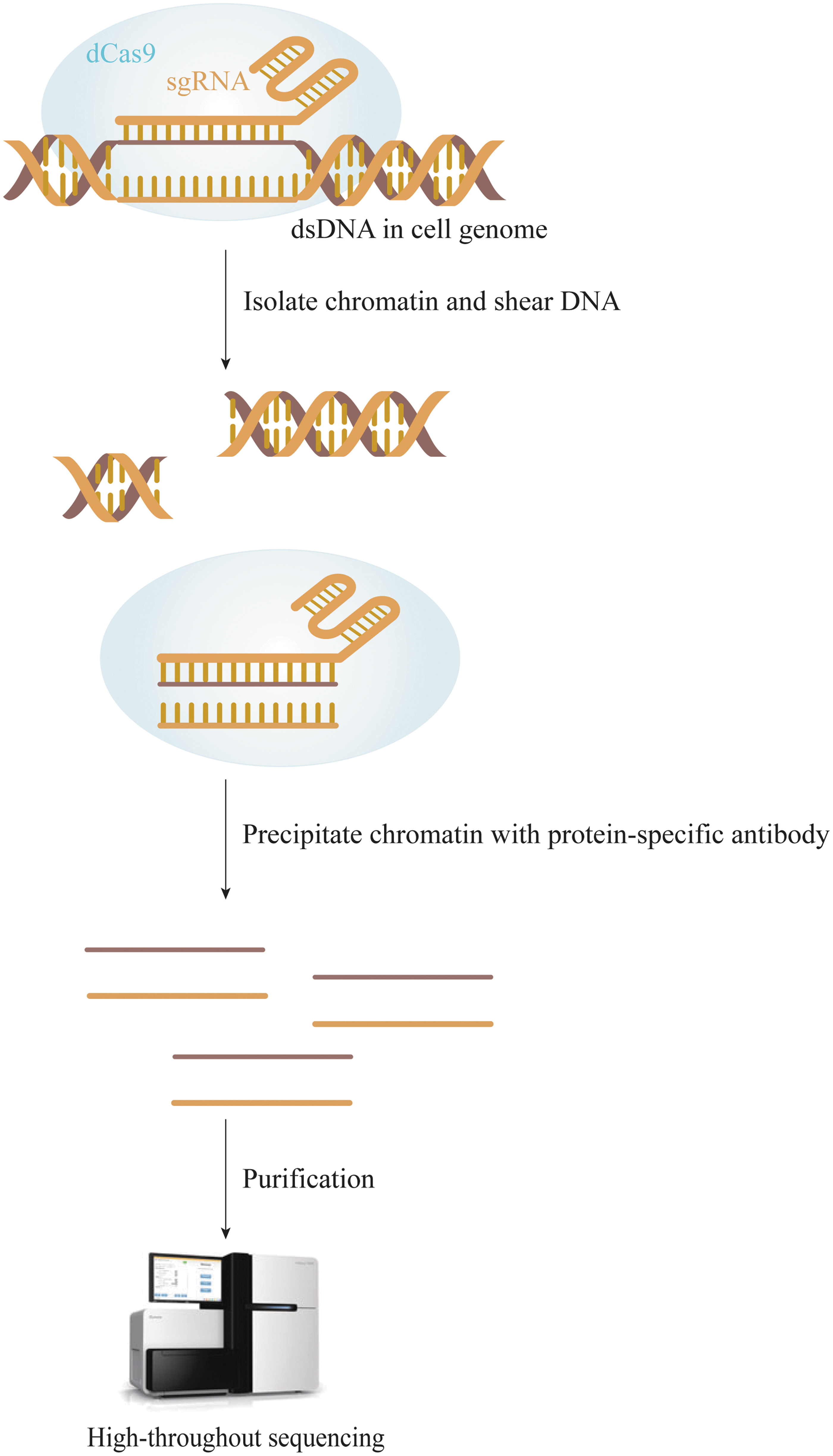
Overview of CHIP-seq workflow. Cellular DNA is crosslinked with dCas9, cells are lysed, chromosomes are separated, and the chromatin is randomly sheared by ultrasound or enzyme treatment. DNA fragments bound to dCas9 are precipitated using antibody for specific recognition of the antigen, and then the DNA fragment bound to the protein is released by reverse crosslinking. Finally, the DNA fragment is sequenced using second-generation sequencing technology. CHIP-seq, Chromatin immunoprecipitation with next-generation sequencing technology; dCas9, dead Cas9.
Since ChIP tends to bind to regions with high expression of polymerase II, polymerase III, and tRNA genes, this technique has a significant false-positive rate. 24 In addition, the Cas9 DNA binding site does not necessarily undergo gene editing. 25,26 Therefore, the off-target sites identified are redundant. 16
Integration-deficient lentiviral vectors
Integration-deficient lentiviral vectors (IDLVs) are the first unbiased assay to be applied to off-target analysis of the action of ZFNs (Fig. 2). 27 In addition to ZFNs, this approach is suitable for the analysis of CRISPR/Cas9, 28,29 TALENs, and megaTALs. 28,29 Unlike intact lentiviral vectors, IDLV does not have a preference for genomic integration; when the nuclease cleaves the genome to generate DSBs, IDLV integrates at both on-target and off-target sites during NHEJ, which is equivalent to tagging the DSB sites. Then, the cell genome is extracted and fragmented, and splice sequences added to both ends of the fragment. Since the IDLVs contain two known long terminal repeat sequences, the fragments on both sides of the IDLV can be amplified using linear amplification-mediated PCR (LAM-PCR) technology, and finally, second-generation sequencing performed. IDLV can detect off target events occurring at rates as low as 1% and identify mismatches of 1–13 bp. 28,29

Overview of GUIDE-seq, IDLV, and BLESS workflow. Cas9 and sgRNA are delivered to the cells then DSBs labeled in different ways and are captured followed by library construction. Finally, the sequences of DNA fragments are obtained using second-generation sequencing technology. BLESS, breaks labeling, enrichment on streptavidin, and next-generation sequencing; DSBs, double-strand breaks; GUIDE-seq, genome-wide, unbiased identification of DSBs enabled by sequencing; IDLV, integrative deficient lentiviral vectors; sgRNA, single-guide RNA.
