
Abstract
Lipoprotein lipase deficiency (LPLD) results from mutations within the lipoprotein lipase (LPL) gene that lead to a complete lack of catalytically active LPL protein. Glybera was one of the first adeno-associated virus (AAV) gene replacement therapy to receive European Medicines Agency regulatory approval for the treatment of LPLD. However, Glybera is no longer marketed potentially due to a combination of economical, manufacturing, and vector-related issues. The aim of this study was to develop a more efficacious AAV gene therapy vector for LPLD. Following preclinical biodistribution, efficacy and non-Good Laboratory Practice toxicity studies with novel AAV1 and AAV8-based vectors in mice, we identified AAV8 pVR59. AAV8 pVR59 delivered a codon-optimized, human gain-of-function hLPLS447X transgene driven by a CAG promoter in an AAV8 capsid. AAV8 pVR59 was significantly more efficacious, at 10- to 100-fold lower doses, compared with an AAV1 vector based on Glybera, when delivered intramuscularly or intravenously, respectively, in mice with LPLD. Efficient gene transfer was observed within the injected skeletal muscle and liver following delivery of AAV8 pVR59, with long-term correction of LPLD phenotypes, including normalization of plasma triglycerides and lipid tolerance, for up to 6 months post-treatment. While intramuscular delivery of AAV8 pVR59 was well tolerated, intravenous administration augmented liver pathology. These results highlight the feasibility of developing a superior AAV vector for the treatment of LPLD and provide critical insight for initiating studies in larger animal models. The identification of an AAV gene therapy vector that is more efficacious at lower doses, when paired with recent advances in production and manufacturing technologies, will ultimately translate to increased safety and accessibility for patients.
INTRODUCTION
Lipoprotein lipase (LPL)
We previously contributed toward the development of Glybera, the first European Medicines Agency (EMA)-approved adeno-associated virus (AAV) gene replacement therapy for LPLD. Glybera was based on an AAV1 capsid and a human gain-of-function LPLS447X (hLPLS447X) transgene driven by a cytomegalovirus (CMV) promoter. 10,11
Preclinical and clinical studies showed long-term safety and efficacy of Glybera. A single dose of Glybera administered at a dose of 1 × 1013 genome copies/kg (gc/kg) through an intramuscular (IM) injection resulted in normalization of plasma Tg in a mouse model of LPLD. 11 Similar potency was observed in a naturally occurring feline model of LPLD. 12 Treatment with Glybera in LPLD patients improved postprandial chylomicron metabolism, lowered the frequency and severity of pancreatitis, and reduced the use of health care resources for up to 6 years. 13 –18
Due to high costs related to the use of suboptimal manufacturing processes, vector design and formulation, Glybera was withdrawn from the market in 2017. Consequently, no curative treatments are available for LPLD. Thus, there is an urgent need to redevelop a superior AAV-based gene therapy for the treatment of LPLD that is more efficacious, administrable at lower doses, and as a result, potentially more cost-effective compared with Glybera.
In this study, we designed and tested preclinical biodistribution, safety, and efficacy of novel AAV gene therapy vectors for the treatment of LPLD delivered through an IM route of administration (ROA) or an intravenous (IV) ROA in LPLD mice. Our AAV vectors were based on an AAV1 or an AAV8 serotype, delivering either a luciferase (luc) transgene or a novel codon-optimized hLPLS447X transgene, driven by a CAG promoter, containing a CMV or a muscle-specific ΔUSEx4 promoter. 19,20
To this end, we identify AAV8-CAG-codon-optimized-hLPLS447X, referred to as AAV8 pVR59, encoding a codon-optimized hLPLS447X transgene driven by a CAG promoter delivered in an AAV8 capsid, which resulted in correction of LPLD at 10- to 100-fold lower doses compared with an AAV1 vector based on Glybera, when delivered through an IM or IV ROA in LPLD mice, respectively. AAV8 pVR59 delivered through IM was well tolerated with no systemic or localized toxicity, whereas IV delivery augmented liver pathology in LPLD mice. AAV8 pVR59 was also more effective at transducing primary human skeletal muscle cells compared with an AAV1 vector based on Glybera.
Overall, our studies show the feasibility of developing a superior AAV gene therapy for LPLD, which, following further validation in larger animal models, can be delivered to patients for the treatment of LPLD through an IM ROA at lower doses, making it safer and more economical compared with Glybera.
MATERIALS AND METHODS
Adenovirus production
Recombinant human adenovirus (AdV) serotype 5 (ΔE1/E3) encoding the hLPLS447X transgene (AdV-hLPLS447X) driven by a CMV promoter was produced by VectorBuilder, with amplification and purification done in-house. 21 –23 AdV-hLPLS447X was used to transiently express hLPLS447X in neonatal LPL−/− mice, allowing for the rescue of LPLD mice to adulthood (Supplementary Fig. S1A), as shown previously (described below in “Animals” section). 24
Design and cloning of novel AAV transgene expression cassettes
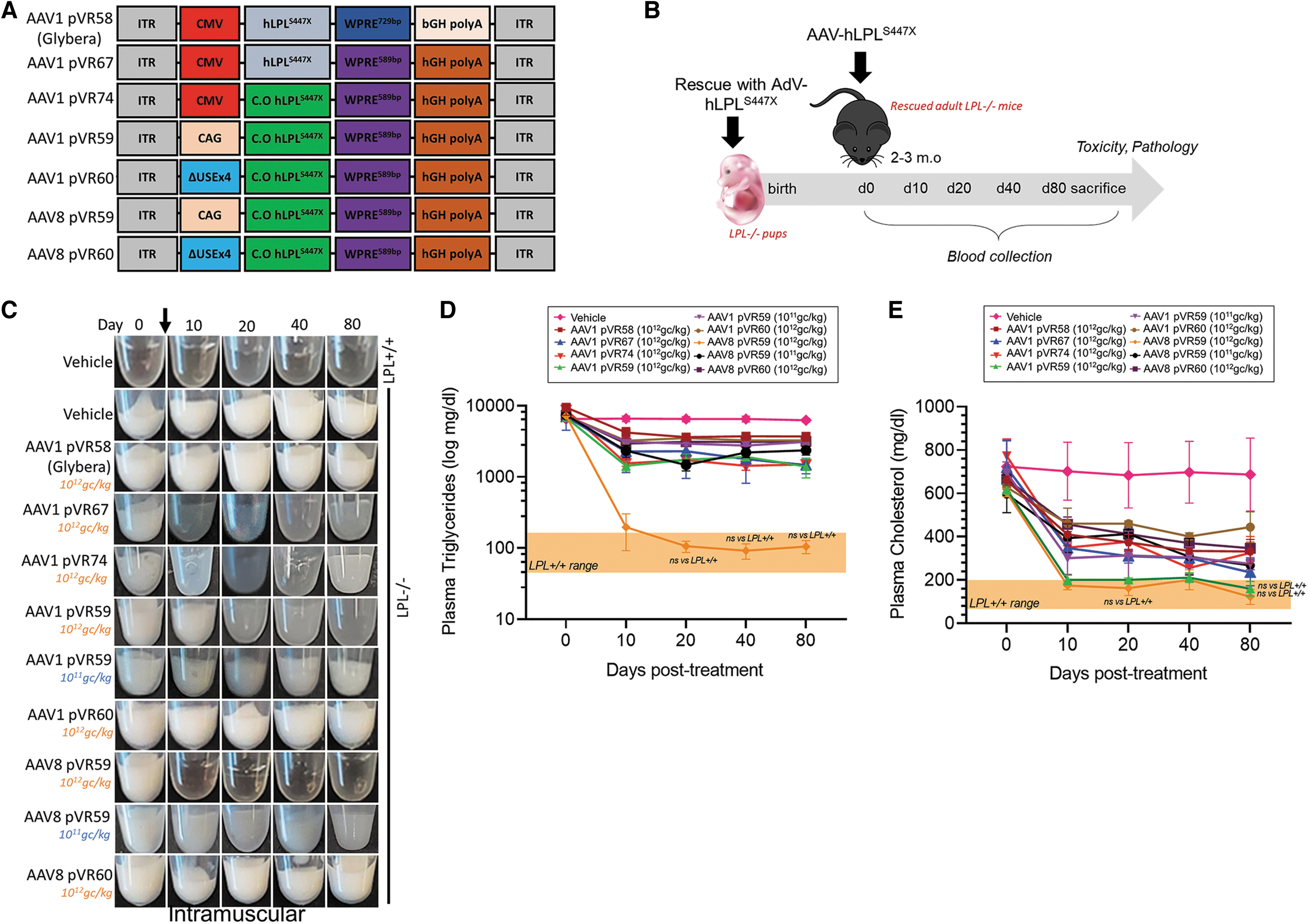
All AAV-luc and hLPLS447X expression cassette designs are provided in Figs. 1A and 2A. AAV expression cassettes encoding a luc transgene (Promega, GenBank: U47298.2) or a novel codon-optimized hLPLS447X transgene were driven by either ubiquitously expressing CMV promoter, a ubiquitously expressing CAG promoter containing a CMV enhancer, and a chimeric intron (chimera between introns from chicken β-actin and rabbit β-globin) or a muscle-specific ΔUSEx4 promoter. 20 WPRE589bp (System Biosciences) and human growth hormone (hGH) polyA were also included. 19,25 The codon-optimized hLPLS447X transgene was synthesized by GenScript. An expression cassette based on Glybera, as described previously, consisting of a CMV promoter, hLPLS447X transgene, WPRE729bp, and a bovine growth hormone (bGH) polyadenylation signal (bGH PolyA), was synthesized by GenScript.

