
Abstract
Endothelial activation and dysfunction are key early steps in atherogenesis. Vascular gene therapy targeting endothelial inflammation and cholesterol accumulation could decrease atherosclerosis progression. ATP-binding cassette subfamily A member 1 (ABCA1) exhibits anti-inflammatory properties and promotes cholesterol efflux. A mouse model showed that systemic endothelial overexpression of ABCA1 decreased diet-induced atherosclerosis. To test if local ABCA1 endothelial overexpression protects against atherosclerosis, we used helper-dependent adenoviral vectors (HDAd) to express ABCA1 or a “Null” control in the carotid endothelium of hyperlipidemic rabbits. Both ABCA1 mRNA and endothelial protein were increased 3 days after vector infusion. After 24 weeks on a high-fat diet, laser-microdissected endothelium showed increased ABCA1 mRNA expression, but whole-vessel ABCA1 mRNA was decreased with HDAdABCA1. Endothelial ABCA1 protein could not be measured at 24 weeks, so its overexpression may be transient. CD68 expression was decreased (–23%, p < 0.001), but ITGAM (–15%, p = 0.3) was unchanged. Macrophage markers for both M1-like macrophages (IL1B: –44% [p = 0.02]; IL6: –40% [p = 0.02]; CCL2: –25% [p = 0.02]) and M2-like macrophages (ARG1: –27% [p = 0.03]; IL10: –23% [p = 0.09]; TGFB1: –13% [p < 0.001]) were also decreased. The inflammatory cytokines IL6 (–100%; p < 0.001) and TNF (p < 0.05) were significantly decreased in the laser-microdissected endothelium, but VCAM1 (+5%, p = 1.0) was unchanged and ICAM1 (+101%; p = 0.03) increased. Lesion size, intimal lipid, and intimal macrophage content were all unchanged (p > 0.5 for all), and vascular cholesterol measured by mass spectrometry (–11%; p = 0.9) also showed no difference. There was a small decrease in the intimal/medial ratio. scRNAseq revealed that vector transcripts were not restricted to endothelial cells after 24+ weeks but were detected in most cell types. The exception was modulated smooth muscle cells, which were found in substantial numbers in larger lesions. Overall, transient overexpression of ABCA1 in the vascular endothelium subtly alters the expression of inflammatory markers, providing only a modest atheroprotection.
INTRODUCTION
Cardiovascular disease (CVD) mortality, after declining for several decades due to improved medical care and management of risk factors, 1,2 is increasing again despite many risk factors such as smoking rate, low-density lipoprotein cholesterol (LDL-C) levels, and blood pressure remaining unchanged or declining. 2 This suggests that current treatments and the control over LDL-C and blood pressure levels that they provide alone are not sufficient as CVD therapies.
Therapies targeting cholesterol efflux to remove lipids from the vessel wall are a promising approach. Previously, we have shown in a hyperlipidemic rabbit model that overexpression of apolipoprotein A-I (apoAI) reduces inflammation and lipid in plaques leading to smaller atherosclerotic lesions. 3 The atheroprotective effect is partial, however, and may be limited by a decrease in the ATP-binding cassette subfamily A, member 1 (ABCA1) protein that binds apoAI to efflux cholesterol. ABCA1 is a ubiquitously expressed transporter 4 that acts in conjunction with apoAI to both remove cholesterol from cells and inhibit inflammatory cellular responses. 5 Targeting ABCA1 may be able to counteract the ABCA1 decrease and work in synergy with apoAI/HDL therapies to improve atheroprotection. Showing that ABCA1 can be overexpressed using the same endothelial cell expression system in vivo and alter cellular signaling and atherogenesis could be a first step toward future synergistic therapies.
Vascular endothelial activation and dysfunction are considered early triggers for atherosclerosis development. 6,7 Stressors activate endothelial cells (EC) causing elevated NF-kB signaling that increases inflammatory cytokines such as TNF, CCL2, and IL6, and adhesion molecules, such as E-selectin, ICAM1, and VCAM1. 8 –12 Together, these changes to the endothelium increase the adhesion of monocytes, which differentiate into macrophages in the vascular wall. 13,14 The inflamed endothelium can increase LDL transport into the vascular wall 15,16 and accelerate cholesterol crystal deposition, 17 –19 spurring atherogenesis. Inflammation contributes to endothelial dysfunction, characterized by a loss of nitric oxide availability that drives atherosclerosis development. 20 –22 Decreasing the cholesterol accumulation and subsequent inflammatory signaling in the endothelium is a possible target for reducing atherosclerosis.
Overexpression of ABCA1 in cultured EC increases cholesterol efflux from the cells and decreases the inflammatory response to insults. 23 In mouse studies, knockout of ABCA1 in the endothelium decreases endothelial cholesterol efflux and vascular eNOS leading to increased macrophage accumulation and atherosclerosis development. 24 Conversely, overexpression of ABCA1 in the endothelium increases endothelial cholesterol efflux leading to atheroprotective transcriptional changes and a smaller lesion. 25 However, in that study by Vaisman et. al, ABCA1 was overexpressed in all EC, not just vascular EC, which led to elevated plasma high-density cholesterol. Thus, it remains unknown whether a localized ABCA1 overexpression in the endothelium of atherosclerotic vessels would diminish endothelial activation and reduce atherosclerosis.
We hypothesized that overexpressing ABCA1 in the carotid endothelium of atherosclerotic rabbits would reduce lipid and macrophage accumulation in the vascular intima leading to reduced lesion areas. Secondarily, we hypothesized that expression of inflammatory markers and adhesion molecules would be reduced in the vascular endothelium, with macrophages taking a more healing, M2-like phenotype. In these studies, we tested whether a viral vector expressing ABCA1 (HDAdABCA1) infused into the carotid arteries of hyperlipidemic rabbits could reduce atherosclerosis 24 weeks after treatment, utilizing laser microdissection and single-cell RNA sequencing (scRNAseq) to specifically examine possible cell-specific roles of endothelial cell activation and macrophage phenotype.
MATERIALS AND METHODS
Viral vectors
We used two helper-dependent adenoviral (HDAd) vectors: HDAdNull (empty CMV-driven expression cassette) 26 and HDAdABCA1(rabbit ABCA1 cDNA with the same CMV promoter. 23 Vector stocks were propagated in human embryonic kidney 293Cre4 cells (Microbix Biosystems, Toronto, Ontario, Canada) 27 and purified as described. 28 All vector preparations have concentrations, measured by spectrophotometry, 29 of 0.6–3 × 1012 viral particles/mL, undetectable E1A-containing genomes (<1 in 106 total vector genomes), and helper virus contamination <1% of total vector genomes. 26 Sequences of primers and probes for detection of E1A, helper virus, and HDAd genomes (pC4HSU 30 -targeted) are in Supplementary Table S1.