The advantage of this technique is that IDLV can efficiently access the nucleus, including in human cells, which are difficult to transfect. However, it can also integrate into other non-nuclease-triggered DSB sites, increasing the false-positive rate and, therefore, requires rigorous controls. Moreover, IDLV merging into DSBs is inefficient and may occur randomly. In addition, IDLV cannot reliably detect off-target sites at low frequencies.
Breaks labeling, enrichment on streptavidin, and NGS
Breaks labeling, enrichment on streptavidin, and NGS (BLESS) technology was designed based on the biotin/streptavidin principle (Fig. 2). 30 The principle of BLESS is to generate DSBs by CRISPR/Cas9-mediated gene editing, followed by direct in situ labeling of unrepaired DSBs using biotin linkers. 31 The biotinylated fragments are subsequently captured by streptavidin-coated magnetic beads, then enriched and sequenced with the labeled DSBs for direct detection. BLESS can be performed without relying on exogenous detection of DNA damage repair mechanisms, enabling transient detection of DSBs. 32
The ability of BLESS and ChIP-seq to detect off-target events during genome editing of mouse and human cells has been compared and the results showed BLESS can detect more off-target sites. 32,33 Thus, the BLESS technology is both more sensitive and more quantitative. In addition, this technique is also suitable for detection of exogenous and endogenous DSB in smaller cell and tissue samples. 34 However, BLESS can only detect OTEs at a certain point in time, and may therefore miss many off-target sites. Furthermore, knowledge of the reference genome is required.
Recently, Biernacka et al have proposed a new direct in situ genomic DSB labeling method known as immobilized-BLESS (i-BLESS), 35 which in principle is applicable to all (particularly small) cells and is able to detect very rare breaks. I-BLESS is a highly sensitive and accurate method that detects DSBs with an average frequency rate as low as 0.001%. However, its shortcoming is the relatively high number of cells required.
Genome-wide, unbiased identification of DSBs enabled by sequencing
Genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq) technology is similar to IDLV and BLESS technologies in that it enables genome-wide unbiased off-target detection through labeling of DSBs (Fig. 2). For GUIDE-seq the CRISPR system plasmid and modified double-stranded oligonucleotides (dsODNs) are first transfected into the cell line, and then DSBs are produced by the Cas protein. It is then possible to integrate the dsODNs transfected into the cells into the genome using NHEJ, after which the extracted genome is detected using second-generation sequencing to identify relevant off-target sites. This process has been used to identify dsODNs by high-throughput sequencing as amplification markers containing DSB gene regions. GUIDE-seq is capable of capturing off-target activities beyond bioinformatic predictions and is able to detect indel mutations occurring at frequencies as low as 0.03%. 26
While it is simple and easy to implement as well as cost-effective, high-target cell transfection efficiency is required and GUIDE-seq cannot be applied to some cell types. 16 However, GUIDE-seq can detect RNA-guided nuclease-independent DSB hotspots within the genome. 36,37 In addition, GUIDE-seq can detect off-target sites that ChIP-seq fails to identify. 26
While GUIDE-seq is relatively simple compared with ChIP-seq, IDLV and BLESS techniques, it can only detect off-target events when dsODNs are introduced immediately after the occurrence of DSBs. 38 –40 In addition, it is not clear whether dsODNs tags can be integrated into all DSBs of the cell, a problem shared with IDLV. Furthermore, the efficiency of GUIDE-seq is limited by chromatin availability and epigenetic factors. 26 GUIDE-seq's low false positivity rate and high correlation of edit signals highlight its applicability. However, GUIDE-seq cannot effectively exclude mispriming artifacts. Therefore, Nobles et al proposed an improvement of the GUIDE-seq method, known as iGUIDE, 41 which includes the same steps as GUIDE-seq, but uses a larger deoxyribonucleotide.
LAM high-throughput genome-wide translocation sequencing
Initially, Chiarle et al developed the high-throughput, genome-wide translocation sequencing (HTGTS) method to elucidate the mechanism of translocation in mammalian cells. 42 Later, it was improved by Hu et al and renamed LAM-HTGTS 43 (Fig. 3) due to the introduction of a step to enrich target DNA fragments before adapter ligation. This improved the efficiency of HTGTS while reducing both the cost and time.