Biodistribution following administrations of AAV vectors encoding luciferase in wild-type mice.
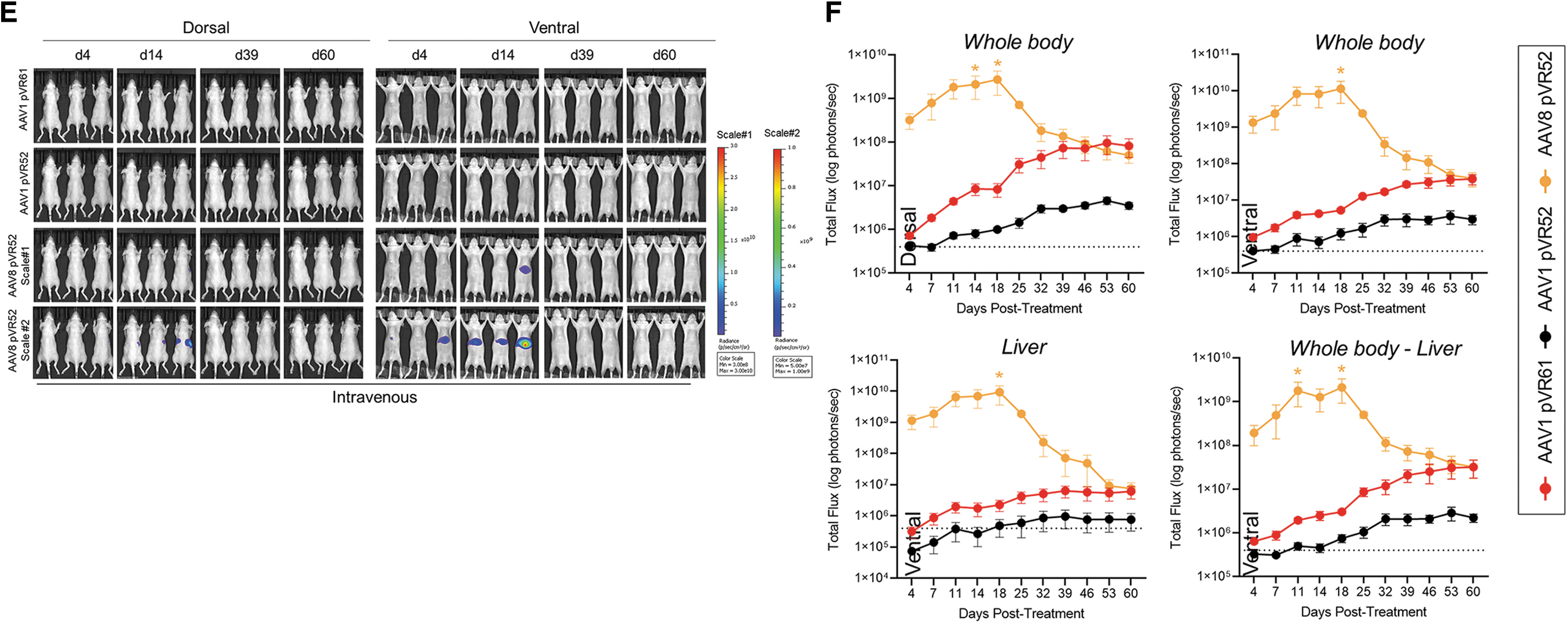
Correction of hyperlipidemia following administrations of AAV-hLPLS447X vectors in LPL−/− mice.
AAV production, lysis, and purification
HEK293SF-3F6 cells were used for AAV production. 26 Cells were triple transfected with AAV production plasmids (pAAV2/1 or pAAV2/8 rep/cap [GENEMEDI], pHelper [Cell Biolabs] and the transgene expression cassette) using PEIpro (Polyplus). Transfected cells were harvested at 72 h post-transfection. AAV lysis was carried out as described previously with slight modifications. 27 Briefly, 1% (v/v) lysing buffer concentrate (200 mM MgCl2, 1% Triton X-100) and 1% (v/v) Benzonase 2.5 U/mL (Merck) was introduced for 2 h at 37°C with agitation, followed by the addition of concentrated MgSO4 (37.5 mM) for 30 min. 28 The lysed harvest was clarified and concentrated with tangential flow filtration and purified by iodixanol gradient ultracentrifugation (Beckman Optima L-80 XP). 29 Kolliphor P 188 was added to a final concentration of 0.001% (w/v).
AAV titer quantification
AAV titers were quantified by droplet digital (ddPCR) using the Bio-Rad QX200 droplet generator and reader, along with the Eppendorf Master Cycler X50s (Thermo Fisher) and QX200 ddPCR EvaGreen Supermix (Bio-Rad). Forward and reverse primer targeting the ITR regions (GGAACCCCTAGTGATGGAGTT, CGGCCTCAGTGAGCGA). Data were acquired and analyzed using QX Manager Software.
Primary human myotube studies
Human primary skeletal muscle cells (ATCC, PCS-950-010) were propagated in Skeletal Muscle Cell Growth Medium (PromoCell) and differentiated into myotubes using Skeletal Muscle Differentiation Medium (PromoCell) on fibronectin-coated plates at 37°C in 5% CO2. Four days post-differentiation, myotubes were pretreated with hydroxyurea (2 mM, 18 h) and incubated with AAV vectors at a multiplicity of infection of 1 × 105 gc/cell. Heparin-treated (Fresenius Kabi) (20 U/mL, 4 h) conditioned media and cells were collected 4 days following AAV treatment for downstream analysis.
Animals
For our studies we used LPLD (LPL−/−) mice generated through gene targeting. 30 LPLD (LPL−/−) mice shortly after birth following suckling, exhibit extreme HTG and cyanosis, resulting in death within 24–48 h. 24,30,31 We have previously shown that transient expression of hLPLS447X through the administration of AdV-hLPLS447X immediately following birth allows for survival of LPL−/− pups to adulthood and the consequent development of profound LPLD phenotypes. 24 LPLD (LPL−/−) pups were rescued immediately following birth by IM administration (1 injection site/hindlimb) of 108 plaque-forming unit (pfu) of AdV-hLPLS447X, as described previously. 24 C57BL/6J wild-type mice used for biodistribution studies were purchased from Charles River. All animals were maintained on a C57BL/6J background and raised on regular rodent diet with free access to water. All procedures involving animals were performed with the approval of the Animal Care Committee at the University of British Columbia and the National Research Council of Canada.
Administration of AAV vectors in mice
C57BL/6J mice were used for biodistribution studies at 1–2 months of age. LPL−/− mice were used for efficacy studies at 2–3 months of age. Mice were administered with AAV vector at either 1 × 1011 gc/kg (L.D) or 1 × 1012 gc/kg (H.D) through IM or IV. IM injections were administered to anesthetized mice within the quadricep (quad) in both legs. IV injections were administered either through the tail vein or retro-orbitally to anesthetized mice. Sterile PBS containing 0.001% Pluronic was used as vehicle.
Bioluminescence luciferase imaging
Bioluminescence luciferase imaging (BLI) in anesthetized mice was performed twice a week for a period of 60 days using an IVIS-Lumina III preclinical imager and analyzed using the Living Image 4.1 software (PerkinElmer).
Plasma lipids assessment
Blood plasma samples were collected from 4-h fasted mice. Plasma Tg was assessed using Infinity Tg Liquid Stable Reagent (Thermo Fisher). Plasma total cholesterol (Tc) and high-density lipoprotein-cholesterol (HDL-c) was assessed using Infinity Cholesterol Liquid Stable Reagent (Thermo Fisher). HDL-c was extracted using 2 × LDL/VLDL Precipitation Buffer (Abcam). Non-high-density lipoprotein-cholesterol (non-HDL-c), including low-density lipoprotein-cholesterol and VLDL were calculated by subtraction of Tc from HDL-c.
