
Abstract
Adeno-associated virus (AAV) based viral vectors are widely used in human gene therapy and form the basis of approved treatments for several genetic diseases. Immune responses to vector and transgene products, however, substantially complicate these applications in clinical practice. The role of innate immune recognition of AAV vectors was initially unclear, given that inflammatory responses early after vector administration were typically mild in animal models. However, more recent research continues to identify innate immune pathways that are triggered by AAV vectors and that serve to provide activation signals for antigen-presenting cells and initiation of adaptive immune responses. Sensing of the AAV genome by the endosomal DNA receptor toll-like receptor 9 (TLR9) promotes early inflammatory response and interferon expression. Thus, activation of the TLR9>MyD88 pathway in plasmacytoid dendritic cells (pDCs) leads to the conditioning of antigen cross-presenting DCs through type I interferon (IFN-I) and ultimately CD8+ T cell activation. Alternatively, pDCs may also promote CD8+ T cell responses in a TLR9-independent manner by the production of IL-1 cytokines, thereby activating the IL-1R1>MyD88 signaling pathway. AAV can induce cytokine expression in monocyte-derived DCs, which in turn increases antibody formation. Binding of AAV capsid to complement components likely further elevates B cell activation. At high systemic vector doses in humans and in non-human primates, AAV vectors can trigger complement activation, with contributions by classical and alternative pathways, leading to severe toxicities. Finally, evidence for activation of TLR2 by the capsid and of additional innate receptors for nucleic acids has been presented. These observations show that AAV vectors can initiate several and likely redundant innate immune pathways resulting in an exaggerated adaptive immune response.
INTRODUCTION
Adeno-associated viruses (AAVs) are small (∼25 nm), nonenveloped, single-stranded (ss)-DNA viruses that belong to the genus Dependoparvovirus, family Parvoviridae, and require a helper virus such as herpes virus or adenovirus for their replication. The ss-DNA genome of AAV mainly comprises four open reading frames, namely rep, cap, assembly activating protein, and membrane-associated accessory protein. These open reading frames encode viral proteins that are required for its replication, viral capsid, capsid assembly, and virus egress, respectively. The genome is flanked by two inverted terminal repeats (ITRs), one at each end of the DNA strand. These are the only viral sequences retained in AAV vectors. AAVs have been isolated from humans and other primates, and currently, 13 different AAV serotypes have been identified. 1 These AAV serotypes infect a wide variety of dividing and nondividing cells and have generally not been associated with severe or chronic disease. 2 A large variety of engineered capsids have been developed to further improve efficacy and expand the range of tropism. 3 The excellent safety profile, coupled with their broad tissue tropism and their ability to infect dividing and nondividing cells, has led to the development of AAV-based recombinant vectors as one of the most prominent and leading platforms for in vivo gene therapy. 4,5 In fact, over the past decade, AAV vector-based therapeutics have seen accelerated clinical development, with eight products having regulatory approval in the United States, Europe, and/or elsewhere. Among the earliest AAV medicines were Voretigene neparvovec (Luxturna) and Onasemnogene abeparvovec (Zolgensma) for the treatment of Leber’s congenital amaurosis and spinal muscular atrophy, respectively. In the last two years, five more AAV gene therapies—Etranacogene dezaparvovec (Hemgenix), Fidanacogene elaparvovec (Beqvez), Valoctocogene roxaparvovec (Roctavian), Eladocagene exuparvovec (Upstaza), and Delandistrogene moxeparvovec (Elevidys)—have secured regulatory approval for the treatment of hemophilia B, hemophilia A, aromatic L-amino acid decarboxylase deficiency, and Duchenne muscular dystrophy (DMD), respectively. 6 –8 Despite tremendous advancement and a bright future, AAV vectors are not yet a “silver bullet” as immunogenicity still remains a major hurdle for the safety, durability, and efficacy of AAV gene therapy. Although AAV is substantially less inflammatory compared with many other viral vectors, treatments may require administration of high vector doses of up to 1014 vg/kg, in particular for applications that depend on systemic vector administration. 9 –11 At such high doses, host antiviral factors such as innate sensors would readily detect molecular signatures associated with AAV or the infection process, thereby initiating a cascade of immunological reactions that may cause immunotoxicity and ultimately render gene transfer ineffective. Furthermore, the complement arm of the immune system has been implemented in recent fatalities in high-dose systemic AAV administration in patients. 12 –17
Initially, innate immune responses to AAV vectors received limited attention because (1) the observed inflammatory/cytokine responses were weak and transient, (2) AAV failed to activate inflammasomes, (3) AAV was found to bind to certain complement factors but failed to activate complement, and (4) no specific innate immune sensors were found. No clear links between innate and adaptive immunity had been identified. This changed over the past 1.5 decades. Since then, steady progress has been made resulting in the identification of multiple innate immune players and a better understanding of AAV immunogenicity. In this review, we attempt to provide a broader overview of the interactions between AAV vectors and the innate immune system of the host and of links between innate and adaptive immunity.
Innate immunity and sensing of viral vectors
Innate immunity constitutes the initial and foremost defense mechanism of the host against invading microbial pathogens such as bacteria and viruses. The components of the innate immune system include physical (e.g., skin) and anatomical barriers (e.g., mucosal surfaces), specialized immune cells [e.g., macrophages, dendritic cells (DCs), granulocytes], cellular receptors [e.g., Toll-like receptor (TLR)], soluble mediators (e.g., cytokines and complement proteins), and antimicrobial peptides (e.g., defensins). All these components of innate immunity work in concert to prevent pathogens from entering and eliminate pathogens that manage to invade the host. Innate immune cells conduct frequent surveillance of the host body and engage with microbes through interaction of pathogen-associated molecular patterns (PAMPs; e.g., microbial nucleic acids, surface glycoprotein, lipoproteins, peptidoglycans, and so on) with various cellular receptors called pattern recognition receptors. 18 These host–pathogen interactions lead to the activation of cellular machinery that induces the production of cytokines (soluble mediators), which activate immune cells, thereby creating a microenvironment conducive to the initiation of antigen-specific adaptive immune responses.