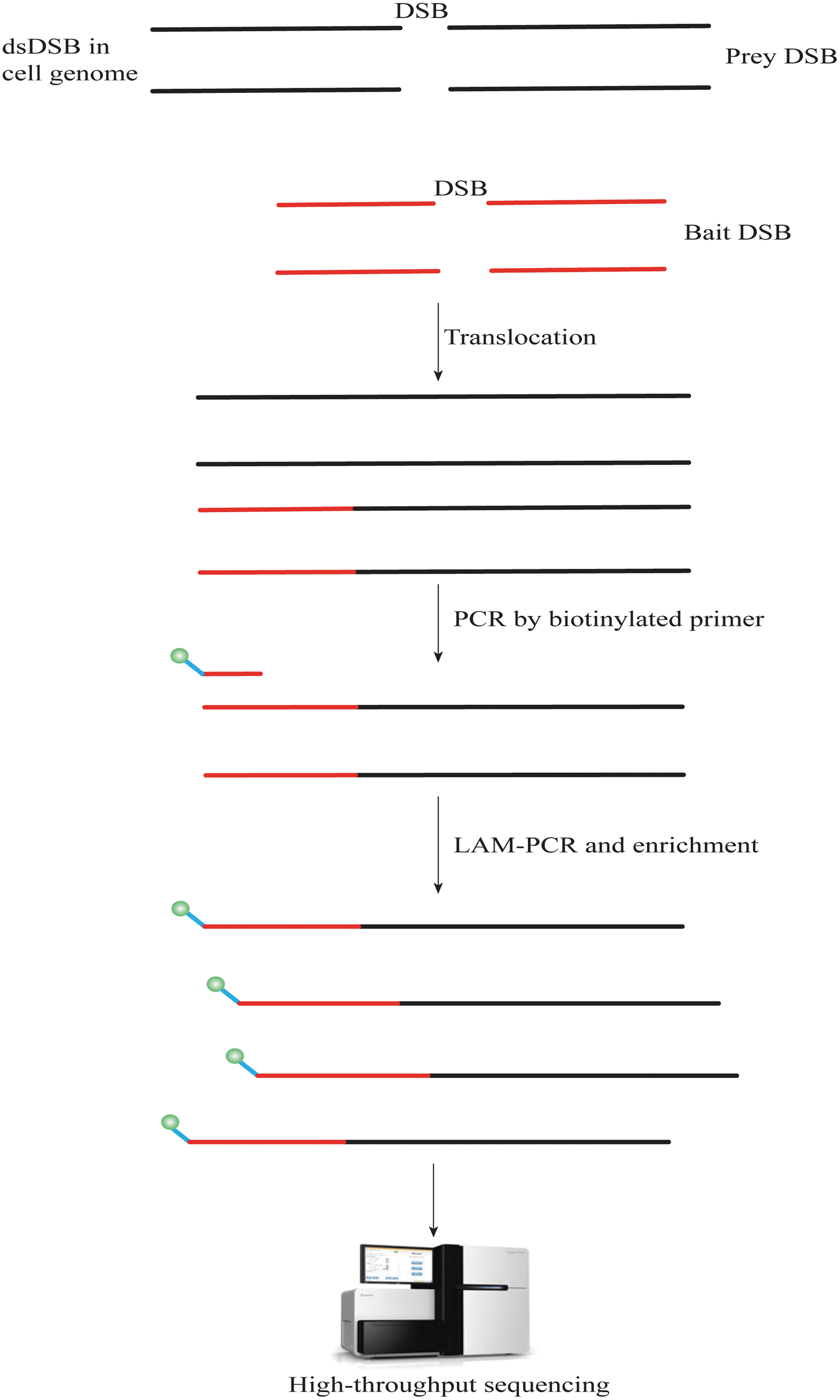
Overview of LAM-HTGTS workflow. DSBs occur in cellular genomes under the action of nucleases triggering gene rearrangements. Genome-wide prey DSBs are identified by translocation of fixed bait DSBs in cultured mammalian cells. The extracted cellular genomes are fragmented, then biotin-tagged capture primers targeting areas near the sgRNA of a gene for OTEs are used for enrichment by LAM-PCR and biotin-strand affinity system. The fragments are then analyzed using second-generation sequencing. HTGTS, high-throughput genome-wide translocation sequencing; LAM-PCR, linear amplification-mediated PCR; OTEs, off-target effects.
DSBs occur in the cell genome in response to nucleases and trigger gene rearrangements. Discovery of genome-wide prey DSBs has been found to occur through translocation of fixed bait DSBs in cultured mammalian cells. The extracted cell genome is then fragmented and capture primers with biotin tags are used to target the region near the sgRNA of each gene to identify off-target events. Sequences with target gene fragments, including those with and without rearrangements, are enriched using the LAM-PCR biotin-strand affinity system, and then analyzed using second-generation sequencing. 43
This method does not require the introduction of additional special sequences, has higher sensitivity than IDLV, lower background values than BLESS, and is relatively economical. The disadvantage is that only loci where rearrangements occur can be detected, 36,43 while the probability of rearrangement is relatively low. 44 Therefore, a large number of off-target sites may be missed. Thus, it can be used in combination with other methods for improved functionality. In addition, LAM-HTGTS is applicable to the human genome, and its efficiency is only limited by chromatin availability or epigenetic factors. 44
Genome-wide off-target analysis by two-cell embryo injection
Genome-wide off-target analysis by two-cell embryo injection (GOTI), a newly reported method by Zuo et al, 45 is a powerful approach to detect off-target events and can assess genome-wide OTEs induced by genome editing tools, such as CRISPR/Cas9 and BEs. 10,46 This method includes injecting a mixture of Cre (distinguishing two cells), gene editing messenger RNA (mRNA) (Cas9/BE3/ABE7.10), and sgRNA into the oocyte of a two-cell mouse embryo (derived from mating Ai9 male mice with wild-type females). When the chimeric embryo reaches embryonic day (E) 14.5, it is digested into a single-cell suspension and then the edited and unedited cells are separated by fluorescence-activated cell sorting. Finally, the two cell populations are processed independently for high-throughput WGS.
GOTI can detect off-target sites in cell populations derived from a single gene-edited oocyte cleavage, as well as in the mouse embryo at an early stage. Whereas previous studies used a large number of cells with varied gene editing results, population averaging resulted in a loss of signal for random OTEs. In addition, since the edited and unedited cells are derived from a single progenitor cell, GOTI minimizes any confounding effects of genetic background and somatic mutations. Therefore, GOTI can be commonly used for genome editing tools without the introduction of DSBs.
Compared with other methods for detecting OTEs, GOTI significantly improves the sensitivity of off-target detection and is a method with better specificity and higher accuracy for detecting OTEs. Therefore, given these advantages, GOTI could be a groundbreaking new approach to assess the safety of gene editing tools. However, GOTI also has limitations, including the long duration of the experiment, 45 which can only be carried out on specific species 45 and cannot be used to evaluate the OTEs in the context of the human genome. In addition, currently the cost of GOTI is prohibitive.