LPL assessment
Blood plasma samples from 4-h fasted mice were collected 10 min after IV injection of 0.1 U/g heparin (Fresenius Kabi). Plasma hLPLS447X protein expression was assessed using a home-made sandwich ELISA specific for detecting human LPL protein. Antibodies and dilutions used are provided in Supplementary Table S1. Plasma hLPLS447X activity was assessed using EnzChek Lipase Substrate (Thermo Fisher), as described previously. 32 LPL-specific activity was calculated by subtraction of total lipase activity (noninhibited samples) from non-LPL-specific activity (samples treated with an human LPL neutralization antibody (5D2)> (Bio-Rad) or recombinant nANGPTL4). 33 hLPLS447X protein expression and activity in conditioned media from cultured cells were assessed as described above.
High-fat load clearance test
Clearance of a high-fat load was assessed following an IV infusion of 10% Intralipid (Sigma). Blood plasma samples were taken preadministration and then at 30 min, 1, 2, and 3 h post-Intralipid infusion for determination of plasma Tg clearance.
Total anti-AAV and anti-hLPLS447X IgG measurement
Total AAV and hLPLS447X IgG plasma antibody levels (neutralizing and non-neutralizing) were measured using ELISA, as described previously. 34 Antibodies and dilutions used are provided in Supplementary Table S1.
Plasma clinical chemistries and Cytokine/Chemokine 44-Plex Array
Plasma liver enzyme levels were assessed by IDEXX laboratories. A plasma cytokine array—Mouse Cytokine/Chemokine 44-Plex Discovery Assay Array (MD44) was carried out by Eve Technologies.
Tissue collection
Anesthetized mice were cardiac perfused with ice-cold PBS. Tissues were either snap-frozen in liquid nitrogen dry ice or fixed in 10% buffered formalin for histological analysis.
Western blotting and In-Cell Western blotting
For western blotting, protein lysates were separated on NuPAGE Bis-Tris Mini Protein Gels (Thermo Fisher), transferred onto 0.45 μm Amersham Hybond P Western blotting membranes (Amersham), followed by blocking with Superblock (Thermo Fisher), incubation with primary antibodies and detection using IRDye 800-labeled secondary antibodies (Li-Cor). For In-Cell Western blotting, cells plated in black, clear-bottom 96-well plates were fixed in 4% PFA, followed by blocking with Superblock (Thermo Fisher), incubation with primary antibodies, and detection using IRDye 800-labeled secondary antibodies (Li-Cor). Antibodies and dilutions used are provided in Supplementary Table S1. Relative target protein expression was normalized against total protein using Revert 700 Total Protein Stain (Li-Cor) or CellTag 700 Stain (Li-Cor). Blots or plates were scanned using the Odyssey Infrared Imaging system (Li-Cor) and quantified using ImageJ.
Quantitative real-time PCR
RNA was prepared using the RNeasy Plus Mini Kit (Qiagen). One-step quantitative real-time PCR was carried out using the QuantiNova SYBR Green RT-PCR Kit (Qiagen) on the Applied Biosystems 7500/7500 Fast Real-Time PCR System (Thermo Fisher). Hs_LPL_1_SG (QT00036771; Qiagen) was used to detect the noncodon optimized hLPLS447X transgene, hLPL_1 (GeneGlobe ID—SCB0420509-200; Qiagen) was used to detect the codon-optimized hLPLS447X transgene and Mm_Rn18s_3_SG (QT0244807518s; Qiagen) was used to detect 18s for normalization using the ΔΔCT method.
Tissue AAV vector genome quantification
DNA from snap-frozen tissues was prepared using the DNeasy Blood & Tissue Kit (Qiagen). AAV viral vector genomes (VGs), presented as AAV VG copies normalized to ng of tissue DNA (gc/ng DNA), were quantified by ddPCR, as described above. Forward and reverse primer, targeting the WPRE (ATCCTGGTTGCTGTCTCTTTAT, GAATTGTCAGTGCCCAACAG) and 250 nM probe (56-FAM/CTGTCAGCT/ZEN/CCTTTCCGGGACTTT/3IABkFQ).
Histological analysis
Formalin-fixed tissues were processed, sectioned, and stained with Hematoxylin and Eosin (H&E) (BC Children's Hospital Histology Core Laboratory). H&E slides were tile-scanned using the Olympus BX61 Microscope and scored by a blinded pathologist (VPC Neuropath, Inc.). LPL expression in formalin-fixed tissues was examined using immunohistochemistry. Antibodies and dilutions used are provided in Supplementary Table S1. All micrographs were captured using the Zeiss Axio Vert. A1 microscope.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9. A Student's t-test or one- or two-way analysis of variance were used to determine statistical significance. Post hoc significance was assessed using Fisher's least significant difference, Tukey's, or Sidak's multiple comparisons test. A p-value ≤0.05 (two-tailed) was considered significant. Data are presented as mean ± standard deviation, unless otherwise stated. The number of biological samples (n) is indicated in the figure captions.
RESULTS
In vivo biodistribution of novel AAV vectors
AAV vectors delivering a luc transgene were designed (Fig. 1A) and tested through an IM or IV ROA for in vivo biodistribution using whole-body BLI (Fig. 1B).
The IM ROA resulted in strong luc expression in the injected skeletal muscle for vectors utilizing a CMV promoter (AAV1 pVR61 vector based on Glybera) or a CAG promoter (AAV1 pVR52 and AAV8 pVR52), and to a lesser extent (5–10-fold lower) for vectors driven by the muscle-specific ΔUSEx4 promoter (AAV1 pRV56 and AAV8 pRV56), reaching peak levels around day 18 and thereafter remaining stable (Fig. 1C, D). In addition to transducing the injected skeletal muscle, the IM ROA for AAV8 pVR52 also resulted in prominent liver transduction, much stronger compared with the other vectors, with peak signal around day 14 and then decreased to day 4 post-treatment levels by endpoint (Fig. 1C). Furthermore, based on significantly weaker transduction of vectors driven by the muscle-specific promoter when delivered through IM compared with the CMV- and CAG-based vectors, in vivo biodistribution of the ΔUSEx4 promoter-based AAV-luc vectors delivered through the IV ROA was not tested (Fig. 1C, D).
Following IV administration, strongest luc expression was observed in the livers of mice treated with AAV8 pVR52, with levels peaking around 1 × 1010 p/s around day 18, ∼100-fold higher compared with AAV1 pVR61 and AAV1 pVR52 (Fig. 1E, F). However, similar to what was observed through the IM ROA, expression within the liver started declining after day 18, reaching levels of 1 × 107 p/s by endpoint, which were comparable to those of AAV1 pVR52 and AAV1 pVR61 at endpoint and still significantly higher than baseline in a vehicle-treated mouse (4 × 105 p/s) (Fig. 1E, F).
In vivo efficacy of novel AAV vectors in LPLD mice
To determine the therapeutic efficacy of our AAV vectors encoding a hLPLS447X transgene for the treatment of LPLD, we utilized LPLD (LPL−/−) mice. 30 We, along with other, have observed that LPL−/− pups exhibit severe plasma HTG and die within 24–48 h of birth (Supplementary Fig. S1B–D). 24,30,31 However, we have previously shown that LPL−/− pups can be rescued to adulthood using AdV-mediated gene transfer of hLPLS447X to skeletal muscle (Supplementary Fig. S1A). 24 Due to the transient nature of AdV-derived hLPLS447X expression, rescued adult LPL−/− mice develop pronounced LPLD phenotypes. 24
To confirm the successful generation of LPLD mice for use in our current studies, we first assessed LPLD phenotypes and measured plasma hLPLS447X expression in rescued LPL−/− mice. AdV-derived hLPLS447X transgene expression was lost 1 month after rescue and consequently rescued adult LPL−/− mice exhibit pronounced LPLD phenotypes, including plasma lipemia and severe HTG (Supplementary Fig. S1B, E–G). LPL−/− mice were treated with novel AAV-hLPLS447X vectors or an AAV1 vector based on Glybera (AAV1 pVR58) at either the L.D or H.D through an IM or IV ROA (Fig. 2A, B).
Summary of plasma lipid-lowering efficacies of all AAV vectors tested in LPL−/− mice are provided in Supplementary Table S2. AAV1 pVR58 utilized a noncodon-optimized hLPLS447X transgene along with a 729 bp WPRE sequence (WPRE729bp) and a bGH PolyA (Fig. 2A). The novel AAV-hLPLS447X vectors utilized a codon-optimized hLPLS447X transgene along with a 589 bp WPRE sequence (WRPE589bp) and a hGH PolyA (Fig. 2A). We first assessed the impact of codon optimization of the hLPLS447X transgene (AAV1 pVR74) and the use of a hGH PolyA sequence along with a shorter WPRE sequence (AAV1 pVR67) compared with AAV1 pVR58 through the IM ROA. Vehicle-treated adult LPL−/− mice exhibit plasma Tg levels of ∼6,200 mg/dL, associated with severe plasma lipemia, with normal levels in vehicle-treated LPL+/+ mice being around 100 mg/dL (Fig. 2C, E). AAV1 pVR58-treated mice had plasma Tg of ∼3,700 mg/dL at endpoint, which was a 60% reduction compared with baseline (Fig. 2C, D).