During a microbial infection, the nature of a particular PAMP dictates which pattern recognition receptor will be engaged. 19 For example, several endosomal TLRs detect various forms of microbial nucleic acids. Specifically, TLR3 detects dsRNA and TLR7/8 detects ssRNA genomes of a variety of viruses, whereas TLR9 is activated upon recognition of DNA, and in particular unmethylated CpG motifs present in microbial DNA. 20 TLRs are type I transmembrane proteins consisting of three domains as follows: (1) an N-terminal extracellular ligand binding domain, (2) a transmembrane region, and (3) a C-terminal cytoplasmic domain. 21 The extracellular domain has a short tandem of leucine-rich repeat motifs providing specificity for a particular PAMP. Under homeostatic conditions, TLRs exist as monomers. However, upon ligand binding, TLRs either form homo- or hetero-dimers thereby bringing their cytoplasmic domain closer. This dimerization of TLRs recruits the downstream adaptor molecule myeloid differentiation primary response 88 (MyD88) or toll/interleukin-1 receptor (TIR) domain-containing adaptor protein inducing interferon beta to its cytoplasmic domain to initiate nuclear factor-κB and mitogen-activated protein kinase signaling that leads to the secretion of type I interferons (IFN-I, i.e., IFN-α and -β) and a variety of pro-inflammatory cytokines. 19 Below, we review TLR and cytokine receptors that have been implicated in innate responses to AAV.
Sensing of the viral genome using TLR9
TLR9 is mainly expressed intracellularly in the endo-lysosomal compartments of specialized innate immune cells such as plasmacytoid dendritic cells (pDCs), B cells, and macrophages. TLR9 is activated in response to foreign DNA containing unmethylated CpG motifs that are more prevalent in microbial than mammalian DNA. TLR9 was the first innate sensor that was identified to be critical for AAV innate sensing (Fig. 1). Zhu et al., using murine model and in vitro cultures of human peripheral blood mononuclear cells (PBMCs) demonstrated that pDCs but not conventional DCs (cDCs) or macrophages respond to the AAV genome through TLR9-MyD88 pathway leading to production of IFN I, which promotes adaptive immune responses against AAV capsid, as well as AAV encoded transgene products. 22 These results came as a surprise to the AAV community as AAVs are known to be poor transducers of antigen-presenting cells (APCs). However, it is possible that AAVs can be endocytosed by APCs, leading to capsid degradation by endosomal proteases, thereby exposing the AAV genome to endosomal TLR9. Since this initial discovery, the TLR9-MyD88 pathway has been consolidated as an important component of innate immune sensing of AAV, driving early innate inflammation and IFN I production in the tissue, promoting CD8+ T cell activation, and modifying antibody responses. 23 –26 A recent study found that CpG motifs in the vector genome promoted the emergence of a pDC-like myeloid cell population in mice, which directly bound an antibody transgene product through Fc-FcγR interactions, thereby reducing early expression of vector-encoded immunoglobulin. 27 Another recent investigation reports TLR9-dependent reduction of dendritic length and complexity and disruption of synaptic transmission in mouse somatosensory cortex in response to AAV gene transfer, which was accompanied by changes in AMPA receptor subunit composition but could be prevented by TLR9 blockade. 28 These observations further illustrate the multifaceted impact of TLR9 activation in response to AAV vectors.

Innate sensors and downstream signaling in AAV recognition and antiviral response. TLR2 mediates recognition of AAV capsid, whereas TLR9 recognizes unmethylated CpG motifs in the AAV genome. TLR2 and 9 recruit the downstream adaptor molecule MyD88, which promotes the secretion of IFN-I and pro-inflammatory cytokines. AAV ITRs have inherent transcriptional activity, which could lead to the formation of dsRNA. Cytoplasmic dsRNA sensors such as RIG-I and MDA5 may recognize and drive the secretion of IFN-I. Some of the internalized AAV capsids are proteolytically degraded into smaller peptides in endosomes, whereas some of those that manage to escape endosomes are degraded in the cytoplasm (by proteasomes). These peptides are loaded onto MHC molecules and transported to the plasma membrane, where they are presented to T cells in order to initiate adaptive immune responses against the capsid. Italicized molecules in gray font are known components of the indicated pathways, but have not been directly studied in the context of AAV immunogenicity. For simplicity, pathways are illustrated within a single cell, whereas these in fact occur in different cell types (such as in different subsets of dendritic cells, macrophages, or nonimmune cells). AAV, adeno-associated virus; TLR9, toll-like receptor 9; IFN-I, type I interferon; RIG-I, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated protein 5.
Besides CpG contents, the configuration of the vector genome has been identified as a factor that determines the magnitude of TLR9 signaling. AAV vectors containing self-complementary DNA were designed to increase transgene expression thereby lowering the effective dose of AAV vectors in patients. However, these vectors may also be more potent in the activation of TLR9-MyD88 mediated immune responses than AAV vectors containing the ss-DNA genome. 23,29 TLR9-mediated innate immune responses appear largely independent of AAV capsid. 23 Since unmethylated CpG motifs are the primary ligand for TLR9, minimizing the number of such motifs in the AAV expression cassette would reduce TLR9 activation and related immune responses. Indeed, preclinical and clinical studies have demonstrated that AAV-associated cellular responses to both AAV capsid and transgene product are inversely related to the number of CpG motifs, thereby further consolidating the role of TLR9 innate sensing of AAV. 24,30 –32 Furthermore, by incorporating a TLR9 inhibitory sequence in AAV expression cassette, Chan et al. have shown a reduced immune response in murine and swine models, representing an alternative strategy to the CpG depletion approach. 33 However, immune responses were still observed at high vector doses, for instance, in macaques injected through intravitreal route. TLR9 inhibition merely delayed immune-mediated clinical uveitis in these experimental animals. Chan et al. concluded that there might be additional pathways mediating these immune responses. 33 This conclusion is supported by muscle gene transfer studies in mice, which showed vector dose-dependent effects on cellular immune responses to the transgene product, likely reflecting activation of multiple innate pathways. 34,35
TLR2 as a sensor of capsid
TLR2 is present on the surface of a variety of cells, including monocytes, macrophages, DCs, microglia cells, Schwann cells, B cells, and T cells. TLR2 recognizes lipoproteins associated with a variety of Gram-negative and Gram-positive bacteria and mediates cellular responses against a variety of infectious pathogens, including enveloped viruses. Unlike other TLRs, TLR2 can form both homodimer, as well as heterodimer with TLR1 and TLR6; however, homodimers of TLR2 are less potent in inducing the secretion of pro-inflammatory cytokines compared to heterodimers. 20 In the context of AAV gene therapy, the AAV capsid is a potential PAMP that could activate the host innate immune system (Fig. 1). In a murine model of AAV liver gene transfer, Martino et al. showed increased TLR2 expression soon after administration with scAAV vector. 23 Using primary cultures of human nonparenchymal cells, Hosel et al. demonstrated that both liver sinusoidal endothelial cells and Kupffer cells (KCs) upregulated TLR2 and could induce cytokines IL-1β, IL-6, IL-8, and TNF-α upon sensing of AAV capsid. 36 Although Kuranda et al. showed induction of pro-inflammatory cytokines IL-6 and IL-1β upon exposure of human PBMCs with empty AAV capsid, they did not directly implicate TLR2 for this pro-inflammatory milieu. 37 A recent study found that TLR2 deficiency had a modest effect on CD8+ T cell and antibody formation against the transgene product in hepatic gene transfer in mice, resulting in somewhat elevated levels of circulating transgene product. 38 However, adaptive immune responses to the capsid were found to be independent of TLR2 in a murine model of muscle gene transfer. 26 It should be emphasized that upregulation of various innate sensors and cytokines is mediated by cytokines such as IFN I, and therefore, a mere upregulation might not be indicative of direct involvement. Thus far, a strong dependency of immune responses to AAV gene transfer on TLR2 has not been documented. 25,26,38,39 In addition, the AAV capsid carries a net negative charge, and systemic doses represent a significant surface area. The negative charge and large surface area of AAV may contribute to the immediate activation of platelets following systemic infusion. The activated platelets subsequently contribute to the innate response and endothelial injury.