In live mice
Verification of in vivo off-target events
Akcakaya et al developed an in vivo off-target validation method, known as verification of in vivo off-target (VIVO), in 2018. 47 The VIVO process can be divided into two steps. The first step is the identification of off-target sites for in vitro shearing using circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq), and the second step is the evaluation of in vivo indel mutations at the sites identified in the first step using targeted amplicon sequencing of genomic DNA isolated from the livers of nuclease-treated mice (mice euthanized at day 4 or week 3 postinfection). VIVO can stably identify the OTEs of CRISPR across the genome in vivo. Moreover, VIVO was used to show that correctly designed gRNAs can be used with minimal off-target sites to efficiently edit the mouse genome. 47
Cell-specific nontarget mutations that are not dependent on the reference genomic sequence (not necessarily reflecting the genetic background of the organism being studied, or the genetic background of the patient in a clinical application) can also be detected using VIVO. Since genomic DNA from patients or specific organisms can be screened in vitro, reads from the sequencing step reflect possible Cas9 targets in the subject's genome.
VIVO is a highly sensitive strategy that efficiently identifies OTEs of CRISPR-Cas nucleases genome-wide in vivo even at frequencies as low as ∼0.13%. Like all existing methods, VIVO's detection limit is defined by the current error rate (∼0.1%) of the indels' NGS. Thus, VIVO has set an important standard for in vivo detection of OTEs caused by editing nucleases.
Discovery of in situ Cas off-targets and VERification by sequencing
Discovery of in situ Cas off-targets and VERification by sequencing (DISCOVER-seq) (Fig. 4) is a commonly used in vivo, unbiased off-target identification method developed by Wienert et al in 2019. 48 The method is based on tracking the precise recruitment of MRE11 to DSB by chromatin immunoprecipitation followed by NGS. A custom open-source bioinformatics pipeline, BLENDER (Blunt END finder), is then used to identify genome-wide off-target sequences. The MRE1 protein was chosen because 90% of the read peaks in the MRE11 ChIP-seq begin or end at the cas9-induced cleavage site. In addition, MRE11 has a high-quality ChIP antibody that crossreacts between human and mouse, allowing interrogation of human-derived cells or mouse models using a single protocol. 48
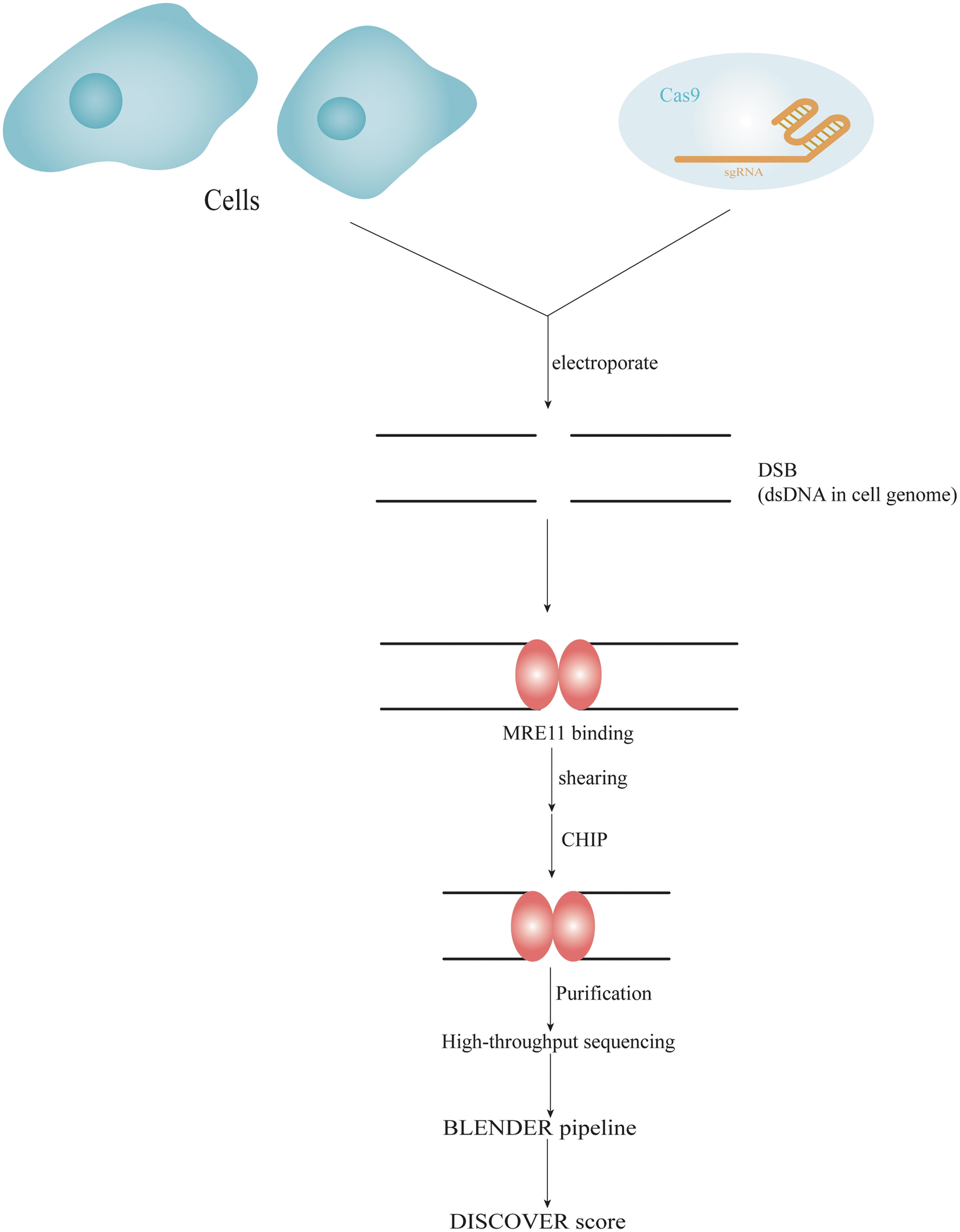
Overview of DISCOVER-seq workflow. This system tracks the precise recruitment of MRE11 to DSB by chromatin immunoprecipitation followed by next-generation sequencing. A custom open-source bioinformatics pipeline, Blunt END finder (BLENDER) identifies off-target sequences genome wide. DISCOVER-seq, discovery of in situ Cas off-targets and VERification by sequencing.