Animal studies
All animal studies were approved by the University of Washington Office of Animal Welfare. The study design, conduct, and reporting comply with the ARRIVE guidelines. 31 Specific pathogen-free male and female New Zealand White rabbits (3.0–3.5 kg, approximately 14 weeks old; Western Oregon Rabbit Company, Philomath, OR) were fed either a normal laboratory diet (Rabbit High Fiber, #5326, Purina Laboratory Diet, Gray Summit, MO), a high-fat diet, including 1.0%, 0.5%, 0.3%, or 0.25% cholesterol, added to the normal diet (all including 3% soybean oil; Dyets Inc., Bethlehem, PA), or a mixture of the cholesterol-containing diet and the normal diet (to achieve diets with <0.25% cholesterol).
Two small studies tested ABCA1 expression after HDAdABCA1 infusion in rabbits on a normal laboratory diet and a high-fat diet. Another study tested flow cytometry to measure ABCA1 protein expression in transduced carotid EC. Methods for these studies are in Supplementary Data.
The main study tested whether HDAdABCA1 infusion prevented atherosclerosis. Rabbits were fed 1% cholesterol for 2 weeks, followed by 0.5% cholesterol for 2 weeks, then received bilateral common carotid vector infusions. This high-fat diet combined with the vector-infusion surgery stimulates the formation of complex carotid atherosclerotic plaques. 32 Postoperatively, rabbits were maintained on a sliding-scale diet (0–0.25% cholesterol) that was adjusted every 2 weeks based on plasma cholesterol levels to maintain plasma cholesterol at 200–800 mg/dL. Sample size calculations using data from a previous study 3 showed that data from 25 rabbits per group would allow detection or exclusion of median differences of 25–30% in intimal lesion area, intimal lipid, and intimal macrophage content (α = 0.05 and β = 0.2). We enrolled 30 rabbits (16 males and 14 females) to accommodate attrition. We used our published protocol for in vivo gene transfer to rabbit carotids (details in Supplementary Data). 33
One rabbit died 2 days after vector infusion surgery from respiratory failure. Twenty-six rabbits were euthanized 24 weeks after vector infusion. Both carotids were removed and cut into eight equal segments (Supplementary Fig. S1A). The segments were processed for histology, laser-capture microdissection/mRNA measurements, measurement of vector DNA and mRNA, cholesterol content, nitric oxide, and RNA analysis on scraped endothelium. RNA analysis was not completed on scraped endothelium because CDH5/GAPDH ratios showed EC were not enriched. Due to low surgical and diet-induced complication rates, our a priori calculated sample size for the primary endpoints was met with several rabbits remaining. Thus, the final three rabbits were euthanized 28–31 weeks after vector infusion and their carotids used for scRNAseq analyses. Because both experimental and control arteries were in the same rabbit, we did not attend to potential confounders such as cage location.
Processing of vessel segments for DNA, RNA, and cholesterol analyses
The three frozen segments from each artery were pooled and pulverized with a liquid nitrogen-cooled mortar and pestle. The pulverized tissue was separated into three aliquots that were stored at –80°C for later measurements of vector DNA (by quantitative PCR, qPCR, ∼3 mg per sample), cholesterol (by mass spectrometry; ∼3 mg per sample), and several mRNA species (by reverse transcriptase-mediated qPCR, RT-qPCR, all remaining tissues: ∼3–30 mg). For two rabbits (four vessels), a low amount of tissue extract was obtained; therefore, cholesterol measurements were not done on these rabbits. Vessel segments were processed by persons who were blinded to treatment.
Measurement of vector DNA
DNA was extracted from pulverized artery tissue with the DNeasy Blood and Tissue kit (Qiagen Sciences, Germantown, MD) and quantified by spectrophotometry (NanoDrop; Thermo Fisher Scientific, Waltham, MA). Vector copies were measured by qPCR amplification of 50 ng of DNA, targeting a sequence of noncoding stuffer DNA in the HDAd backbone. 34 Vector copy number was measured with reference to a standard curve generated by serial dilution of the pC4HSU plasmid 30 that contains the stuffer DNA sequence 26 and then normalized to a number of vascular wall cells. Primer and probe sequences are in Supplementary Table S1.
Measurement of RNA
Pulverized tissue was homogenized with a Polytron PT3100 (Polytron Devices, Paterson, NJ) in RLT buffer (RNeasy mini kit, Qiagen Sciences) with 1% added β-mercaptoethanol. RNA extraction was continued as per the manufacturer’s protocol. Total RNA was quantified by spectrophotometry (NanoDrop). Target mRNAs were measured by RT-qPCR performed on 50 ng of total RNA, with results normalized to GAPDH mRNA measured in the same extracts. Primer and probe sequences are in Supplementary Table S1.
Measurement of artery wall cholesterol
Carotid artery cholesterol was measured in 24 rabbits as described previously, 35 with minor modifications. Details are in Supplementary Data.
Histochemical and immunohistochemical staining
Sections (6-μm thick) were cut from OCT blocks at two steps (∼150 μm) and stained with hematoxylin and eosin, Oil Red O (ORO), and with antibodies detecting macrophage-associated antigen (RAM-11; 0.37 µg/mL; #M0633, Dako, Carpinteria, CA), muscle actin (HHF-35; 5 µg/mL; #MA1-82599, Thermo Fisher Scientific), ICAM1 (1:200 dilution; from Dr. Myron Cybulsky, University of Toronto, Toronto, ON, Canada), and VCAM1 (1:50 dilution; also from Dr. Cybulsky). 36 Isotype-matched mouse IgG was a negative control. RAM-11 was detected with a fluorophore-tagged secondary antibody (6.7 µg/mL; #a32727, Invitrogen, Waltham, MA) and Fluoroshield/DAPI mounting medium (F6057, Sigma, St. Louis, MO). Other antibodies were detected with biotinylated goat anti-mouse IgG (7.5 µg/mL, #BA-9200) followed by avidin/biotin complex formation (VECTASTAIN ABC HRP Kit), and visualization with the ImmPACT® NovaRED (all reagents from Vector Laboratories, Burlingame, CA). Additional details on sectioning and analysis are in Supplementary Data.