Role of interleukin-1-Receptor-1 in B and T cell activation
The interleukin-1 (IL-1) family consists of 10 receptors and 11 cytokines. 40 IL-1 receptors are present on the surface of a variety of cells, including endothelial cells, hepatocytes, fibroblasts, keratinocytes, monocytes, neutrophils, T cells, and B cells. Similar to TLRs, IL-1 receptors contain the cytoplasmic Toll-IL-1-Receptor (TIR) domain. Interleukin-1-receptor-1 (IL-1R1) binds to both cytokines IL-1α and IL-1β and IL-1R accessory protein (IL-1RAcP, also called IL-1R3). 41 Binding of IL-1R1 to either of its ligand IL-1α or IL-1β prompts IL-1RAcP to bind to IL-1R1. This association brings the TIR domains of IL-1R1 and IL-1RAcP in close proximity providing the necessary signal to adaptor protein MyD88 and interleukin-1 receptor–activated protein kinase 4 to bind to the TIR domain. Binding of MyD88 and interleukin-1 receptor–activated protein kinase 4 to the TIR domain initiates a cascade of downstream signaling leading to the activation of nuclear factor-κB, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase pathways inducing secretion of pro-inflammatory cytokines. 40,41 In the context of AAV gene therapy, various studies have reported the upregulation and secretion of both IL-1α and IL-1β cytokines in different disease models. 23,33,36,37 For example, Kuranda et al. demonstrated that monocyte-derived DCs (moDCs) in human peripheral blood mononuclear cells (PBMCs) secrete IL-1β upon stimulation with AAV. 37 Until recently, priming of CD8+ T cell responses against AAV capsid or transgene product was thought to strictly depend on TLR9 signaling (Fig. 2). In a murine model of liver gene transfer, we found that CD8+ T cell responses against a transgene product expressed in the liver can occur independent of TLR9 or IFN I and instead rely on IL-1R1>MyD88 signaling (Fig. 2). 38 Both IL-1α and IL-1β contribute to this response (with IL-1α having a perhaps greater contribution), which can be blunted by blocking IL-1 signaling. 38 Interestingly, these responses did not require inflammasomes, consistent with earlier observations that AAV vectors are ineffective in activating inflammasomes. 23 In summary, at least two innate signaling pathways exist that promote the activation of CD8+ T cell responses in AAV gene transfer, namely TLR9>MyD88>IFN I>IFNaR and IL-1α/β>IL-1R1>MyD88. The extent to which these are activated and contribute to the response depends on target tissue and vector dose. Interestingly, dampening IL-1R1 signaling had a mild effect on transgene-specific antibodies but not on capsid-specific antibodies.
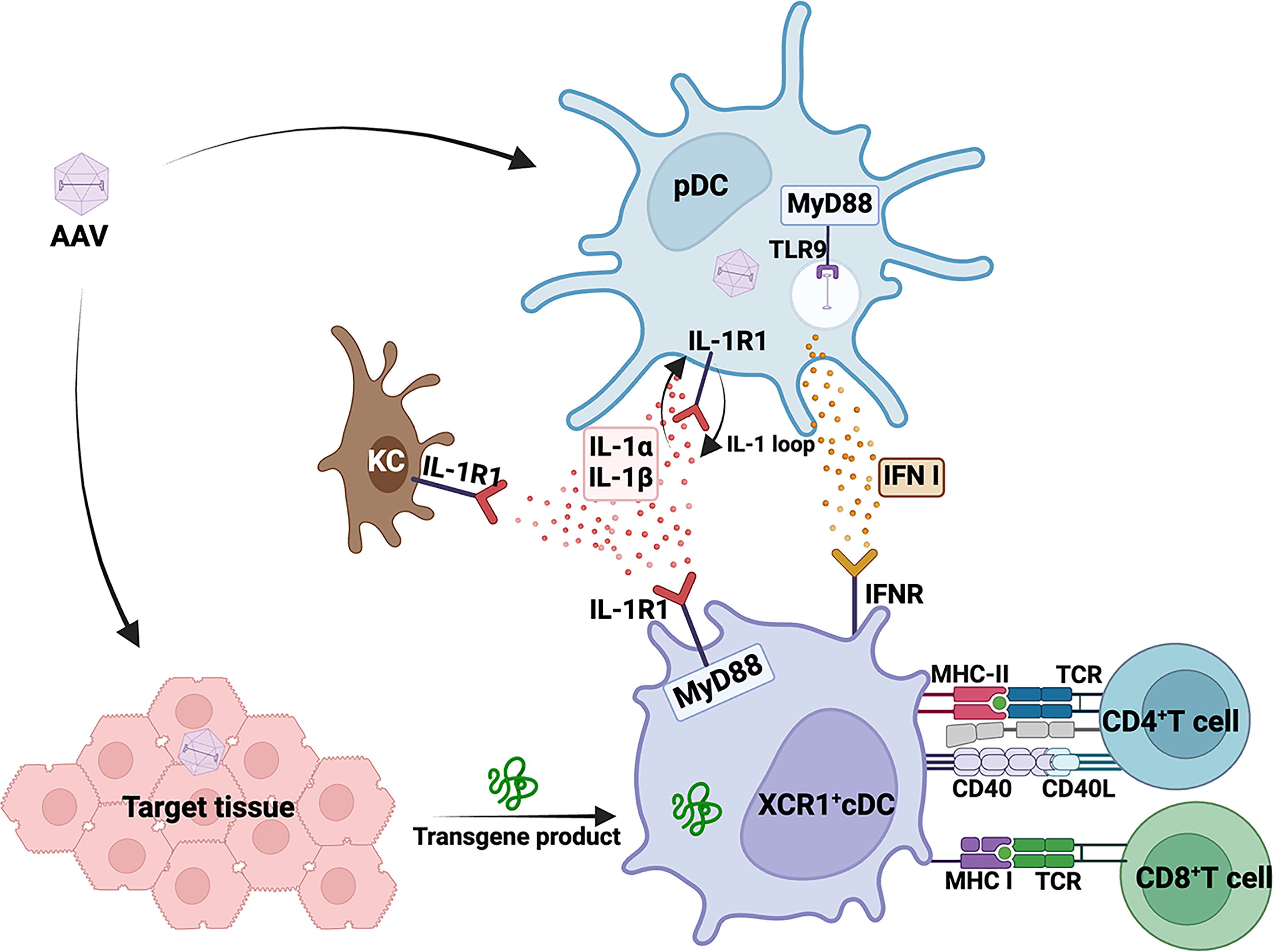
Multiple innate sensing pathways mediate cellular responses to AAV-encoded transgene product. TLR9>MyD88 and IL-1R1>MyD88 mediated signaling is critical for cellular responses to the AAV-encoded transgene product in liver. pDCs are central to the activation of both TLR9 and IL-1R1 signaling upon AAV liver gene transfer. Other cell types, including KCs, cDCs, and T cells, also play an important role in mediating these cellular responses. AAV, adeno-associated virus; TLR9, toll-like receptor 9; pDC, plasmacytoid dendritic cell; IL-1R1, interleukin-1-receptor-1; KC, Kupffer cell; cDC, conventional dendritic cell.