The DISCOVER-seq method allows for unbiased identification of OTEs in cells and organisms that rely on the natural repair process of cells (through recruitment of DNA repair factors) to identify incisions. As a result, it is less invasive and more reliable. The DISCOVER-Seq method truly enables genome-wide analysis of the off-target risk of CRISPR systems, with simplified operational steps and significantly better accuracy than the VIVO system, and therefore may have greater potential for clinical application. It utilizes the MRE11 subunit of the MRN DNA repair protein complex to bind the DNA peaks before insertional deletions (Indels). Used in combination with CHIP-seq and the custom software BLEENDER analysis, the result is highly specific identification of off-target mutations. 48
It should be noted that DISCOVER-seq can detect only 0.3% of indels 48 and is time consuming. In addition, there is a low false positive rate because genome editing requires the binding of DNA repair enzymes. This method has universal applicability to all systems and was the first method to find cleavage sites directly from the CRISPR editing tool in living tissue. In addition to testing the reliability of this method in induced pluripotent stem cells and mouse models, a comparative analysis with the VIVO system was also performed to confirm the superiority of DISCOVER-seq. However, DISCOVER-seq does have shortcomings, including a relatively high detection limit, which results in a requirement for more starting material (≥5 × 106 cells), 48 as well as the requirement for a higher read depth. Theoretically, not all DNA damage sites may recruit MRE11. In addition, this method focuses on capturing DSB loci in the genome, but is not suitable for detecting genomic single nucleotide variants (SNVs). 49
In vitro detection
Digested genome sequencing
Digested genome sequencing (Digenome-seq) is an off-target detection technique performed entirely in vitro (Fig. 5). 25,50 The cell genome is first extracted and the fragments are digested by a nuclease with identical splice sequences at both ends of the fragments. 50 Cas9 protein and sgRNA are then mixed with the digested fragments (which are partially cleaved and partially uncleaved) for cleavage, and this in vitro digestion produces sequence reads with identical 5′ end cleavage sites that can be read using second-generation sequencing and compared with the genomic sequences. Digenome-seq is currently used to assess the genome-wide specificity of Cas9 and Cpf1 nucleases. 25,50,51

Overview of Digenome-seq, CIRCLE-seq, CHANGE-seq, and SITE-seq workflow. Cas9 and sgRNA are mixed with genomic DNA, incubated in vitro, then ligated with different adaptors followed by library construction. Sequences of the DNA fragments are obtained using second-generation sequencing technology. CHANGE-seq, circularization for high-throughput analysis of nuclease genome-wide effects by sequencing; CIRCLE-seq, circularization for in vitro reporting of cleavage effects by sequencing; Digenome-seq, digested genome sequencing; SITE-seq, selective enrichment and identification of tagged genomic DNA ends by sequencing.
This method is highly sensitive and can detect indels present at <0.1% with high reproducibility and homogeneity. This technique directly detects cleaved sites rather than bound sites, and the coverage of off-target sites is relatively large. However, there is an efficiency problem with the tag introduction approach, as not all of these tags can be integrated into the DSBs. This is especially true for low-frequency off-target sites. Moreover, second-generation sequencing analysis is performed without separating the fragments that are cleaved from those that are not. Therefore, a large read length of up to 400 million is required to detect low-frequency loci. Moreover, this is an off-target assay technique performed entirely in vitro, so it may not truly reflect the situation in vivo, and there may be a false-positive rate because it is not influenced by chromatin structure. In addition, Digenome-seq is not suitable for screening large amounts of gRNA. 50,52
To investigate whether or how chromosomes in eukaryotic cells affect CRISPR/Cas9 on-target and off-target activity, Kim et al improved upon the Digenome-seq to generate the Digenome-seq using cell-free chromatin DNA (DIG-seq) approach. 53 Unlike Digenome-seq, which obtains histone-free DNA in vitro by nuclease digestion, DIG-seq separates cytoplasmic and nuclear pellets using lysis buffer, generating chromosomal DNA. Cas9 protein/sgRNA is then incubated with chromatin DNA in vitro and compared with the reference genome using second-generation sequencing. Thus, this enables detection of genome-wide off-target activity. While DIG-seq is also an in vitro method, it does preserve the chromosome structure in eukaryotic cells. Therefore, DIG-seq retains all the advantages of Digenome-seq, but with enhanced accuracy.
To detect the specificity of BE3, Kim et al modified Digenome-seq. 54 For this approach, genomic DNA is treated with a mix of the BE and in vitro DNA-modifying enzymes to produce DNA DSBs in uracil-containing loci. The off-target site was then identified from the WGS data, thereby demonstrating the high specificity of BE3 deaminase. Later, Kim et al adapted Digenome-seq using a similar approach to detect the specificity of the ABE. 55
Digenome-seq is a highly sensitive method capable of identifying BE3 off-target sites present at a frequency of 0.1%. However, Digenome-seq in its current form cannot be used to analyze the specificity of BE1 and BE2, because BE1 and BE2 are composed of dCas9, not nCas9.
Kim et al developed Nickase-based Digenome-seq (nDigenome-seq) in 2020, 56 an in vitro assay that identifies single-strand breaks caused by CRISPR Cas9 nickase using WGS. nDigenome-seq is based on the principle of extracting DNA from HEK293T cells, followed by incubation with Cas9 H840A nickase and in vitro transcription of sgRNA, fragmentation, end-repair and splice ligation of DNA to create the library, followed by WGS to identify genome-wide off-target sites. nDigenome-seq can be used to analyze CBE, ABE, and hRad51-Cas9 and PE genome-specific indels.