Laser-capture microdissection
Tissue was collected by laser-capture microdissection from both carotid arteries in 13 out of the 26 rabbits. Endothelial-rich and macrophage-rich regions of intima (strong RAM-11 staining on adjacent slides) were collected with a Leica DM6000B laser microdissection scope and RNA was extracted as described above. RT-qPCR targets were measured with multiplexing (#95132, QuantaBio, Beverly, MA). Primer and probe sequences are in Supplementary Table S1. See Supplementary Data for details.
scRNAseq
Three rabbits were used for scRNAseq using the 10 × Chromium Next GEM Single Cell 3′ Kit v3.1 (10 × Genomics; Pleasanton, CA). One sample was removed because of a visible failure of GEM formation accompanied by detection of a low number of genes/cell. Sequenced transcripts were aligned to a custom rabbit reference genome, and analyzed to identify cell clusters, differentially expressed genes (DEGs), and regulated pathways, as described in Supplementary Data.
Statistical analysis
For the expression studies and flow cytometry of ABCA1, data were analyzed with a t-test, unless data failed the normality or equal variance tests, in which case a rank-sum test was used. In the main atherosclerosis gene therapy treatment study, intimal lipid (ORO stain), cholesterol (mass spectrometry), and intimal macrophage accumulation (RAM-11 immunostaining) were prospectively chosen as the primary endpoints of the study. Because lesion masses in the left and right carotids of the same rabbit are highly correlated and lesion size is highly variable among rabbits, 3 we prospectively selected an analytic approach that compares HDAdABCA1 and HDAdNull treatments in the same rabbit. We used a paired t-test, or a Wilcoxon signed-rank test when the paired differences failed the Shapiro–Wilk normality test. When an assay value was below the limit of detection, the sample was assigned a value of zero. All statistical tests were performed with the SigmaPlot 13 program (Systat, San Jose, CA). Data are reported as group medians, or the median paired difference, as indicated. Analysis of sex as a biological variable is included in Supplementary Table S8.
RESULTS
Expression of ABCA1 mRNA and protein in HDAdABCA1-transduced arteries
We measured ABCA1 mRNA 3 days after transduction of carotids of rabbits fed a normal laboratory diet. Using ABCA1 transgene-specific primers and whole-vessel RNA extracts, we detected ABCA1 transcripts in 100% (8 of 8) of HDAdABCA1-transduced arteries and 0% (0 of 6) of HDAdNull-transduced arteries (p < 0.001; Supplementary Fig. S1B). Using primers that detect both endogenous and transgenic (i.e., total) ABCA1 mRNA, we detected ABCA1 mRNA in all arteries, with an approximately twofold increase in HDAdABCA1-transduced arteries (p = 0.07; Supplementary Fig. S1C).
We repeated this study in high-fat diet-fed rabbits. Three days after transduction, ABCA1 transgene RNA was again detected in 100% (7 of 7) of HDAdABCA1-transduced and 0% (0 of 7) of HDAdNull-transduced arteries (p < 0.001; Fig. 1A), with total ABCA1 mRNA approximately twofold higher in HDAdABCA1-transduced arteries (p = 0.04; Fig. 1B).

ATP-binding cassette subfamily A member 1 (ABCA1) mRNA and protein expression. Carotids of rabbits on a high-fat diet were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1) and harvested 3 days later.
Western blotting of luminal endothelial cell extracts (Supplementary Fig. S1G) from this same set of arteries showed a mean 1.6-fold increase in ABCA1 protein in HDAdABCA1-transduced carotids (p = 0.06; Fig. 1C, D, Supplementary Fig. S1H, I). Although this increase in ABCA1 normalized to the preselected membrane housekeeping gene was only borderline significant, this was at least partially due to the loss of the highest ABCA1-expressing sample (see Supplementary Data). ABCA1 protein without normalization beyond equal loading (Supplementary Fig. S1F) and normalized to total protein (Fig. 1E, Supplementary Fig. S1J, K), both of which included all samples, were both significant, convincing us that ABCA1 protein was elevated at 3 days posttransduction.
Persistent HDAd genomes and ABCA1 transgene expression in high-fat diet-fed rabbits
To test whether ABCA1 overexpression in arterial endothelium reduces atherosclerosis, rabbits were fed a high-fat diet for 4 weeks, then treated with local intraluminal infusion of HDAdABCA1 in one carotid and HDAdNull in the contralateral carotid (Fig. 1). Rabbits were maintained on a high-fat diet and arteries were harvested 24 weeks later. This combination of hyperlipidemia and carotid artery surgery/infusion reliably produces focal complex atherosclerotic lesions at the site of vector infusion. 3,32,37 Mean plasma cholesterol at the time of vector infusion was 1,420 mg/dL, dropping to 379 mg/dL at 4 weeks after vector infusion and remaining stable until artery harvest (Fig. 2A). Because HDAdABCA1- and paired control HDAdNull-infused arteries were in the same rabbit, variability in plasma cholesterol levels among rabbits cannot account for effects of the ABCA1 transgene.