Cytoplasmic DNA sensors
For AAV vectors to reach the nucleus, they need to escape the endosomal compartment and traffic through the cytoplasm (for a comprehensive review of cellular AAV trafficking, see Riyad and Weber). 42 AAV capsids undergo proteosome-mediated degradation in the cytoplasm, which may expose the packaged genomes to cytoplasmic DNA sensors. 43 –45 Several cytoplasmic sensors of microbial/damaged DNA can initiate downstream signaling. One such molecule is cyclic GMP-AMP synthase (cGAS), which detects DNA and DNA-RNA hybrids in the cytoplasm. The resulting signaling is mediated by downstream molecules that include stimulator of interferon genes (STING) and TANK-binding kinase-1, which in turn initiate interferon regulatory factor 3 mediated secretion of cytokines. 46 Chandler et al. found significant upregulation of mRNA transcripts of multiple innate sensors, including TLR9, cGAS, and STING, 7 days postsubretinal administration of AAV in mice. 47 Furthermore, using mouse embryonic fibroblasts, the authors showed improved AAV transduction in fibroblasts derived from cGAS-/- mice. Although a wider role for the cGAS-STING pathway in the innate response to AAV or links to adaptive responses has not yet been established, emerging data from human induced pluripotent stem cell–derived 2- and 3-dimensional cultures of central nervous system cells point toward cGAS-STING pathway mediated upregulation of pro-inflammatory cytokines. 48 Interestingly, authors found that activation of cGAS-STING pathway is downstream of p53 (a tumor suppressor gene) upregulation, and inhibition of STING with an antagonist (H151) abrogated the pro-inflammatory response. Several other studies showed no effect of STING deficiency on B or T cell responses. 26,35 However, activation of this pathway may impact gene editing applications that use AAV vectors to deliver DNA templates, for example, in ex vivo editing of hematopoietic cells. 49
Cytoplasmic RNA sensors
Cytoplasmic RNA sensors are DExD/H box RNA helicases that are classified under retinoic acid-inducible gene I (RIG-I) like receptors (RLRs) family. RLRs are expressed ubiquitously in almost all tissues at low levels during homeostasis. Expression is increased many fold after virus infection or upon exposure to IFN I, thereby mounting an effective immune response against RNA viruses. 50 Of the three members of RLR family, RIG-I and melanoma differentiation-associated protein 5 (MDA5) are the best characterized. Although they share structural similarities, they differ in their ability to detect and interact with RNAs of different sizes. RIG-I preferentially interacts with small RNAs, whereas MDA5 recognizes larger size RNAs. 51 AAV ITRs are known to have intrinsic low-level transcriptional activity, 52,53 therefore could potentially generate complementary strands of RNAs. These complementary RNA strands could theoretically form dsRNA (Fig. 1). Recombination between plus and minus strands during AAV replication may also form genomes that may express dsRNA upon transduction and second-strand formation. 54 Shao et al. provided in vitro evidence for transcription of the opposing strand, albeit at lower frequency compared to the transgene mRNA. 55 HeLa and hepatocyte-derived cell lines had upregulated MDA5 and IFN-β expression 6 days following AAV transduction. Furthermore, using chimeric mice in which mouse livers were partially (∼70%) reconstituted with human hepatocytes, the authors showed about a 2-fold increase in gene expression of MDA5 and RIG-I. Transient inhibition of MDA5 and its downstream adaptor mitochondrial antiviral signaling (MAVS) in HeLa cells was able to increase the transgene expression, presumably by preventing IFN-β expression. Furthermore, emerging data from Verdera et al. demonstrate that RNA sensing through MAVS may drive AAV-induced antiviral responses in certain cell types, as inhibition of MAVS in cultures of human induced pluripotent stem cell–derived astrocytes comprehensively reduced expression of interferon stimulated genes. 48 Whether dsRNA expression plays a significant role in AAV gene transfer is not yet clear, and no links to adaptive immune responses or tissue inflammation have been established. Reichel et al. demonstrated a significant increase in expression of MDA5 and Laboratory of Genetics and Physiology 2 (the third member of RLR family) upon subretinal injection with AAV8 in nonhuman primates, 56 albeit it is unknown whether this relates to dsRNA expression from the vector.