The high specificity of PEs was demonstrated by Kim et al. For this, they used nDigenome-seq to detect potential genome-wide off-target sites for Cas9 H840A nickase and then sequenced targeted amplicons of off-target candidate genes identified by nDigenome-seq. Furthermore, although the cut caused by Cas9 H840A nickase is essential for PE2-assisted genome engineering, Cas9 H840A nickase-mediated nDigenome-seq was found to be an indirect method to analyze the genome-wide specificity of PEs. More direct methods, such as GOTI 49 and transcriptome sequencing, 57 can be used to complement nDigenome-seq-based analysis of the specificity of PEs.
Circularization for in vitro reporting of cleavage effects by sequencing
The CIRCLE-seq technique is similar to Digenome-seq in that it takes advantage of the ability of the Cas9 protein to digest DNA in vitro, using hairpin connectors and other labeled DNA fragments to achieve screening of off-target sites by sequencing or other means (Fig. 5). 58 CIRCLE-seq first fragments the genome followed by ligation cyclization to enrich the DNA in the sample, before removal of linear genomic DNA using exonuclease to obtain circular DNA. This circular DNA is then treated with Cas9 complex followed by PCR amplification and attachment of an adapter, and finally second-generation sequencing.
CIRCLE-seq can detect low-frequency off-target mutations using in vitro nuclease-mediated, highly enriched genomic DNA cleavage, 59 and can detect <0.1% of off-target sites with high sensitivity. Using the example of targeting the HBB gene, the researchers showed that CIRCLE-seq identified 26 of the 29 loci detected by Digenome-seq, and also detected 156 new loci that could not be detected by other methods. Moreover, CIRCLE-seq does not require a reference genome. The degradation step before the use of Cas9-sgRNA virtually eliminates the presence of high background DNA to improve sensitivity. 58 However, since the technique is performed in vitro, it may not be a true reflection of what is happening in vivo. In addition, a large amount of genomic DNA (∼25 μg) is required for each CRICLE-seq sample.
Selective enrichment and identification of tagged genomic DNA ends by sequencing
Selective enrichment and identification of tagged genomic DNA ends by sequencing (SITE-seq) technology is an in vitro, unbiased off-target assay (Fig. 5). 60 First, the high-molecular-weight cell genome is obtained and then cut in vitro with Cas9/sgRNA ribonucleoprotein (RNP). Biotinylated connector sequences are added to the cut site, and then the genome is broken up, and streptavidin is used for enrichment. After the connector sequences are added to the other end of the segment, PCR and second-generation sequencing are performed.
All Cas9 cleavage sites in the genome can be mapped using the SITE-seq method. Compared with Digenome-seq, this technique enriches and detects off-target sites, improving the sensitivity of the assay. As a result, SITE-seq requires very small NGS read depth compared with Digenome-seq. However, SITE-seq requires cleaved DNA and biotinylated adapters for ligation, and SITE-seq is performed entirely in vitro. Furthermore, as the concentration of Cas9/sgRNA RNP increases, it detects far more off-target sites than those detected in vivo, so it may not truly reflect the off-target situation in cells, which is time and concentration dependent, 61 but can provide a guideline for the selection of editing sites.
Circularization for high-throughput analysis of nuclease genome-wide effects by sequencing
Circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq) is an easy-to-use, sensitive, high-throughput, scalable method for determining the location of unintended DNA DSBs caused by genome editors, such as CRISPR-Cas9 (Fig. 5). CHANGE-seq works by first randomly labeling an average of ∼400 bp with a custom Tn5 transposon ligated to genomic DNA through an uracil-containing adapter. The 9 nt DNA gap generated by Tn5 is then filled with high-fidelity uracil-resistant U+ polymerase and sealed with Taq DNA ligase. A mixture of USERase and T4 PNK is then used to release 4 bp overhangs to enrich for circular DNA molecules. Unwanted linear DNA is degraded with a nucleic acid exonuclease mixture and then the purified circular DNA is treated with Cas9/sgRNA RNP and the sheared DNA ends are released at on-target and off-target sites. Finally, off-target events are detected using second-generation sequencing technology.
Tsai et al used the CHANGE-seq technique 62 to reveal genetic and epigenetic factors affecting CRISPR-Cas9 genome-wide activity. This is a scalable, tagmentation-based measure of Cas9 genome-wide activity in vitro. Furthermore, CHANGE-seq analysis of six targets in eight independent genomes showed that human SNVs have a significant effect on activity at ∼15.2% of the off-target sites. 62 In addition, this method outperforms CIRCLE-seq in sequencing efficacy and parallel testing.
OFF-TARGET ANALYSIS FOR BASE EDITORS
With respect to BE, there are several special detection methods that can be utilized for off-target detection in addition to the aforementioned WGS and GOTI methods.
EndoV-seq
In 2019, Liang et al developed the first method to detect genome-wide OTEs of the ABE system, EndoV-seq (Fig. 6). 63 Similar to Digenome-seq, EndoV-seq relies on the processing of modified DNA enzymes in vitro. The principle of this method is to obtain ABE7.10 protein by prokaryotic expression and in vitro purification, then incubate it with gRNA and genomic DNA in vitro. Under the guidance of gRNA, ABE7.10 cleaves the complementary strand of the target site on the genomic DNA and converts A to I on the noncomplementary strand. Endonuclease V (EnodV) is then used to cut the genomic DNA containing I base, causing the DNA DSBs, and finally, WGS combined with bioinformatics analysis is used to detect the DNA DSBs, thus identifying the off-target sites in the whole genome.