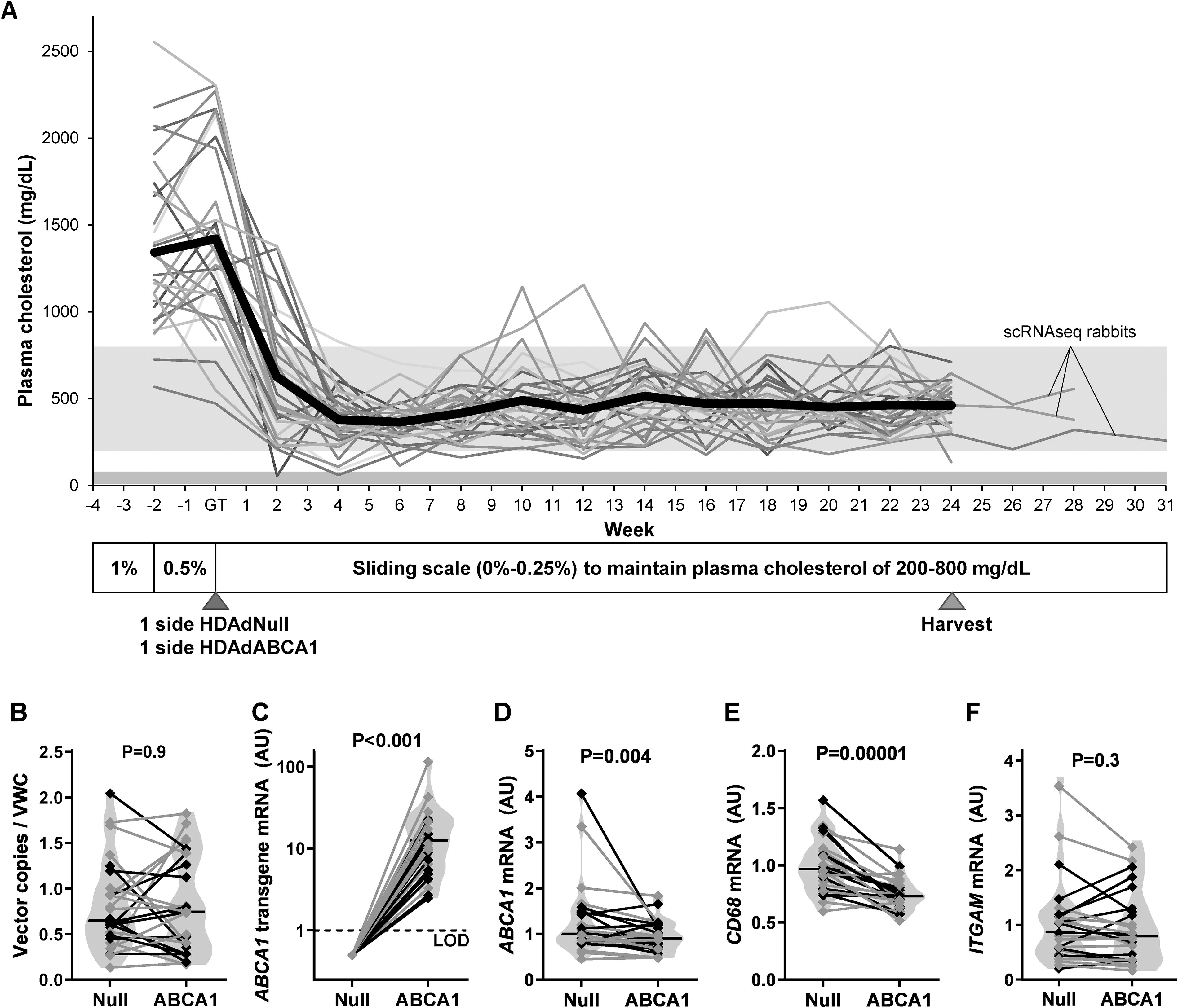
Effect of endothelial ABCA1 expression on macrophage markers in hyperlipidemic rabbits.
HDAdNull and HDAdABCA1 arteries both had a median of 0.7 vector genomes per vascular wall cell, with no significant difference between the groups (p = 0.9; Fig. 2B). Because we performed most of the statistical analyses on differences between paired arteries in the same rabbit (see the Materials and Methods section), here and elsewhere (unless otherwise noted) we present results as the median percentage differences between HDAdABCA1 carotids and their paired HDAdNull controls. ABCA1 transgene mRNA was present in 100% (24 of 24) of HDAdABCA1 arteries and 0% (0 of 24) of HDAdNull arteries (p < 0.001; Fig. 2C). Surprisingly, total ABCA1 mRNA was slightly lower in extracts of full-thickness HDAdABCA1 arteries (–10%, p = 0.004; Fig. 2D).
ABCA1 transgene expression decreases mRNA encoding CD68, cytokines, and ICAM1
In whole-artery RNA extracts of fat-fed rabbits, mRNA encoding the macrophage marker CD68 was decreased by a median of 23% in HDAdABCA1 arteries (p < 0.001; Fig. 2E), suggesting reduced macrophage accumulation. However, another macrophage marker (ITGAM) was not significantly decreased (15% lower, p = 0.3; Fig. 2F). To determine if macrophage polarization was altered by HDAdABCA1 transduction, we measured mRNA encoding markers of M1- and M2-like macrophages in whole-artery RNA extracts (Fig. 3A–I). Several markers of M1-like macrophages were decreased in HDAdABCA1 arteries, including IL1B (–44%, p = 0.02), IL6 (–40%, p = 0.02), and CCL2 (–25%, p = 0.02), while IFNG and TNF mRNA levels were not significantly different (+8%; p = 1.0 and –2%, p = 0.5, respectively). Expression of markers of M2-like macrophages was also lower in HDAdABCA1 arteries, including TGFB1 (–13%, p < 0.001) and ARG1 (–27%, p = 0.03), with IL10 also trending lower (–23%, p = 0.09). MRC1 expression was unchanged (–1%, p = 0.8). HDAdABCA1 arteries had lower expression of ICAM1 (–14%, p = 0.006) but no significant change in VCAM1 expression (+8%, p = 0.3; Fig. 3J, K). NOS3 (+4%, p = 1.0; Fig. 3L) was also unchanged.
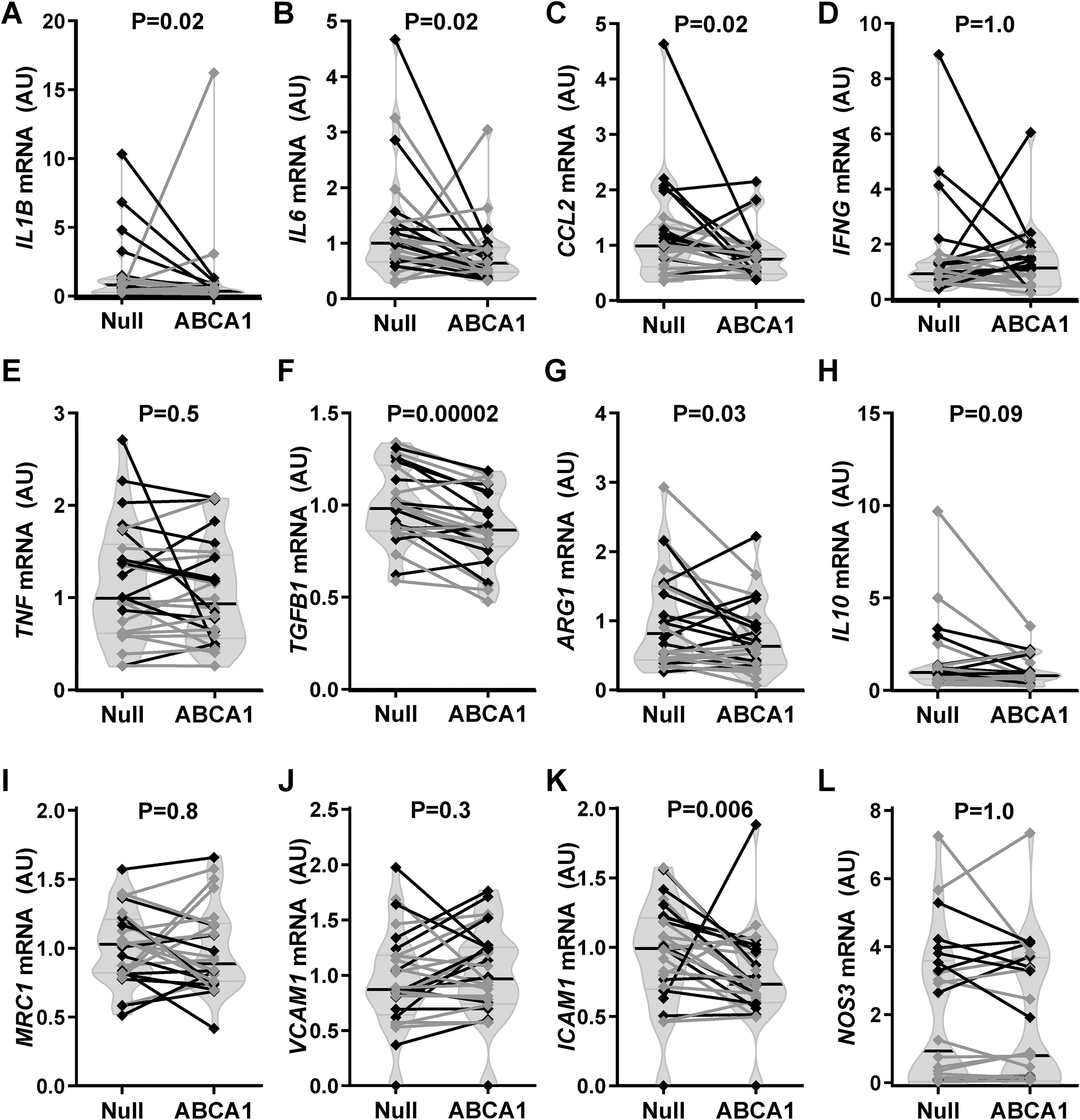
Carotid mRNA expression of selected targets. Carotids of rabbits on a high-fat diet were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1) and harvested 24 weeks later.