Cells that mediate innate sensing, immunity, and the transition to adaptive CD8+ T cell responses
In a seminal study, using in vitro cultures of human immune cells and mouse models, Zhu et al. showed that TLR9>MyD88 mediated innate sensing of AAV genome takes place in pDCs, which then produce IFN I, whereas macrophages and cDCs do not play a major role in sensing of AAV. 22 Furthermore, the study found TLR9 signaling to be requisite to subsequent CD8+ T cell responses to the transgene product. Later studies found that the effect on antibody formation is more of a modulatory (affecting titers and IgG subclasses) rather than a strong requirement. Removal of potentially immune stimulatory CpG motifs or addition of TLR9 inhibitory sequences has shown effectiveness in reducing CD8+ T cell activation against transgene product and capsid in preclinical studies. 24,33,57 Comparison of clinical trial outcomes in hepatic AAV gene transfer to hemophilia revealed a higher likelihood for sustained expression if CpG motifs were removed and a tendency for loss of expression if CpG enriched. 30 –32,58,59 Interestingly, a hemophilia B patient, who—unlike other study participants—showed sustained therapeutic factor IX expression after having received a CpG-rich vector construct, had a mutation in one allele of IL-6R likely resulting in a lack of functionality of this receptor (IL-6 is one of the cytokines that may be induced by TLR9 signaling in response to AAV, see further below). 30
Using capsid-specific CD8+ T cell responses as a model, we demonstrated in mice that although pDCs are critical to provide an innate activation signal through TLR9, cDCs were equally important because they perform MHC I cross-presentation of the antigen. 26 Consistent with observations on other viruses, pDCs and cDCs cooperate through IFN I to mount a CD8+ T cell response against AAV capsid. In addition to conditioning of cDCs by cytokines, costimulation by CD4+ T helper cells through CD40-CD40L is crucial for cross-priming of capsid-specific CD8+ T cells. 60,61 Cross-presentation of AAV antigen depended on XCR1+ DCs, which are thought to be, in general, the principal subset of cDCs that performs this function during antiviral T cell priming. 57 In the case of AAV, priming of CD8+ T cells occurs independent of natural killer cells (NK cells), but requires the receptor for IFN I (IFNaR) to be expressed on cDCs, implying a direct effect of pDC-produced IFN I on the cDCs. 26,60
Perhaps the best-studied example of an innate immune response within a tissue transduced with an AAV vector is gene transfer to mouse liver (Fig. 3). Within 2–6 h, expression of multiple chemokines and cytokines is induced, including IFN I. This is accompanied by infiltration of innate immune cells, including neutrophils, macrophages, and NK cells. These responses depend on TLR9, are largely dependent on KCs, but are subtle and self-limiting and resolve within less than 1 day. 23,62 Coadministration of an oligonucleotide that inhibits TLR9 abolishes this response. Furthermore, TLR9 deficiency or TLR9 inhibition modestly increases the efficacy of gene transfer by 1.5- to 2-fold. 23,33 Depending on vector design and dose, a spike in systemic IL-6 may be observed, which is TLR9 dependent in the mouse and has also occurred in a patient that received AAV liver gene transfer. 23,30
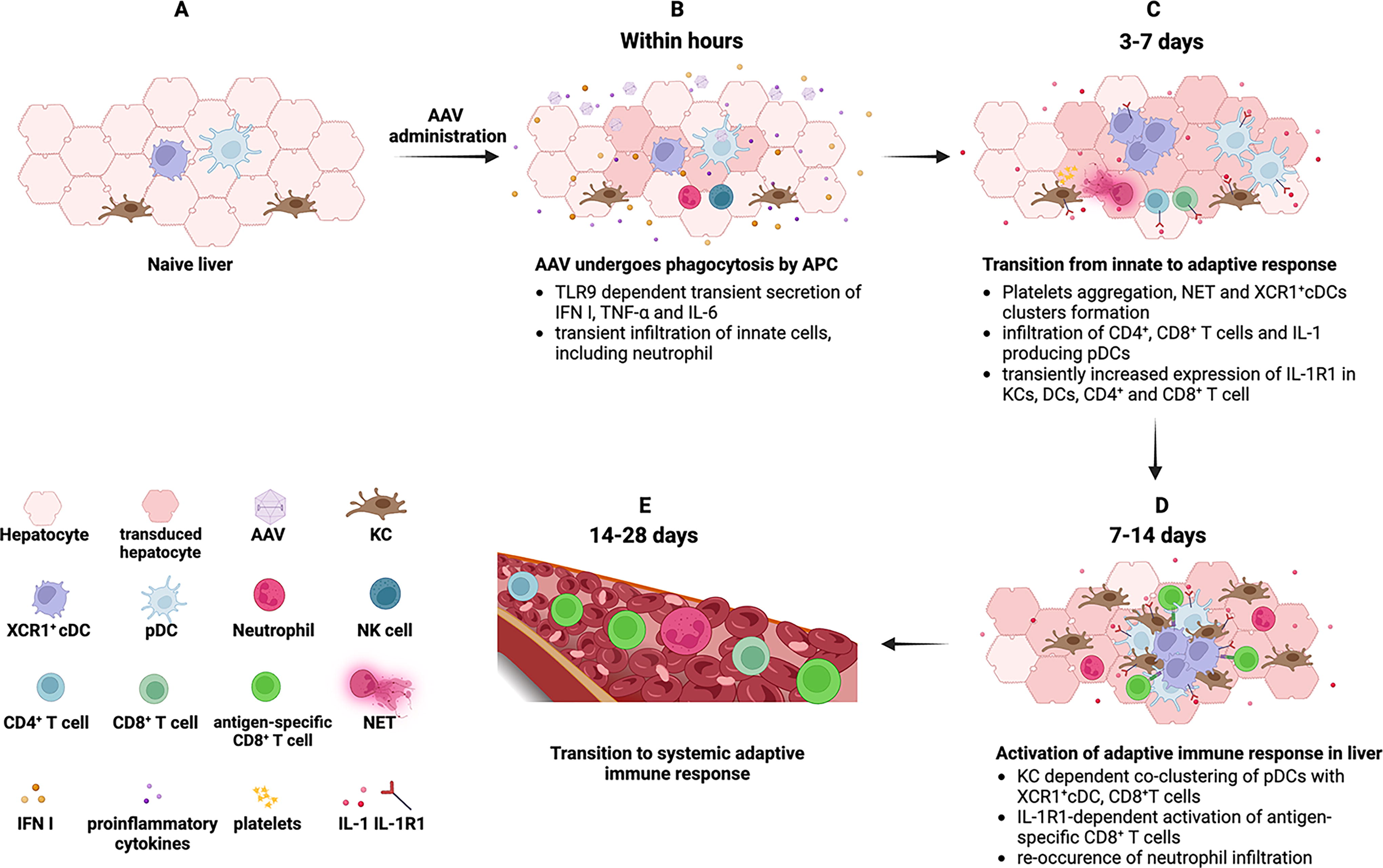
A perspective timeline of innate and adaptive immune responses to AAV vectors within liver. Within hours of AAV administration, endosomal TLR9 in pDCs recognize the AAV genome and drive the transient expression of IFN I and pro-inflammatory cytokines such as TNF-α and IL-6. This transient antiviral environment is dependent on KCs and involves infiltration of innate cells, including neutrophils and NK cells
Unlike adenoviral and lentiviral vectors, IFN I only has a modest effect on AAV transduction and transgene expression. 63 While inflammation of parenchymal tissue and cytokine expression return to baseline within one day, innate responses continue on subsequent days. Carestina et al. in a mouse model of liver gene transfer used intravital microscopy to demonstrate platelet aggregation and neutrophil extracellular trap formation along sinusoidal walls. 64 By day 3, various cell types show upregulation of IL-1R1 expression, and intrahepatic T cells appear nonspecifically activated. 38 At the same time points, these outcomes are not observed in liver-draining lymph nodes, indicating a response that is orchestrated within the liver itself. Within one week, pDCs expressing IL-1α and IL-1β infiltrate the liver and subsequently cocluster with XCR1+ cDCs, KCs, and CD8+ T cells. KC depletion does not prevent pDC infiltration, but disrupts cluster formation. 38 Carestina et al. and Kumar et al. studies in mice are in agreement that KCs take up AAV particles and are required for the immune response within the liver but differ in that Carestina et al. report a transient depletion of KCs, whereas Kumar et al. found KC numbers to increase during the first two weeks. While both studies utilized AAV serotype 8, this discrepancy may be due to the substantially different vector dose applied (Carestina et al. used a 100-fold higher dose).