Treatment with AAV1 pVR67 or AAV1 pVR74 resulted in a slight improvement in plasma lipemia and plasma Tg lowering compared with AAV1 pVR58, however, neither resulted in normalization to wild-type levels (Fig. 2C, D). Thus, the combination of a hGH PolyA sequence along with a shorter WPRE sequence resulted in increased efficacy compared with AAV1 pVR58, with codon optimization of the hLPLS447X transgene having no additional impact.
Following testing our AAV-hLPLS447X vectors delivered through an IM ROA, we identified AAV8 pVR59 as the most efficacious vector at clearing plasma lipemia and normalizing plasma Tg in LPL−/− mice. More specifically, AAV8 pVR59 treatment at the H.D resulted in complete plasma lipemia clearance within 10 days, associated with normalization of plasma Tg levels to 105 mg/dL by endpoint (Fig. 2C, D). Intermediate plasma lipemia clearance was observed following treatment with AAV8 pVR59 at the L.D, which was a significant improvement compared with AAV1 pVR58 administered at the H.D (Fig. 2C, D). No other vectors normalized plasma lipemia and plasma Tg to wild-type levels (Fig. 2C, D). Plasma Tc was normalized in mice following IM treatment with AAV1 pVR59 and AAV8 pVR59 at the H.D, with intermediate efficacies observed with AAV1 pVR58, AAV1 pVR59, AAV1 pVR60, and AAV8 pVR60 (Fig. 2E).
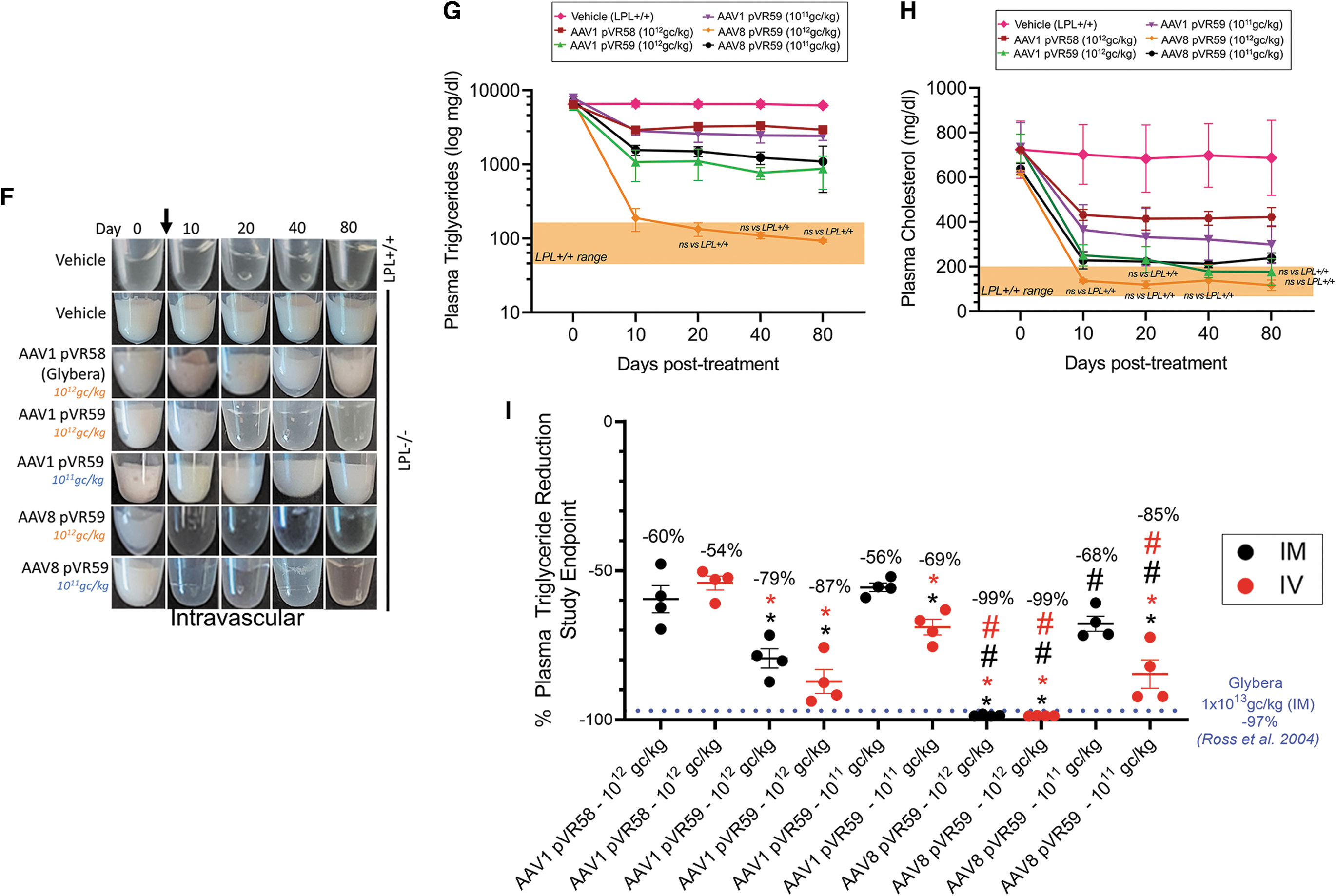
Next, we examined the efficacy of our AAV-hLPLS447X vectors delivered through an IV ROA. Subtle clearance in plasma lipemia was observed following treatment with AAV1 pVR58 at the H.D associated with plasma Tg of ∼2,945 mg/dL at endpoint (Fig. 2F, G). AAV8 pVR59 treatment at the H.D resulted in complete clearance of plasma lipemia within 10 days of treatment, associated with normalization of plasma Tg levels to 93 mg/dL at endpoint (Fig. 2F, G). Complete plasma clearance was also observed following IV treatment with AAV8 pVR59 at the L.D (Fig. 2F), which was a significant improvement compared with AAV1 pVR58 administered at the H.D and equally effective as AAV1 pVR59 administered at the H.D (Fig. 2G). Plasma Tc was normalized in mice following IV treatment with AAV8 pVR59 at both doses, with intermediate efficacies observed with AAV1 pVR58 and AAV1 pVR59 (Fig. 2H).
When comparing efficacy between the IM and IV ROA, IV delivery of both AAV1 pVR59 and AAV8 pVR59 at the L.D was found to be more efficacious compared with IM delivery (Fig. 2I). Furthermore, AAV8 pVR59 delivered through IM at the current H.D of 1 × 1012 gc/kg was equally effective at lowering plasma Tg compared with Glybera delivered IM at ∼10-fold higher doses of 1 × 1013 gc/kg in previous studies (Fig. 2I). 11 Similarly, AAV8 pVR59 delivered through IV at the current L.D of 1 × 1011 gc/kg showed comparable potency to Glybera through IM at a 100-fold higher dose of 1 × 1013 gc/kg in previous studies (Fig. 2I). 11
AAV VG and hLPLS447X transgene biodistribution in the plasma and tissues of LPLD mice following treatment with AAV8 pVR59
Since LPL is primarily secreted, we assessed plasma hLPLS447X transgene expression and corresponding lipolytic activity in LPL−/− mice treated with AAV-hLPLS447X vectors. IM treatment with AAV8 pVR59 at the H.D resulted in 3-fold and 3.5-fold higher plasma hLPLS447X protein expression and activity levels compared with AAV1 pVR58, respectively, at endpoint (Fig. 3A, B). IV treatment with AAV8 pVR59 at the H.D resulted in 4.5-fold and 5-fold higher plasma hLPLS447X protein expression and activity levels compared with AAV1 pVR58, respectively, at endpoint (Fig. 3C, D).

hLPLS447X transgene assessment and VG biodistribution following administration of AAV8 pVR59 compared with AAV1 pVR58.