Effects of ABCA1 transgene expression on intimal and medial mass, intimal cellularity and lipid, intimal adhesion molecule expression, and whole-vessel cholesterol content
Histological staining was used to assess intimal lesions (Supplementary Fig. S2). Neither intimal area nor medial area was significantly different in HDAdABCA1 carotids (–9%; p = 0.3 and –1%; p = 0.8, respectively; Fig. 4A, B). Medial thickness was also unchanged (–1%; p = 0.9; Supplementary Fig. S3A). Nevertheless, there was a small borderline significant decrease in the intimal/medial ratio of HDAdABCA1 arteries (–11%; p = 0.06; Fig. 4C) and a corresponding increase in the calculated luminal area (+10%, p = 0.06, Supplementary Fig. S3B).

Histological analyses and vascular cholesterol. Carotids of rabbits on a high-fat diet were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1) and harvested 24 weeks later, sectioned, and stained.
HDAdABCA1 arteries had no significant differences in intimal area stained for neutral lipids (11% less ORO-stained area; p = 0.6; Fig. 4D), macrophages (8% less RAM-11-stained area; p = 0.5; Fig. 4E), or smooth muscle cells (8% more HHF-35-stained area; p = 0.6, Fig. 4F). HDAdABCA1 arteries had no significant differences in intimal area stained for ICAM1 (23% decrease; p = 0.5; Fig. 4G) or VCAM1 (37% decrease, p = 0.2, Fig. 4H). Normalization of these measurements to the total intimal area of each section also revealed no significant differences between HDAdABCA1 and HDAdNull arteries (Supplementary Fig. S3C–G).
Cholesterol and nitric oxide were measured in whole carotid segments. Mass spectrometry showed that total artery wall cholesterol was not significantly lower in HDAdABCA1 arteries (11% median decrease; p = 0.9; Fig. 4I). Unesterified cholesterol (15% mean increase; p = 0.6), cholesterol ester (14% median decrease; p = 0.9), and percentage of esterified cholesterol (0.1% median decrease; p = 0.4) were all unchanged (Supplementary Fig. S3H–J). Nitric oxide measured by electron paramagnetic resonance (11% increase, p = 0.2; Supplementary Fig. S3K) was also unchanged.
Measurement of mRNA in laser-capture microdissected arteries
Transduction in this animal model is largely confined to luminal EC. 38,39 Because ABCA1 overexpression in EC in vitro significantly reduces inflammatory cytokine and adhesion molecule mRNA, 23 we used laser-capture microdissection (n = 13 pairs of HDAdABCA1 and HDAdNull arteries) to isolate endothelial-enriched mRNA and test whether ABCA1 overexpression has the same effects in vivo. Increased ABCA1 transgene mRNA in endothelial-enriched tissue in HDAdABCA1 carotids (p = 0.03; Fig. 5A) led to a 46% higher median total ABCA1 mRNA; this was of borderline statistical significance (p = 0.07; Fig. 5B). IL6 mRNA was far lower in HDAdABCA1 carotids (100% decrease; p < 0.001; Fig. 5C), and TNF mRNA was largely undetectable in HDAdABCA1 (p = 0.047, Fig. 5D). VCAM1 mRNA was equally abundant in HDAdABCA1 and HDAdNull carotids (p = 1.0; Fig. 5E), while ICAM1 was increased in HDAdABCA1 carotids (101% increase; p = 0.03; Fig. 5F).

mRNA expression in endothelial cell-enriched extracts. Carotids of rabbits on a high-fat diet were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1) and harvested 24 weeks later and sectioned. mRNA expression normalized to GAPDH, quantified in endothelium-enriched tissue isolated by laser-capture microdissection for
Reductions of RNA from several cytokines in whole-vessel extracts of HDAdABCA1 carotids (Fig. 3) could be explained by lower macrophage numbers, effects on macrophage gene expression, altered gene expression in other cell types, or a combination of these factors. To help differentiate among these possibilities, we used laser-capture microdissection to isolate macrophage-rich mRNA (using RAM-11-stained adjacent sections as a guide) from HDAdABCA1 and HDAdNull carotids (n = 13 each). We found no significant differences in levels of mRNA encoding any M1 (CCL2, IL1B, IL6, TNF) or M2 (IL10, TGFB1) cytokine (Supplementary Fig. S4A–F), suggesting that HDAdABCA1 transduction does not alter cytokine expression in artery wall macrophages. Interestingly, transgenic ABCA1 transcript was detected within the macrophage-enriched tissue (Supplementary Fig. S4G), although total ABCA1 mRNA was unchanged (Supplementary Fig. S4H).
scRNAseq reveals modulated smooth muscle cells and the presence of HDAd vector in multiple cell types
To further explore cell-specific effects of atherosclerosis and HDAdABCA1 treatment, we performed scRNAseq on HDAd-transduced carotids from rabbits, all harvested 28–31 weeks after transduction (Fig. 2A). Cells from one HDAdABCA1 artery could not be analyzed because of a technical problem (see the Materials and Methods section), leaving 2 HDAdABCA1 arteries and 3 HDAdNull arteries. After applying quality-control metrics, analyses of transcriptomes of 13,687 cells from HDAdABCA1 arteries and 17,332 cells from HDAdNull arteries revealed cell clusters (Fig. 6A), identified using standard cell-type markers (Fig. 6B), representing all major vascular cell types (Supplementary Table S2). The identified cell types were equally represented in the HDAdABCA1 and HDAdNull arteries (Fig. 6C).