The formation of coclusters between different subsets of DCs and T cells to link innate and antigen-specific responses against viruses was originally described in lymphoid organs. 65 We now know that the liver is able to form analogous structures and acquire functions in T cell priming normally attributed to lymphoid organs. 38 Priming within the somewhat immune suppressive hepatic environment likely explains the delay in the generation of a fully functional T cell response to gene transfer or to viral infections of the liver. 66 One could speculate that similar mechanisms are responsible for CD8+ T cell responses against AAV capsid that were observed in humans only one month or later after liver gene transfer. 67 The resulting CD8+ T cell response against the transgene product is first detectable in the liver before lymph nodes or spleen, consistent with T cell activation within the hepatic tissue. 38 Thus, early detection of immune responses that start in the target tissue using peripheral blood samples is challenging. Of particular significance for gene therapy is the observation that the hepatic CD8+ T cell response was TLR9 independent. Therefore, the TLR9-dependent early innate response seen in the first few hours after vector administration is not required for the later wave of responses that ultimately promotes CD8+ T cell activation. Higher levels of transgene expression may, however, activate immune suppressive mechanisms that suppress T cell responses and promote tolerance to the transgene product. 65,66,68 –70
In summary, at least two distinct pathways exist that promote CD8+ T cell activation upon AAV gene transfer by conditioning of cDCs with cytokines (Fig. 2). Both are mediated by pDCs, which produce IFN I in response to TLR9 signaling or produce IL-1 within the tissue environment. In either case, help by CD4+ T cells is required in addition to cytokine signaling. The signals that draw pDCs into the liver tissue and that are responsible for their TLR9-independent activation have not yet been identified.
Innate signals leading to B cell activation
As explained below, certain components of complement bind to AAV particles, likely resulting in enhanced B cell activation. However, the requirements for innate signals in antibody formation against the transgene product or the AAV capsid are less clear than those that link to CD8+ T cell activation. Kuranda et al. observed differential responses in PBMC cultures from seropositive and seronegative individuals. 37 Pulsing cells from seropositive humans directed B cell differentiation into anti-AAV antibody-producing cells, whereas NK cell responses were seen in samples from seronegative individuals. AAV capsid activated IL-1β and IL-6 cytokine secretion in moDCs and IL-1β and IL-6 blockade inhibited antibody formation. In gene transfer to murine skeletal muscle, coadministration of a TLR9 agonist (but not agonists for other TLRs) increased antibody formation against a secreted transgene product (unless Treg were depleted, in which case CD8+ T cell rather and B cell responses were elevated). 71,72 This result correlated with increased frequencies of moDCs, which are known to enhance T follicular helper cell activation, thereby strengthening germinal center formation and antibody production. Kuranda et al. implicated capsid-derived peptides in moDC activation in response to AAV. 37 We found that IL-1 blockade prevented CD8+ T cell responses to the transgene product in hepatic gene transfer but had little effect on antibody formation against either transgene product or capsid. 38 IL-6 blockade may be a more promising target. Although TLR9-deficient mice failed to produce antibody against capsid at low vector doses, elimination of TLR9 signaling has mostly been ineffective in preventing antibody formation. Even MyD88 deficient mice may form antibodies against capsid, despite their inability to signal through TLR9 or IL1-R1. 25,39 These mice tend to have delayed antibody formation with altered IgG class switching due to a B cell intrinsic role of MyD88. It should also be pointed out that some transgene products (therapeutic proteins) may contain intrinsic activation signals that may increase the likelihood of immune responses. 73,74
Complement activation