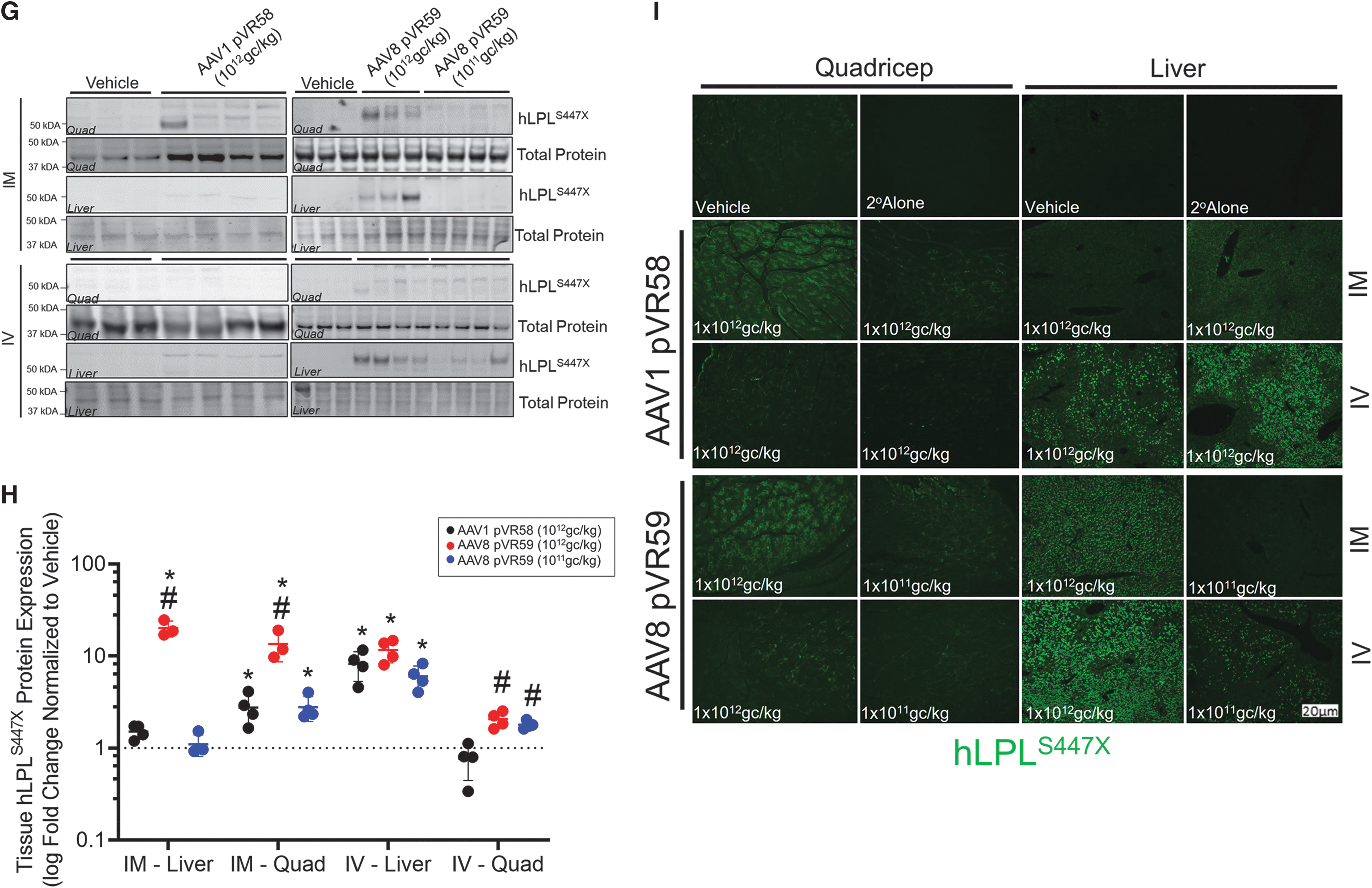
We next assessed AAV VG and hLPLS447X transcript expression within the liver and injected skeletal muscle (quad) following treatment with AAV8 pVR59 and AAV1 pVR58 in LPL−/− mice at endpoint. AAV VG were not significantly different within the skeletal muscle of mice treated with AAV1 pVR58 or AAV8 pVR59 through IM (Fig. 3E). However, the livers of AAV8 pVR59-treated mice through IM at the H.D had ∼100-fold higher VG present compared with AAV1 pVR58-treated mice (Fig. 3E). AAV8 pVR59-treated mice through IM at the H.D exhibited 70-fold and 50-fold higher levels of hLPLS447X mRNA in the liver and quad, respectively, compared with mice treated with AAV1 pVR58 (Fig. 3F). AAV8 pVR59 treatment through IV also resulted in ∼15-fold higher accumulation of AAV VG within the liver compared with AAV1 pVR58 at the H.D (Fig. 3E). These mice also expressed ∼90-fold and 6-fold higher levels of hLPLS447X mRNA in the liver and quad, respectively, compared with AAV1 pVR58 (Fig. 3F).
Using western blotting and immunohistochemistry, we confirmed that hLPLS447X protein expression was readily detected within livers of LPL−/− mice treated with AAV8 pVR59 regardless of the ROA, with the IM ROA being significantly more effective with either AAV8 pVR59 or AAV1 pVR58 at transducing skeletal muscle compared with the IV ROA (Fig. 3G, quantified in Fig. 3H, I).
Assessment of non-Good Laboratory Practice toxicity and immunogenicity following treatment with AAV8 pVR59 in LPLD mice
Pathological assessments of the liver are summarized in Supplementary Table S3. Pathological examination of livers of adult LPL+/+ mice not treated at birth with AdV-hLPLS447X were unremarkable, however, the livers of LPL−/− and LPL+/+ mice treated at birth with AdV-hLPLS447X revealed mild hepatitis. Livers from vehicle-treated LPL−/− mice also revealed mild hepatic degeneration, lipid accumulation, along with two mice (20%) exhibiting severe hepatocellular necrosis. Importantly, the presence of a specific liver pathology and/or its severity following treatment with either AAV1 pVR58 or AAV8 pVR59 through IM in LPL−/− mice was improved compared with vehicle-treated LPL−/− mice. However, liver pathology was worsened following treatment with AAV8 pVR59 through the IV ROA compared with the IM ROA.
Liver enzymes were not significantly affected in vehicle-treated LPL−/− mice compared with LPL+/+ mice (data not shown) or following treatment with AAV8 pVR59 or AAV1 pVR58, regardless of the ROA at the H.D tested, suggesting no severe impact on liver function at endpoint (Fig. 4A–F).
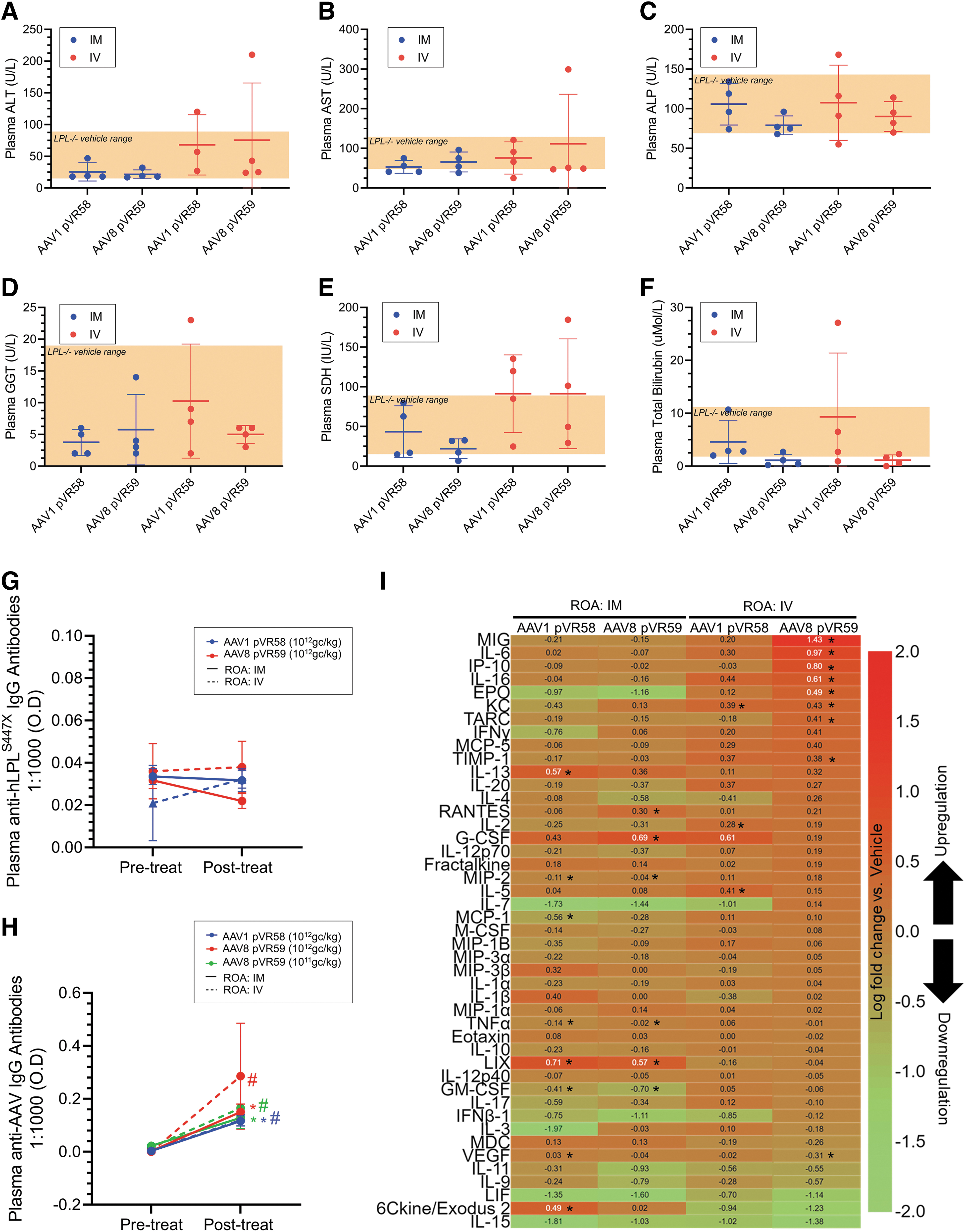
Liver function and immune responses following treatment with AAV8 pVR59 compared with AAV1 pVR58.