Identification of cell types and vector transcript-containing cells by scRNAseq. Carotids of rabbits on a high-fat diet were transduced with either HDAdNull or HDAdABCA1 and harvested 28–31 weeks later. Cells from five carotids (three Null and two ABCA1) were analyzed by single-cell RNA sequencing.
A cluster of modulated smooth muscle cells (modSMC) 40,41 was identified that had lower expression of contractile genes and higher expression of markers of plaque SMC, including LGALS3 and proteoglycans (Supplementary Fig. S5A), as well as high expression of FN1 and VCAM1. IHC for VCAM1 appears to show modSMC in many lesions based on strong VCAM1 expression in the intima adjacent to the media (Supplementary Fig. S5B). The VCAM1-positive regions near the medial layer have low muscle actin staining (Supplementary Fig. S5C) but do stain positively for a macrophage marker (Supplementary Fig. S5D).
Mapping of transcripts of interest (Fig. 3) onto the identified cell clusters revealed ABCA1 expression in most cell types. CD68 expression was most highly expressed in macrophages, but was also expressed in fibroblasts and SMC, while ITGAM transcripts were predominantly in macrophages and NOS3 transcripts were largely confined to EC (Supplementary Fig. S6). Expression of most M1-like (CCL2, IL1B, IL6, TNF, and IL10) and M2-like macrophage markers mapped predominantly to macrophages, whereas other cytokines were expressed more widely (IFNG and TGFB1). VCAM1 and ICAM1 were also expressed by multiple cell types.
HDAd transcripts were identified in 581 cells (2% of the total cells). Surprisingly, most of the cells in which HDAd transcripts were identified (467; 80%) were classified as fibroblasts, while only 5% of cells with HDAd (27 cells) were classified as EC (Fig. 6D, Supplementary Table S3). Previously, we have found that in vivo transduction with HDAd was primarily limited to the EC at 3 days. 38 HDAd transcripts were also found in SMC and macrophages, but not in modulated SMC.
No robust alteration of signaling pathways by HDAdABCA1 by scRNAseq
The small “n” for the scRNAseq analyses likely limits the sensitivity and reliability of comparisons between the Null and ABCA1 groups, particularly with differing atherosclerotic burdens in the two ABCA1 rabbits, but for exploratory purposes we performed differential expression testing within cell-type clusters comparing HDAdABCA1- versus HDAdNull-transduced vessels. There were 5 DEGs in EC (all EC clusters were combined for analysis), 78 DEGs in macrophages, 48 DEGs in SMC1, 1 DEG in SMC2, and 29 DEGs in modSMC (Supplementary Table S4). For all clusters, a sizable portion of the DEG were ribosomal genes (RPS or RPL) or genes without an identified symbol (LOC genes). Unfortunately, with the limited sample size, the top DEGs did not show a clear biological link to atherosclerosis or ABCA1 signaling in any of the cell types. Both overrepresentation analysis and gene set enrichment analysis (GSEA) were used to determine if there were any up- or downregulated gene sets in the cell clusters. Due to the abundance of ribosomal DEGs, overrepresentation analysis primarily identified ribosomal pathways in all cell types (data not shown). GSEA using a higher cutoff false discovery rate of <0.25 for pathway discovery (Supplementary Table S6) identified pathways that also lacked a clear biological significance related to ABCA1 or atherosclerosis signaling. For all scRNAseq data, the small “n” greatly limited the power of the analyses especially with the high variability between rabbits and particularly compared with the main analysis with 26-paired rabbit vessels.
Single-cell analysis on only cells expressing transgenic RNA (Null or ABCA1, across all cell types) revealed 31 upregulated genes and 1 downregulated gene (Supplementary Table S7). The top upregulated gene was ABCA1, which expressed a 4.3-fold higher expression in ABCA1-transduced cells versus Null-transduced cells, lending support that ABCA1 mRNA is still being overexpressed in the transduced cells 24 weeks after transduction.
DISCUSSION
We tested if transduction of the carotid endothelium with HDAdABCA1 would increase endothelial ABCA1 expression and reduce atherosclerosis in hyperlipidemic rabbits. We found that: (1) ABCA1 RNA and protein expression were increased in the endothelium 3 days after treatment; (2) ABCA1 transgene was still expressed after 24 weeks; (3) endothelial ABCA1 overexpression did not significantly reduce lipid and macrophage area, but the intimal/medial area ratio trended lower; (4) endothelial ABCA1 overexpression reduced several markers of macrophages, including both M1 and M2 markers, but did not shift the macrophage phenotype; (5) endothelial ABCA1 overexpression decreased inflammatory cytokine expression, particularly in EC; (6) modulated SMC are abundantly present in larger lesions and may be reduced by endothelial ABCA1 expression; (7) 24 weeks after treatment, transgene is present in many cell types despite past evidence that predominantly EC are transduced in this model. Intriguingly, a many sex differences were also detected in endpoints (Supplementary Table S8), particularly in the polarization of macrophages, that are beyond the scope of this study but warrant investigation.