The complement system is an integral part of the innate immune system and plays a critical role in the detection and elimination of pathogens, removal of pathogen-immune complexes, cellular debris, and dead cells. As the name suggests, the complement system “enhances or complements” the immune system to overcome an invading pathogen. It consists of about 50 proteins, which are mainly produced in liver and include soluble serum proteins, membrane-bound regulatory molecules, and complement receptors. 75 Generally, complement proteins exist in pro/inactive form. However, activation through one of the three pathways [the classical pathway, the alternative pathway, or the lectin pathway; Figure 4] leads to a cascade of reactions culminating in opsonization, phagocytosis, destruction of the pathogen, and activation of pathogen-specific adaptive immune responses. 76,77 These three pathways are triggered by different activation signals/PAMPs. For example, the classical pathway is mainly activated by pathogen-antibody (IgG and IgM) complexes, and the lectin pathway recognizes certain carbohydrate moieties on microbial surfaces. Although no specific PAMP has been identified for the activation of the alternative pathway, this pathway always remains active at low levels by spontaneous hydrolysis of the thioester bond in complement protein C3, thereby acting as a surveillance system for pathogens. 78 Despite different means of activation, all three pathways converge at the C3 convertase stage (C4b2a for classical and lectin pathways; C3bBb for the alternative pathway), which subsequently catalyzes C3 to anaphylatoxin C3a and opsonin C3b. Association of C3b to C3 convertase leads to the formation of C5 convertase (C4b2a3b for classical and lectin pathways; C3bBb3b for the alternative pathway), which cleaves C5 to anaphylatoxin C5a and opsonin C5b. C5b then recruits other complement proteins, C6-C9, to form the lytic membrane attack complex on the surface of the pathogen. Anaphylatoxins (C3a, C5a) generated during complement cascade are generally pro-inflammatory and are responsible for chemotaxis and activation of granulocytes, mast cells, and macrophages to the site of infection 79 through opening of endothelial tight junctions leading to tissue edema. Tissue level complement activation may increase interstitial fluid and lead to organ dysfunction, especially in the heart. Because the complement cascade leads to generation of highly potent inflammatory molecules, its activity is tightly regulated by a variety of soluble and surface-bound regulatory proteins, thus preventing unnecessary damage to host tissues and organs. 80,81
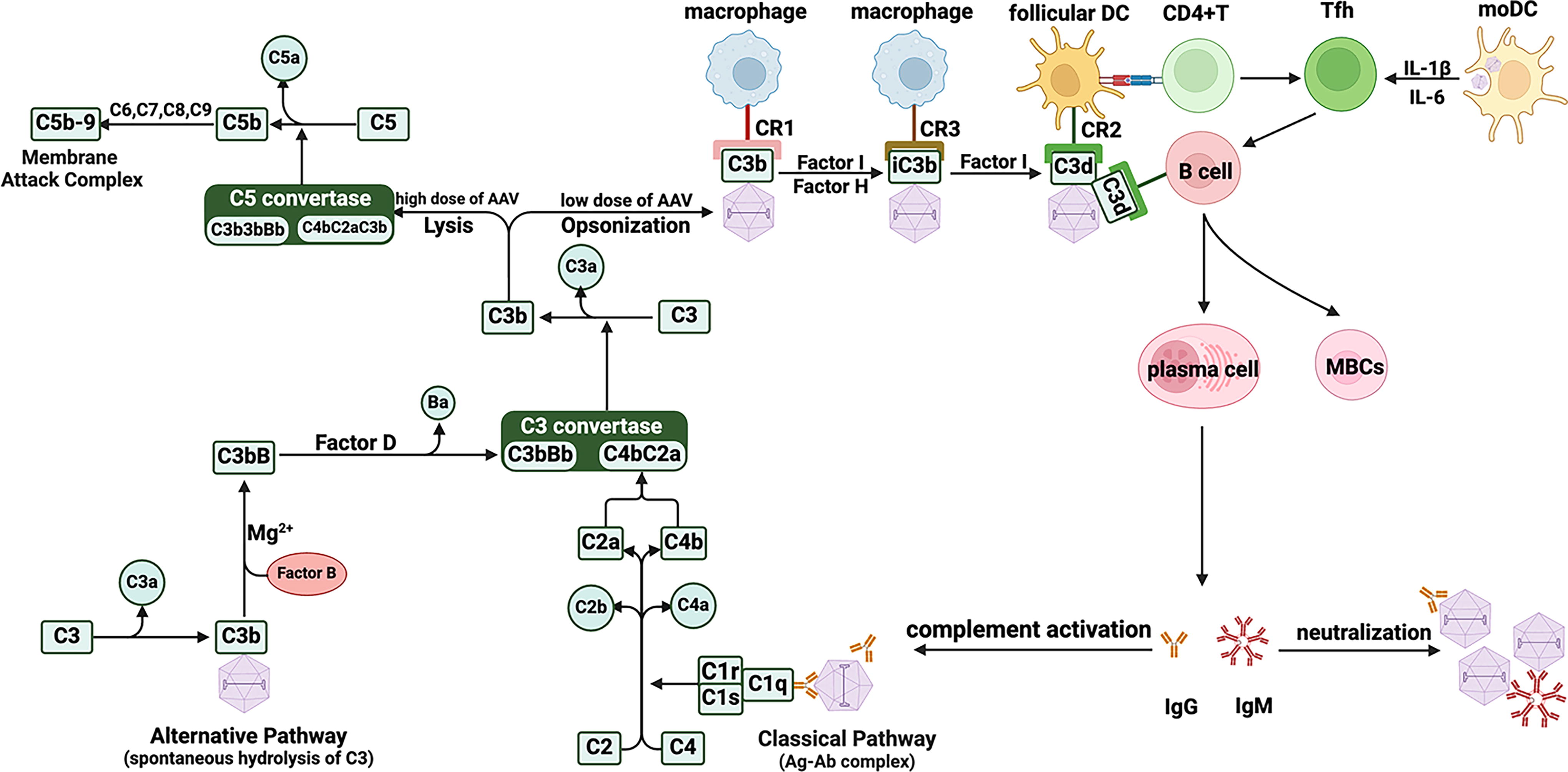
Schematic illustration of AAV-mediated activation of complement pathways. The classical pathway of complement is activated upon recognition of AAV-antibody complexes by C1q, whereas binding of hydrolyzed C3b on AAV activates the alternative pathway. Although these pathways utilize different C3 convertases, they follow the same subsequent steps leading up to the formation of the membrane attack complex. AAVs opsonized with C3b are also recognized by various complement receptors present on the surface of a variety of APCs (including DCs, macrophages, and B cells). This process facilitates AAV uptake, processing, and presentation of AAV-derived antigens to T cells leading to the generation of an effective humoral and cellular response to AAV. AAV, adeno-associated virus.
In 2008, Zaiss et al. first reported that AAV could interact with complement proteins. 82 The authors found that complement may mediate the uptake of AAV vector and trigger pro-inflammatory cytokines in primary murine macrophages and a human monocytic cell line. 82 Although C3 was found to bind directly to AAV capsid, further studies suggested that complement activation by AAV is antibody dependent and follows the classical pathway. AAV capsid lacked cofactor activity for factor I-mediated degradation of C3b to iC3b and instead also bound complement regulatory protein, factor H. The latter may limit complement activation through the alternative pathway but enhances B cell activation through generation of C3d, resulting in engagement of complement receptors on B cells and DCs. This pathway may explain, at least in part, why AAV vector administration tends to induce long-lived plasma cells, which produce sustained high-titer antibodies against capsid.