Pathological assessments of the quads are summarized in Supplementary Table S3. Injected quads of adult vehicle-treated LPL−/− and LPL+/+ mice treated at birth with AdV-hLPLS447X revealed mild diffuse atrophy, hyperplasia of the cellular interstitium, along with selective myofiber hypertrophy. The quad from LPL+/+ mice not treated at birth with AdV-hLPLS447X were unremarkable. In addition, 3 out the 11-vehicle-treated LPL−/− mice exhibited mild-to-moderate myositis and myofiber damage. Treatment with AAV1 pVR58 or AAV8 pVR59 through IM resulted in an overall improvement in skeletal muscle pathology, as noted by a reduction in the severity and/or occurrence of myofiber damage or myofiber loss. Similar improvement was observed following AAV8 pVR59 treatment through the IV ROA at the L.D. Representative micrographs are presented in Supplementary Fig. S2.
All other organs examined, including the heart, pancreas, spleen, and kidneys, had no pathological abnormalities (data not shown). AAV8 pVR59 treatment also did not negatively impact the growth of the mice, as indicated by a net gain in body weight over the study duration (Supplementary Fig. S3).
No plasma IgG antibodies (neutralizing and non-neutralizing) against the hLPL transgene were detected following treatment with AAV1 pVR58 or AAV8 pVR59 (Fig. 4G). However, plasma IgG antibodies against AAV1 or AAV8 capsid, were significantly elevated following treatment with AAV1 pVR58 or AAV8 pVR59 at endpoint compared with baseline, regardless of dose or ROA (Fig. 4H).
Assessment of plasma cytokines at endpoint revealed that AAV1 pVR58 or AAV8 pVR59 treatment through IM resulted in the upregulation of LIX, IL-13, 6Ckine/Exodus 2, RANTES, G-CSF, and VEGF compared with vehicle-treated mice (Fig. 4I). AAV1 pVR58 treatment through IV resulted in the upregulation of KC, IL-2, and IL-5, while AAV8 pVR59 treatment through IV resulted in the upregulation of a greater number of cytokines, including MIG, IL-6, IP-10, IL-16, EPO, KC, TARC, and TIMP-1 compared with vehicle-treated mice (Fig. 4I).
Long-term therapeutic efficacy and functional improvement following IM administration of AAV8 pVR59 in LPLD mice
Long-term efficacy for 180 days following treatment with AAV8 pVR59 delivered through IM at the H.D was assessed in LPL−/− mice (Fig. 5A). We observed complete plasma lipemia clearing for up to 180 days following IM treatment with AAV8 pVR59, associated with 98% and 82% reduction in plasma Tg and plasma Tc at endpoint, respectively (Fig. 5B–D). Furthermore, significantly depleted plasma HDL-c in LPL−/− mice was also normalized to LPL+/+ levels following treatment with AAV8 pVR59 (Fig. 5E). Plasma non-HDL-c, significantly elevated in vehicle-treated LPL−/− mice, also showed complete normalization in these mice (Fig. 5F). Accordingly, plasma hLPLS447X expression (Fig. 5G) and lipolytic activity (Fig. 5H) was found to peak, significantly higher in AAV8 pVR59-treated mice compared with AAV1 pVR58, around day 30 and then remaining stable for 180 days. LPL−/− mice exhibit a profound impairment in their ability to respond to a high-fat load, associated with ∼11% clearance of a plasma Tg load within 3 h post-Intralipid infusion (Fig. 5I, J).
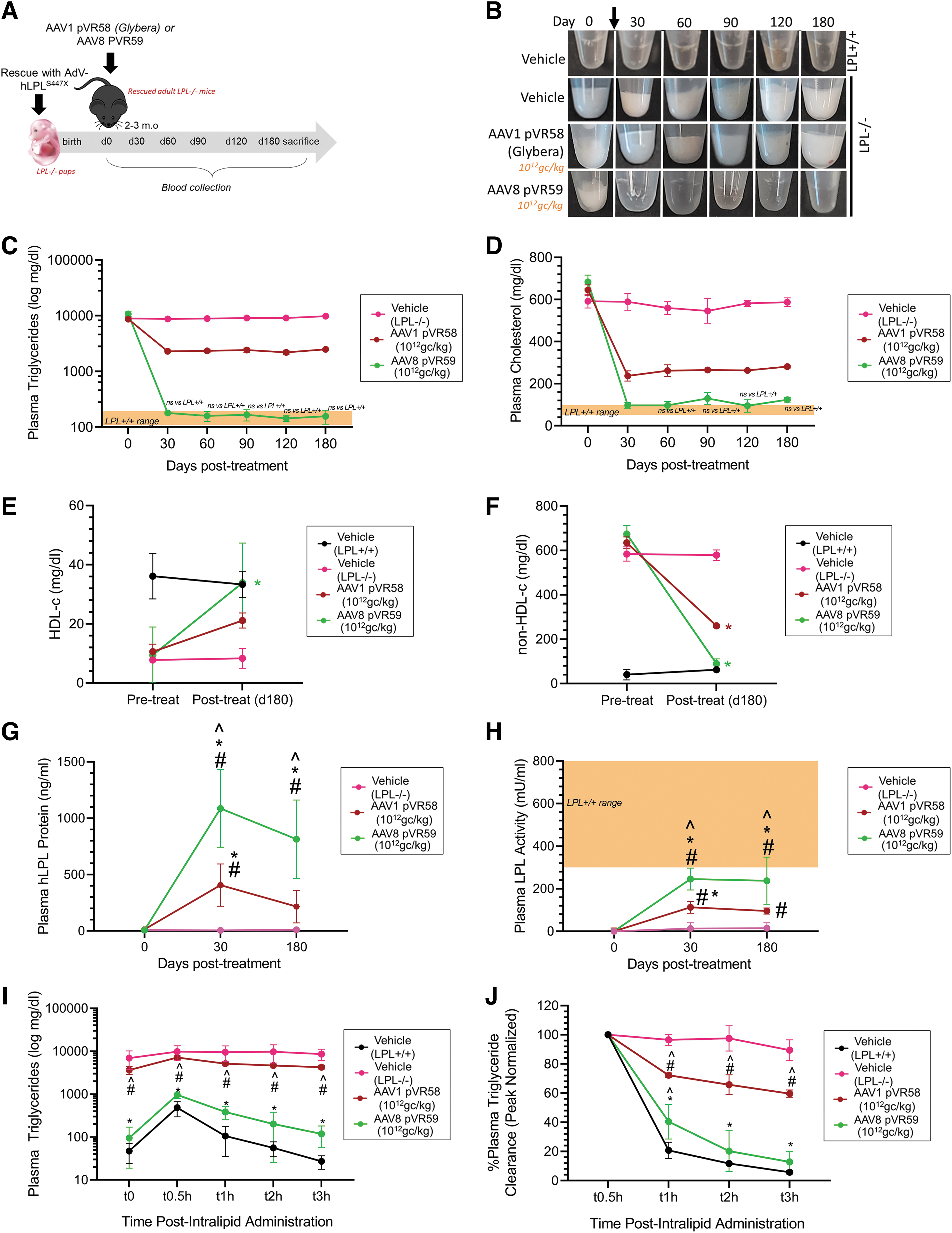
Long-term therapeutic functional improvement in AAV8-pVR59 treated LPL−/− mice compared with AAV1 pVR58.
LPL−/− mice treated with AAV8 pVR59 exhibited normalization of high-fat load clearance, associated with a significant improvement of 86% plasma Tg clearance compared with AAV1 pVR58-treated mice (Fig. 5I, J).
Assessment of AAV8 pVR59 in human muscle cells
We utilized primary human myotubes to determine the ability of AAV8 pVR59 to transduce differentiated human myotubes in vitro (Fig. 6A). AAV8 pVR59 resulted in twofold higher intracellular hLPLS447X protein expression (Fig. 6B, C), which was associated with twofold higher secreted hLPLS447X expression (Fig. 6D) and 1.5 higher lipolytic activity (Fig. 6E) in the human myotubes. These results support the potential clinical translatability and superiority of AAV8 pVR59 in transducing human skeletal muscle compared with AAV1 pVR58.