ABCA1 is highly expressed in many cell types and is both transcriptionally and posttranslationally regulated. 42 This makes confirmation of increased ABCA1 expression important, but also difficult. We initially confirmed overexpression of ABCA1 RNA 3 days after transduction with our HDAdABCA1 by RT-qPCR. However, testing overexpression of ABCA1 protein requires endothelium-specific methods because of the high expression of ABCA1 in other vascular wall cell types. We tried several methods to measure endothelial ABCA1, including IHC, en face IHC, and flow cytometry without success. This was probably caused by inefficient binding of ABCA1 antibodies to the native protein form, as in vitro and ex vivo controls with large ABCA1 increases by Western blot would show minimal or no differences with methods detecting the native protein. We showed increased ABCA1 protein in HDAdABCA1 vessels 3 days following in vivo transduction by mechanically swabbing the endothelium from the luminal surface followed by Western blot on the lysate. Combined with consistent RNA overexpression, this strongly suggests ABCA1 is overexpressed at short time points. However, this method requires most of the transduced vessel to obtain sufficient lysate for quantification making it unviable to measure ABCA1 protein (while also measuring the primary endpoints) in the 24-week atheroprotection study. Even developing a custom antibody to rabbit ABCA1, which greatly improved sensitivity for detecting rabbit ABCA1, and made in vitro flow cytometry successful (Supplementary Fig. S8), did not allow us to detect protein differences in vivo or ex vivo by flow cytometry or other methods we tested that did not require destruction of most of the vessel. We considered a separate 24-week rabbit atherosclerosis experiment solely to measure ABCA1 protein expression at 24 weeks, but concluded that even if we could not detect increased ABCA1 protein, it would only indicate that the increase in ABCA1 protein dropped below our detection limit during the atherosclerosis progression between 3 days and 24 weeks, but would not indicate at when the overexpression was lost. It would still be feasible that early elevation of EC ABCA1 could still cause changes that have lasting effects at 24 weeks. We believe that the many mRNA changes that we see, both in EC-enriched tissue and in the whole-vessel extracts, at 24 weeks support that there are lasting, if not ongoing, effects of transgenic ABCA1 expression. A limitation of this study is that although we can show expression of ABCA1 transgene mRNA and increased ABCA1 RNA in the endothelium and vector-transduced cells at 24 weeks in the atheroprotection study, we cannot confirm that the ABCA1 protein remains elevated at this time. Higher or longer lasting ABCA1 protein expression from the gene therapy could give stronger effects than observed in this study.
Endothelial dysfunction is believed to drive atherosclerosis 20 ; however, therapeutic approaches for targeting endothelial dysfunction are currently lacking. 21 Because lipid accumulation can cause endothelial dysfunction, targeting lipid removal from the vascular endothelium is a desirable target. In vitro overexpression of ABCA1 in EC increases cholesterol efflux out of the cells, leading to a decrease in inflammatory cytokines and adhesion molecules. 23 In our rabbit hyperlipidemic model, we found that endothelial overexpression of ABCA1 decreased endothelial levels of IL6 and TNF but not the adhesion molecules VCAM1 or ICAM1. Previous atherosclerosis mouse studies have shown that endothelial ABCA1 deficiency increases lesion area, 24 while conversely, overexpression of ABCA1 in the endothelium decreases lesion size. 25 Because the mouse study overexpressed ABCA1 in all EC, they saw an increase in system HDL-C that might have been responsible for the atheroprotection they observed. We only targeted a small region of vascular endothelium for ABCA1 overexpression and more importantly used a paired analysis (comparing a treated carotid with a control within the same rabbit) that eliminates systemic factors such as plasma HDL-C as confounding variables. This allows our study to attribute all effects we observed to be due to the local expression of ABCA1 in the vascular endothelium. In our rabbit model, we did not see a significant decrease in the lesion area, although the intimal/medial ratio decrease had p = 0.06. It is possible with improved expression of endothelial ABCA1 that local expression alone would be sufficient to significantly decrease lesion size along with the decreased inflammatory cytokines in the vascular endothelium.
We expected that ABCA1 overexpression in the endothelium could affect subendothelial cells via multiple mechanisms. ABCA1 regulates apo A-I transcytosis across the endothelium into the vascular space. 43 Cytokines and adhesion molecules expressed in an activated endothelium increase monocyte entry into the vascular wall, and cytokines, including IFNG, may polarize macrophages derived from those monocytes toward proinflammatory phenotypes. Thus, we hypothesized that protecting the endothelium would lead to both a reduction in the number of macrophages entering the vascular wall and shift of the polarization away from proinflammatory, M1-like macrophages toward a more resolving, M2-like macrophage phenotype. While we saw a significant 23% reduction in the macrophage marker, CD68, the 14% reduction in CD11b was not significant. It is possible that CD68 is decreased due to a change in its expression from macrophages rather than fewer macrophages. CD68 expression in macrophages can be upregulated due to inflammatory stimuli, 44 so decreased inflammation due to ABCA1 overexpression could also explain the decreased CD68 expression in macrophages. The RAM-11 IHC (–15%) and scRNAseq data (–15%) both showed small, but nonsignificant decreases in macrophages. Expression of both M1 and M2 macrophage-like markers were decreased in the vascular wall, lending further support that the total number of macrophages was likely decreased, but suggesting that the macrophage phenotype was not altered. While a shift in macrophage phenotype has been shown to occur during atheroprotection, 45,46 it often requires extreme changes to promote rapid regression. 45 Additionally, the M1 and M2 macrophage phenotypes as defined in vitro are typically seen as an oversimplification in vivo, where a diverse set of less well-defined and overlapping macrophage polarizations exist 47 at different times and in different parts of the lesion. 48
scRNAseq analysis was pursued to examine cell-specific transcription and provide unbiased discovery of novel signaling pathways. The small “n” limited the power of the scRNAseq analysis, particularly for comparisons between the Null and ABCA1 vessels. Variability in lesion size and the large number of ribosomal genes detected likely further weakened the power of the analysis. We did not consider these limitations to be fatal flaws, as the scRNAseq analysis was undertaken purely in an exploratory capacity and was not part of the original study design. Unfortunately, we did not identify DEGs or regulated pathways that had a clear biological significance related to ABCA1 signaling. Due to the limitations previously mentioned, this should not be interpreted as a lack of effects. The scRNAseq analysis did provide invaluable data regarding cell types present in the rabbit lesions and the cell specificity of transcripts of interest, including the transgenic Null and ABCA1 mRNA.
Using scRNAseq, we detected modulated SMC characterized by a decrease in SMC contractile proteins along with an increase in markers of a synthetic phenotype. This is consistent with other groups 40,41 that have reported proliferation and dedifferentiation of medial SMC into multipotent intermediate cells that transition to multiple cell types. VCAM1, which is expressed primarily in EC and modSMC, indicates that a substantial proportion of these modSMC are near the medial layer (Supplementary Fig. S5B). Both scRNAseq and VCAM1 IHC data showed modulated SMC to be much more abundant in larger lesions, consistent with modulated smooth muscle cells contributing to more advanced stages of atherosclerosis. Modulated smooth muscle cells can continue down a pathway toward protective fibromyocytes or harmful macrophage-like cells. 40,41 Our modSMC cluster had both fibromyocyte markers and macrophage-like cell markers, suggesting the cluster might be an intermediate cell or a mixture of SMC-derived cell types. A strong muscle actin staining (HHF35, Supplementary Fig. S5C) suggestive of a substantial protective fibrous cap is detected but appears to be independent of the modSMC identified by strong VCAM1 expression. The areas containing modSMC, identified by VCAM1 IHC, do stain positively for a rabbit macrophage marker (RAM11, Supplementary Fig. S5D).