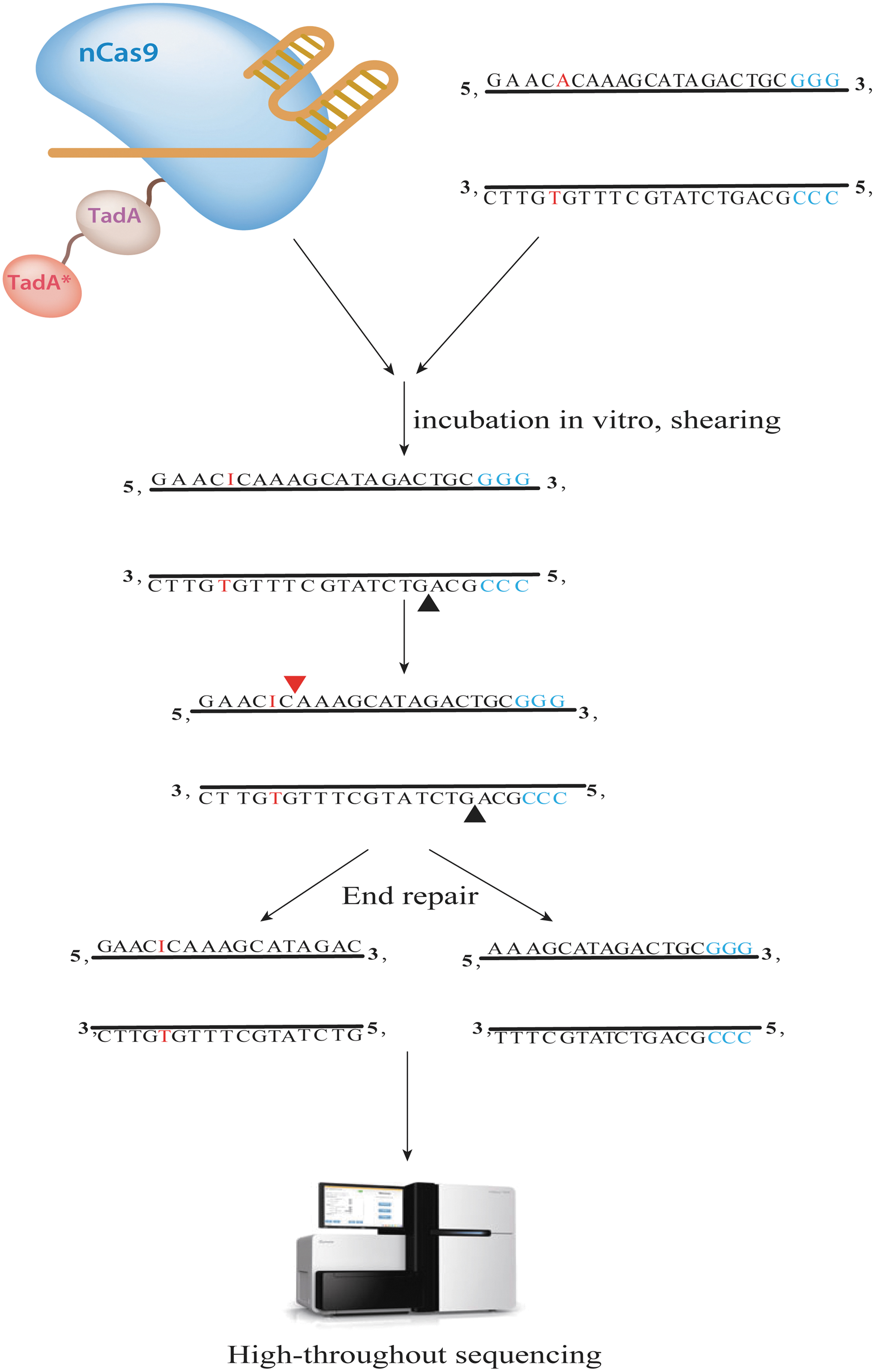
Overview of EndoV-seq workflow. By incubating nCas9-TadA with gRNA and genomic DNA in vitro, under the guidance of gRNA, nCas9 cleaves the complementary strand of the target site on genomic DNA and converts A to I on the noncomplementary strand. Endonuclease V (EnodV) is then used to cleave the genomic DNA containing I bases resulting in DNA DSBs. DNA DSBs are then detected using whole-genome sequencing combined with bioinformatics analysis to detect off-target sites genome wide.
EndoV-seq is able to assess on-target and off-target deamines by ABE. In addition, EndoV-seq can be multiplexing. For example, Liang et al incubated six gRNAs, ABE7.10, and genomic DNA, and detected off-target sites within the whole genome of six gRNAs by single sequencing (MultiplexEndoV-seq). 63 This approach can provide information about how the specificity of ABE increases.
EndoV-seq can be used to detect different ABE variants (e.g., ABE6.3/7.8/7.9 and xCas9-ABE) 5,64 and is a powerful tool for in vitro analysis of ABE off-target events. In addition, EndoV-seq is highly sensitive and can identify off-target sites generated by ABE deamination in vivo at a frequency as low as 0.13%, 63 which is very close to the detection limit of deep sequencing (0.1%). 58 However, EndoV-seq may overestimate the number of potential off-target sites. In addition, because EndoV-seq eliminates the possible complications of spatial obstruction, detected off-target events are not necessarily editable in vivo. 65,66 Furthermore, EndoV-seq is unable to detect deamination at sites on the complementary strand not cleaved by Cas9 nickase.