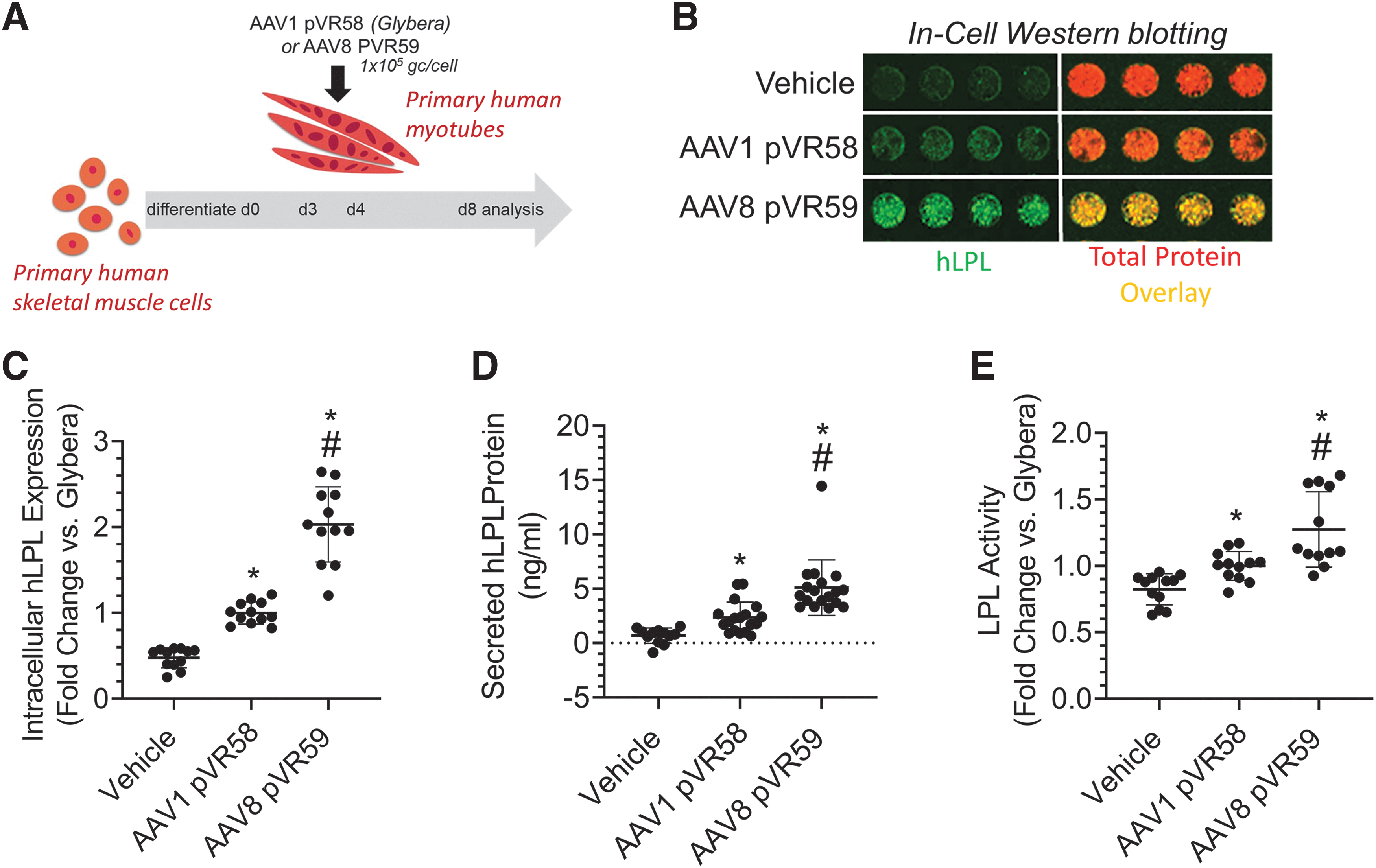
Assessment of AAV8 pVR59 compared with AAV1 pVR58 in primary human myotubes.
DISCUSSION
There are currently no curative therapies available for LPLD. AAV-based gene replacement therapy is a promising approach. Using Glybera, a previously EMA-approved AAV-LPLD gene therapy as a benchmark, we developed and tested the biodistribution, efficacy, and safety of novel AAV gene therapy formulations for the treatment of LPLD. We tested the impact of AAV serotype, promoter, therapeutic transgene, and ROA on treatment efficacy and safety in a mouse model of LPLD (LPL−/− mice). Our studies revealed an AAV8 serotype-based vector encoding a codon-optimized hLPLS447X transgene driven by a CAG promoter, AAV8 pVR59, with superior long-term therapeutic efficacy at 10- to 100-fold lower doses compared with Glybera (AAV1 pVR58).
For the design of our novel AAV vectors, as a therapeutic transgene, we utilized either the native hLPLS447X transgene, which is present in Glybera, or a novel codon-optimized hLPLS447X transgene. Our previous studies suggest that hLPLS447X is significantly more effective at normalizing plasma Tg compared with wild-type hLPL in LPL−/− mice. 24 The hLPLS447X mutation is a gain of function variant present within 20–25% of the general population, which is associated with enhanced lipolytic function through increased lipoprotein uptake, along with an antiatherogenic and cardioprotective lipid profile. 10,24,35 –37 For the promoter to drive hLPLS447X transgene expression, we tested either a CMV promoter, CAG promoter, or a muscle-specific ΔUSEx4 promoter. AAV1 pVR58 (Glybera) delivered a non-codon-optimized hLPLS447X transgene driven by a CMV promoter, while all of our novel AAV vectors, including AAV8 pVR59, delivered a codon-optimized hLPLS447X transgene, driven by either a CAG promoter or a muscle-specific ΔUSEx4 promoter.
We observed that AAV vectors utilizing the CAG promoter resulted in increased transgene expression and in turn significantly increased potency compared with vectors utilizing a ubiquitous CMV promoter or the ΔUSEx4 muscle-specific promoter. 19,20
For delivering the therapeutic hLPLS447X transgene, we utilized either an AAV1 serotype, as utilized in Glybera, or an AAV8 serotype. AAV8 has been extensively tested as a gene delivery vector for the treatment of a variety of diseases, including glycogen storage disease, X-linked myotubular myopathy, ornithine transcarbamylase deficiency, hemophilia, and familial hypercholesterolemia in several preclinical animal models, including mice, dogs, nonhuman primates, and in humans through clinical trials. 38 –40
In addition to the AAV serotype, the ROA also influences the biodistribution and transduction efficiency of AAV vectors in vivo. In previous studies, Glybera was delivered to human patients through a series of 30–40 IM injections under epidural anesthesia. 13 –16 In this study, we investigated the biodistribution and efficacy of novel AAV1 and AAV8-based AAV-LPLD vectors through an IM and IV ROA in LPL−/− mice. Systemic AAV delivery through an IV ROA has not yet been tested for the treatment of LPLD. IV delivery is also expected to be less painful and burdensome in a clinical setting, thus making it a potentially attractive ROA compared with IM delivery.
Our in vivo mouse biodistribution studies revealed that following an IM injection, AAV8 pR59 in addition to transducing the injected skeletal muscle is also effective at transducing the liver. This is in accordance with studies in rhesus macaques demonstrating skeletal muscle and liver transduction following IM delivery of AAV8-based vectors. 41 In contrast, AAV1-based vectors such as AAV1 pVR58 (Glybera), delivered through IM, most prominently resulted in localized transgene expression within the injected skeletal muscle. We also observed more effective transduction of primary human myotubes with AAV8 pVR59 compared with the AAV1 pVR58 vector based on Glybera, suggesting potential application and transability of AAV8 pVR59 for the successful transduction of human skeletal muscle vector administration. This is in accordance with other studies showing that AAV8-based vectors are highly effective at transducing human skeletal muscle xenografts in mice following IM delivery. 42
Furthermore, a recent phase I clinical trial utilizing an AAV8 vector delivered through an IM ROA encoding a neutralizing HIV-1 antibody (AAV8-VRC07) to adults living with HIV, has also shown that the AAV8 capsid is safe and effective for the delivery of therapeutic cargo in human skeletal muscle (NCT03374202).
When delivered through an IV ROA, AAV8-based vectors, in addition to possessing strong liver tropism in mice, have also been shown to exhibit a more uniform tissue transduction pattern throughout the hindlimb, abdominal, and thoracic regions compared with other serotypes, including AAV1, with the hindlimb skeletal muscle being the next most common area of transduction after the liver. 41,43 However, the affinity of AAV8 for transducing the liver is known to significantly differ between mice, nonhuman primates, and humans. 44 Specifically, AAV8-based vectors delivered through IV to nonhuman primates in preclinical studies and patients with hemophilia in clinical trials have revealed ∼10-fold to 100-fold lower therapeutic transgene expression compared with mice, respectively. 40,45 –48 Thus, to what extent AAV8 pVR59 results in liver transduction in humans and what impact this has on therapeutic efficacy compared with an AAV1-based vector such as Glybera remains to be determined.