Worldwide, a high level of AAV seropositivity is observed (40–70% depending on the serotype, geographical location, or ethnicity). 83 –85 In addition, administration of AAV vectors induces highly potent de novo IgM and IgG antibodies. Formation of immune complexes with AAV capsid would generate potential targets for C3b deposition and therefore complement activation. More recent studies by Smith et al. and West et al. further consolidated a contribution of preexisting anti-AAV antibodies in mediating complement activation. 86,87 Smith et al. found that higher titers of preexisting anti-AAV antibodies (≥1:100) were highly efficient in activation of the complement system and increased secretion of pro-inflammatory cytokines and activation of cDCs, independent of the vector genome (i.e., empty vs. full particles). 86 Complement activation could be inhibited using a second-generation C3 modulator, APL-9. Similarly, West et al. also observed activation of the complement system when serum samples had higher levels of preexisting anti-AAV antibodies (IgG1), which were significantly reduced when immunoglobulins were depleted. 87 Comparative analysis of mass spectrometry data from seropositive and seronegative samples showed that anti-AAV antibodies enhanced the binding of viral particles with complement proteins associated with the classical pathway. Enzymatic degradation of IgG (using IdeS enzyme from Streptococcus pyogenes) significantly reduced complement activation. This strategy may be helpful in preventing complement-related immunotoxicities in clinical settings. However, IdeS does not impact IgM, which is equally capable of activating complement system and readily forms within days after AAV infusion. Clinical studies in patients with Duchenne muscular dystrophy (DMD), where seronegative patients were administered with high dose AAV, suggest that IgM formation could still cause significant immunotoxicities. 88,89
Complement activation following AAV administration had not been observed in clinical trials until very high doses were used for systemic delivery, which we now know may trigger life-threatening immunotoxicities. 12,13,88,90 –92 Clinical symptoms that may be related to complement activation included thrombotic microangiopathy (TMA), acute kidney injury involving atypical hemolytic uremic syndrome, thrombocytopenia, elevated transaminases, and endothelial injury. TMA has typically occurred 1–2 weeks after vector administration. 16 For example, a clinical study on AAV gene transfer to patients with Fabry disease reported atypical hemolytic uremic syndrome and TMA associated with elevated levels of complement proteins of both classical and alternative pathways. 93 Salabarria et al. evaluated the kinetics and effects of immune modulation on complement activation in groups of patients who received high systemic doses of AAV989. One group of patients were given only corticosteroids, whereas the other group in addition to corticosteroids received rituximab (B cell depleting antibody) and sirolimus (to prevent T cell activation and proliferation). Patients receiving only corticosteroids had increased levels of IgM and IgG on days 5 and 7, respectively. Some patients in this group experienced thrombocytopenia, likely linked to complement activation. Indeed, levels of complement proteins associated with classical (C3a, C4a, C5a) and alternative (Ba, Bb) pathways along with terminal complement complex SC5b-9 were found to be elevated in patients who only received corticosteroids. Patients treated with rituximab and sirolimus in addition to corticosteroid had no elevations of IgM or IgG and lacked activated complement proteins. Based on these findings, the authors proposed that in humans, the classical pathway is critical for the initiation of complement activation and the occurrence of associated pathologies/toxicities following high-dose systemic delivery of AAV, whereas the alternative pathway may serve as an amplification loop. Thus, complement-mediated immunotoxicities could be minimized in clinical settings using B cell depletion. An alternative protocol targeting CD20 and the B cell survival factor BAFF may also be useful in this regard. 94
Currently, monoclonal antibody eculizumab (an inhibitor of C5) is being used in an attempt to lessen complement-related immunotoxicities. 13,95 However, strategies and modulators that could prevent the activation of multiple complement pathways would be more suitable. For instance, C1 inhibitor for blocking of classical pathway is being evaluated in clinical trial, whereas C3 inhibitor theoretically could block the activation of all three complement pathways.
A recent study by Hordeaux et al. reported high-dose related immunotoxicities in nonhuman primates within 3 days of systemic vector administration. 96 Vector-related severe adverse events included severe thrombocytopenia, transaminase elevations, edema, acute endothelial injury, cholestatic, and hepatotoxicity, among others, and showed some similarities to the ones observed in clinical studies of DMD, spinal muscular atrophy, and X-linked myotubular myopathy. 12,13,17,97,98 This time point would be early during IgM formation and may therefore not reflect classical pathway activation. Further analysis of complement proteins indicated alternative pathway activation, as elevated levels of Bb (complement protein associated with alternative pathway) and terminal complement complex SC5b-9 but not C4a were observed. In contrast to the human experience, the primates exhibited a wider spectrum of dysregulation of coagulation, ranging from disseminated intravascular coagulation to TMA. Nonetheless, this study shows the potential for the alternative pathway to contribute to severe toxicities, including liver injury.
CONCLUSIONS
Although AAV vectors contain weaker inflammatory signals than many other viruses or vector systems, they nonetheless elicit innate responses that can have a significant impact on gene therapy. A deeper understanding of innate immune response mechanisms against AAV has led to the engineering of vectors with reduced immunogenicity and the development of superior immune suppression protocols. Innate immunity links to adaptive responses and to immunotoxicities. Therefore, further study of innate immunity to AAV remains critical. More information is needed on the role of the target tissue and the route of administration. Additional pathways may be identified, and redundancy depending on tissue and vector dose may pose a requirement to circumvent or block more than one pathway for some applications. It may not be possible to prevent adaptive responses entirely by blocking innate signaling. For example, the signaling requirements for reactivation of memory responses are usually lower than for primary responses. The role of the viral capsid in complement activation is not fully understood. While most capsids are able to activate complement in in vitro assays in the presence of antibodies, differences in their biodistribution and half-life in circulation could possibly make a difference in vivo. Animal models aid in the identification of pathways, but never entirely recapitulate human treatment, as illustrated by various discordant immune responses to AAV vectors: CD8+ T cell responses against capsid, complement activation, and associated toxicities, effects of preexisting immunity from natural infection, or cross-reactivity of antibodies formed against capsid.
Footnotes
ACKNOWLEDGMENTS
BioRender software was used for all the graphical illustrations.
AUTHORS’ CONTRIBUTIONS
All authors contributed to writing the article.
AUTHOR DISCLOSURE
R.W.H. is serving on scientific advisory boards for Pfizer, Spark Therapeutics, and Biomarin, and also received research funding from Hoffman LaRoche. D.D. is a member of the scientific advisory board for Solid Biosciences and an equity holder of Solid Biosciences. D.D. is a member of the scientific advisory board for Sardocor Corp. B.J.B. is the inventor of AAV-related intellectual property (US 2022/0347297 A1) owned by the University of Florida and may be entitled to licensing revenue as determined by the University of Florida inventor policy.
FUNDING INFORMATION
This work was supported by National Institutes of Health grants