MuTect2
In addition to targeting DNA, BEs cause genome-wide RNA cytosine deamidation in human cells due to overexpression effects. This results in the generation of RNA editing activity, thereby causing RNA off-target editing affecting the expression of up to 38–58% of genes. 57 Recently, the RNA OTEs of CBE have been greatly reduced, while the off-target activity of ABE is still abundant. Although ABEs do not have detectable guide RNA-independent DNA off-target editing, thousands of A-to-I RNA edits can be induced across the transcriptome in human cells in a guide RNA-independent manner. 57,67
The GATK HaplotypeCaller is a tool for assessing germline SNPs and indels 68 that can be used to analyze RNA A-to-I editing. However, this method generates false negatives because it cannot detect RNA editing with 0–10% efficiency. 57,69,70 Therefore, Li et al 71 proposed that it is more reasonable to assess RNA off-target editing using GATK MuTect2 (for detecting somatic mutations in cancer), asMuTect2 is more sensitive and accurate. Compared to HaplotypeCaller, MuTect2 recovered 2.7 to 11-fold of RNA editing induced by ABEs, 71 especially for RNA editing with 0–10% editing efficiency.
However, the ability of different tools to detect RNA mutations depends on the sequencing depth, the region detected, and the frequency of the variant allele, 72 so different detection tools may lead to different detection results. Using GATK MuTect2 alone is not ideal for detection of RNA OTEs. Therefore, two or more complementary strategies can be used simultaneously when performing off-target detection, such as HaplotypeCaller and MuTect2, depending on their individual advantages and disadvantages. 72,73
dU-detection enabled by C-to-T transition during sequencing
dU-detection enabled by C-to-T transition during sequencing (Detect-seq) is a new method established by Abudayyeh et al and Nishida et al for assessing CBE off-target sites by identifying dU production. CBE converts deoxycytidine (dC) to deoxyuridine (dU) and eventually to deoxythymidine (dT) in bases, enabling C to T editing of DNA. 13,74 Detect-seq takes advantage of the fact that CBE produces the intermediate product, deoxyuracil (dU), during the process of target editing, and biotin is then used to label and enrich the dU produced by CBE. At the same time, cytosine C is replaced with d5fC in the vicinity of the dU site, which, in combination with subsequent chemical labeling reactions generates a tandem C-to-T signal, thereby localizing dU with high confidence and increasing the sensitivity of detection of OTEs. 75 –77
Detect-seq technology is a method that enables highly sensitive, specific, and preference-free detection of CBE off-target sites in cells. In contrast to GUIDE-seq, Detect-seq can identify most loci identified by GUIDE-seq in EMX1 and VEGFA site-2. In addition, Detect-seq identified a large number of off-target sites that were not identified by GUIDE-seq. Furthermore, similar results were obtained with Detect-seq as with WGS-based tools for OTE studies. Meanwhile, Detect-seq captures 30–50% of the off-target sites predicted in silico. These data suggest that Detect-seq is an excellent tool for studying the OTEs of CBE with exceptional specificity and sensitivity. In addition, CBE was also found by Detect-seq to cause a large number of off-target site out-of-protospacer, as well as OTEs on protospacer adjacent motif (PAM)-free strands. Moreover, the off-target sites identified by Detect-seq are significantly enriched in open chromatin regions such as actively transcribed gene regions and gene promoter regions, presenting a sharp contrast with the in vitro experiments and computer predictions.
OFF-TARGET ANALYSIS FOR PRIME EDITORS
Currently, there are no specific off-targeting assays for PEs. In addition to the aforementioned WGS and nDigenome-seq, a new, universal off-targeting method, CRISPR off-targeting single-stranded DNA (ssDNA) sequencing (CROss-seq), has recently been reported. 78 The method is based on detection of ssDNA of the R-loop produced by target recognition to identify on-target and off-target sites of the CRISPR-based gene editing tool.
SUMMARY AND OUTLOOK
Currently, many methods are also applied to clinical off-target detection during CRISPR-based gene editing (Table 3). For example, WGS has been applied for off-target detection during correction of nonsense mutations in a cystic fibrosis organoid biobank by ABEs and CRISPR-Cas9 for refractory non-small-cell lung cancer, 79,80 GUIDE-seq has been used for off-target detection during CRISPR-Cas9 treatment of sickle cell disease, 81 iGUIDE applied to CRISPR-Cas9 off-target assays in CAR-T cell therapy, 82 and CIRCLE-seq for off-target detection during CRISPR-Cas9 treatment of β-thalassemia 83 etc.
Application of CRISPR/Cas in clinical trials
While the emergence of different methods for detecting OTEs may be instrumental in facilitating the translation of genome editing from research to the clinic, more research and evaluation is necessary to address the issues surrounding CRISPR-based gene editing technology OTEs.
The OTE detection methods can be divided into in vitro and in vivo assays. While in vivo assays are the preferred method for detecting OTEs due to the ability to truly reflect the occurrence of off-target events, few methods exist for detection of OTEs in vivo. Thus, the development of high-sensitivity and unbiased in vivo assays will be necessary to improve current off-target detection methods.
Currently, there are several off-target assays for Cas9, and few for BE and PE assays. This is because neither the BEs nor PEs produce DSBs and thus, Cas9-based off-target detection methods for DSB generation are not applicable. Development of Cas9 protein tagging-based genomes would make it possible to detect BE and PE OTEs.
Additionally, the off-target assays currently available are mostly focused on the multicellular level. The ability to detect OTEs of the Cas9 protein at the single-cell level is central to utilization of this method in gene editing.
Most available NGS-based off-target detection methods use PCR amplification to build libraries, which leads to the loss of some off-target sites due to the preferential nature of PCR. One possible alternative would be to ligate the T7 promoter to both ends of the library. In vitro transcription could then be used to amplify the library, thus avoiding the preferences associated with PCR.
In conclusion, future implementation of precision sequencing technology coupled with the introduction of more accurate detection technology will greatly advance this field.
Footnotes
AUTHORs' CONTRIBUTIONS
H.W. and Y.W. conceived and designed the structure of this review; Y.W., X.L., M.L., and Z.L. wrote the article; W.Z., H.S., and F.W. revised the article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2018A030313860, 2020A1515010889, 2018A030313114), the Guangzhou Science and Technology Project (202002030477), and the Guangdong project of graduate education innovation (2019JGXM65).