LPL is primarily synthesized within skeletal muscle cells and adipose tissue and then secreted and bound to the luminal surface of blood vessels, where it breaks down Tg within chylomicrons and VLDL. 2 –4,6,8,49 However, endogenous LPL transcript is absent in healthy adult livers. Thus, the role of liver-localized LPL is not well understood.
Overexpression of LPL within the livers of high-fat-fed mice and rabbits has been shown to be effective at improving insulin resistance, glucose metabolism, transiently normalizing HTG and fat tolerance, while conferring protection against hypercholesterolemia and atherosclerosis. 50 –52 Thus, it is plausible that AAV8 pVR59, through enhanced transduction of the liver and skeletal muscle when delivered through IV or IM, respectively, allows for increased hLPLS447X production and secretion into the plasma, leading to enhanced therapeutic efficacies compared with an AAV1-based vector such as Glybera. Indeed, following AAV8 pVR59 treatment through either ROA, we observed increased hLPLS447X transgene production within the liver and skeletal muscle, which was associated with on average 2–4-fold higher plasma hLPLS447X expression, compared with the AAV1 pVR58 vector based on Glybera.
LPL−/− mice used in these studies show severely elevated plasma Tg levels (>5,000 mg/dL). 24,31 In humans, plasma Tg levels >1,000 mg/dL are strongly associated with high risk of pancreatitis, a life-threatening manifestation of LPLD. 53 –55 However, LPL−/− mice do not exhibit pancreatitis. Thus, we assessed the therapeutic efficacy of our novel AAV vectors by evaluating plasma Tg reduction following vector administration. Treatment with AAV8 pVR59 but not AAV1 pVR58 (Glybera), or any other novel AAV vector tested, administered at doses ranging from 1 × 1011 to 1 × 1012 gc/kg, resulted in normalization of plasma Tg (<100 mg/dL) to levels that are no longer considered a risk factor for the development of pancreatitis in humans.
When comparing between the two ROA, we observed superior efficacy with AAV8 pVR59 using doses as low as 1 × 1011 gc/kg through IV or 1 × 1012 gc/kg through IM, a 100-fold lower dose and a 10-fold lower dose, respectively, compared with the 1 × 1013 gc/kg dose of Glybera required to achieve a similar plasma Tg-lowering efficacy (>90% plasma Tg lowering) from previous studies. 11
Furthermore, our studies showed long-term transgene expression, up to 180 days, along with significantly improved, stable therapeutic plasma lipid-lowering efficacies following IM delivery of AAV8 pVR59 compared with Glybera.
Due to the highly pro-tolerogenic nature of the liver, there is evidence suggesting that some degree of gene transfer to the liver in combination to other key target organs such as the skeletal muscle may be beneficial. 56 –59 Mechanistically, liver-directed gene transfer has been shown to induce systemic transgene tolerance through the activation of transgene product-specific regulatory T cells in a dose-dependent manner. 56 –58 Thus, IM administration of AAV8 pVR59, through transduction of a combination of the liver and skeletal muscle, may evoke a favorable tolerogenic response that improves transgene longevity compared with the use of Glybera or another AAV1 vector that does not as effectively transduce the liver.
Humoral antibody response against a therapeutic transgene, in combination with high vector doses, contribute to rapid loss of transgene expression following administration, often being a primary cause of clinical failure. 59,60 In our current studies, no antibodies against the hLPLS447X transgene were detected following treatment with AAV1 pVR58 or AAV8 pVR59, potentially attributed to the fact that our LPL−/− mice have been tolerized to hLPLS447X at birth following the AdV rescue procedure. Conversely, humoral responses against AAV1 and AAV8 capsids were developed following treatment, however, while these responses do not allow for readministration, they do not have any impact on therapeutic efficacy upon initial administration. 60
Systemic IV delivery of AAV vectors at very high doses (>1 × 1014 gc/kg) has been seen as a potential safety concern. Serious adverse events, including death, most often related to hepatotoxicity and liver failure, have been recently observed in IV-delivered high-dose AAV gene therapy clinical trials for X-linked muscular dystrophy (NCT03199469) and Duchene muscular dystrophy (NCT03362502). Furthermore, two children with spinal muscular atrophy have recently died from liver failure 5 weeks after treatment with Zolgensma, an FDA-approved AAV9-based gene therapy, at high doses of >1 × 1014 gc/kg. While it has been postulated that pre-existing liver conditions in some of these patients may have been responsible for exacerbating hepatotoxicity following AAV gene therapy, it is of critical importance to use as low of a vector dose as possible to obtain required therapeutic treatment efficacies.
To this end, in our mouse studies with AAV8 pV59, maximal treatment efficacy was observed at AAV vector doses of no more than 1 × 1012 gc/kg, a 100-fold lower dose compared with 1 × 1014 gc/kg, which has been associated with hepatotoxicity in the aforementioned clinical trials and up to 10-fold lower than those used with Glybera in clinical trials.
In our mouse studies the occurrence and severity of skeletal muscle and liver pathology was improved following IM treatment AAV8 pVR59 compared with vehicle-treated LPL−/− mice. These results are in accordance to what has been observed previously in preclinical mouse studies with Glybera. 11,61 However, the IV ROA with AAV8 pVR59 resulted in increased occurrence of liver pathology, including hepatic lipid accumulation and occurrence of hepatocellular necrosis in a dose-dependent manner. Interestingly, there have been some reports in mice suggesting a deleterious effect of tissue-specific overexpression of LPL that is associated with increased lipid accumulation, insulin resistance, and localized pathology..62,63 Based on our studies, due to the lack of augmentation of skeletal muscle or liver pathology following IM administration of AAV8 pVR59, it is plausible that IV administration results in supraphysiologic hepatic hLPL expression that is associated with increased localized hepatic Tg hydrolysis, resulting in excessive lipid accumulation and hepatotoxicity.
Whether the adenoviral-based rescue procedure of LPL−/− mice at birth, which is known to result in transient hLPL expression within the liver and whether excessive dyslipidemia and pre-existing liver pathology in LPL−/− mice before AAV-hLPLS447X treatment augments liver pathology needs to be further examined. Future toxicity studies in larger animal models will be carried out to validate these findings. Nonetheless, our results collectively suggest that IM delivery of AAV8 pVR59 is a safe and potentially clinically translatable approach for the treatment of LPLD.
CONCLUSIONS
In summary, we developed and tested the efficacy and safety of several novel AAV-hLPLS447X gene therapy vectors in a mouse model of LPLD. Our studies revealed that AAV8-CAG-codon-optimized-hLPLS447X (AAV8 pVR59) possessed superior long-term therapeutic efficacy compared with AAV1 pVR58, a vector based on Glybera, in mice with LPLD, with no apparent toxicity when delivered through an IM ROA. Our studies collectively highlight the feasibility and support the redevelopment of a safe and superior intramuscularly delivered AAV-based gene therapy product for the treatment of LPLD that can ultimately be delivered at lower doses and in turn be available to patients at lower costs compared with Glybera. Our future efforts will focus on refinement and optimization of AAV8 pVR59 for utilization and validation in larger animal models and consequently in clinical studies along with the development of more efficient manufacturing processes and scale-up.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Dr. Silvia Borrelli, Jacqueline Slinn, Mark Wang, Qingwen Xia, Steven Zhou, and Yun Ko for technical assistance and support. The authors thank Dr. Saskia B. Neher for providing the pNIC-Bio3 ANG4 26-164 plasmid to produce recombinant nANGPTL4.
AUTHORs' CONTRIBUTIONS
N.M., R.G., P.S.C., M.J.M., S.M.E., C.J.D.R., D.B.S., and M.R.H. designed research; N.M., N.N., N.C., N.N-.M., V.L., M.M., D,.F., S.M.E., W.L., and E.M.H. performed research; N.M., R.G., P.S.C., M.J.M., and L-.H.Z. analyzed data; N.M. wrote the article; R.G., P.S.C., M.J.M., W.L., C.J.D.R., D.B.S., and M.R.H. edited the article. All authors read and approved the final article.
DATA AVAILABILITY STATEMENT
Any additional information related to the article is available upon reasonable request.
AUTHOR DISCLOSURE
N.M., R.G., P.S.C., M.J.M., N.C., C.J.D.R., D.B.S., and M.R.H. are inventors on a patent application related to this work filed by the University of British Columbia and the National Research Council Canada. M.R.H. currently serves on the public boards of Ionis Pharmaceuticals, Xenon Pharmaceuticals, Aurinia Pharmaceuticals, AbCellera, and 89bio. The other authors declare no competing interests.
FUNDING INFORMATION
This work was supported by the Cell and Gene Therapy Challenge Program at the National Research Council of Canada (C.J.D.R and M.R.H) (CGT-101), Canadian Institutes of Health Research (N.M) (201910MFE-430323-FPP-CAAA-267763), and the Michael Smith Foundation for Health Research (N.M) (RT-2020-0381).
SUPPLEMENTARY MATERIAL
Supplementary Figure S1
Supplementary Figure S1
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.