As expected, we found ABCA1 transgene in whole-vessel extracts and endothelial-enriched extracts (laser microdissection) 24 weeks after infusion, however, despite HDAd primarily transducing EC, 38 ABCA1 transgene was also increased in macrophage-enriched lesion (laser microdissection)—transgene detected in Null samples is likely due to cross-contamination, possibly due to static on the laser microdissection scope, as it is never detected in Null samples that do not involve laser microdissection. Additionally, scRNAseq showed transgene expression (ABCA1 or Null) in EC and many non-EC cell types, with most being in fibroblasts. While we have previously found transduction primarily in the EC 3 days following transduction in normolipidemic rabbits, 38 there are several possible explanations for why hyperlipidemia and the longer time after transduction may have altered the transgene expression. First, atherosclerosis and the associated endothelial dysfunction have been shown to increase endothelial permeability, which could lead to more HDAd being able to pass through the endothelial barrier and transduce subendothelial cells. Also, endothelial-to-mesenchymal cell transition (EndMT) that is known to occur in atherosclerosis 49 –51 could explain why transgene was detected in multiple non-EC cell types. Dedifferentiated EC can have protective or deleterious effects as the EC transition into multiple cell types, including SMC and fibroblasts, 49 –51 both of which contained transgene expression in the scRNAseq analysis. The lack of transgene in the modSMC cell cluster, which is derived from SMC, further supports that the transgene expression in nonendothelial cell types is due to endothelial transition to other cell types rather than an initial transduction of nonendothelial cells. The detection of transgene in non-EC compared with EC could be further enhanced by the loss of EC both during the experiment and the analysis. EC apoptosis and turnover are known to be increased with proatherosclerotic conditions. 52 Additionally, the low percentage of EC in the scRNAseq analysis (∼2%) compared with histological analysis (∼9%) suggests a potential bias toward EC loss during single-cell processing that may accentuate the appearance of non-EC transgene expression. The loss of EC might also explain the low percentage of total cells that contained transgene in the scRNAseq data, although since we were targeting only the endothelium, which are less than 10% of the vascular wall cells, we would not expect more than ∼4% of cells would contain transgene even with a high transduction efficiency and normal (healthy cell) turnover of EC. Despite this, both here in this study and previously, 3 we have shown that the presence of viral vectors and transgene expression remains elevated at 24 weeks.
As a lipid transporter, ABCA1 therapy could prove particularly useful in conjunction with therapies utilizing apoAI or small HDL particles that promote cholesterol efflux via the ABCA1 transporter, providing a synergistic protective effect. This is especially true given the regulation of ABCA1 expression that occurs in cholesterol-loaded cells and in advanced plaques. Increased cellular cholesterol increases ABCA1 expression via liver X-receptor (LXR)-dependent upregulation. 53 We saw this in our preliminary studies, with ABCA1 expression being higher in the carotids of rabbits on a high-fat diet (Fig. 1B) than those on a normal diet (Supplementary Fig. S1C). In previous therapeutic studies overexpressing apoAI in the vessel wall of hyperlipidemic rabbits, we found that the apoAI-treated vessels, with ∼30% reduction in lesion size and plaque lipid, had a significant decrease in vascular wall ABCA1 expression, 3 perhaps due to the removal of lipid altering LXR-related regulation. This downregulation of ABCA1 likely limits the effectiveness of the therapeutic treatment and is one of the reasons we sought to target ABCA1 overexpression in the vascular wall. Even still in the present study, overexpressing ABCA1 in the endothelium, we saw a decrease in ABCA1 mRNA in the whole vessel wall. Because laser microdissected endothelium showed higher ABCA1 mRNA in vessels with HDAdABCA1, the decrease in the whole vessel is likely due to a downregulation of ABCA1 in nonendothelial cells, perhaps due to a nominal decrease in plaque lipid (Fig. 4D) or maybe due to other compensatory ABCA1 regulatory mechanisms activated by overexpression in the endothelium. Thus, while overexpressing ABCA1 in EC is likely beneficial and should continue to be explored, upregulating ABCA1 throughout the vessel wall should be pursued to counteract the downregulation within other vascular wall cells. Therapy targeting ABCA1 expression in macrophages and SMCs could also be beneficial to treat the downregulation of ABCA1 protein that has been observed in advanced plaques. 54 The potential synergistic effect of ABCA1 overexpression alongside apoAI or small HDL therapy also warrants further exploration, especially in the context of extending ABCA1 overexpression to the lipid-laden smooth muscle cells and macrophages in atherosclerotic plaques.
As with all therapeutic studies, it is unknown if a higher dosage/expression would provide different results. This is particularly relevant in our study because while transgenic mRNA was still elevated at 24 weeks, technical limitations prevented us from testing whether ABCA1 protein remained elevated at the 24-week endpoint, so it may have only had a transient increase. The alteration of inflammatory and macrophage markers that we see by RT-qPCR suggests that ABCA1 expression was sufficient to cause an effect, but it is unknown if this is due to ongoing overexpression of ABCA1 protein or a lasting effect from the early increase in ABCA1 protein, which was shown at 3 days after transduction. Also, while there was a near-significant decrease in the intimal/medial ratio, and nominal decreases in lipid and macrophage markers, this protection could be improved by a higher or longer lasting expression of ABCA1 in the endothelium. We have optimized the viral vectors we use for long-term vascular endothelial expression, but the best vector, used in this study, still has expression that declines significantly after an early peak in our rabbit studies. 55 Increased endothelial cell turnover caused by atherosclerosis could exacerbate the loss of long-term expression. An integrating vector, combined with a better expression cassette, could improve the long-term vascular expression of therapeutic targets, although the size of genes such as ABCA1 presents a challenge for the limited capacity (especially relative to HDAd) of integrating vectors.
In summary, we found that endothelial ABCA1 overexpression provides long-term ABCA1 mRNA expression, but perhaps only a transient increase in ABCA1 protein, which decreases inflammatory cytokines in the endothelium and likely alters the number or expression of macrophages. These subtle changes did not lead to a significant decrease in the lipid or macrophage area within the lesion and only had a small effect on the intimal/medial ratio. Because macrophages and macrophage-like cells derived from SMC are the most lipid-laden cells in the lesion, it may be more atheroprotective to target these cells with ABCA1 overexpression. In the future, we hope to test gene therapy with small RNAs, which can be transported between cells in extracellular vesicles, to alter the expression of ABCA1 in subendothelial plaque cells.
Footnotes
ACKNOWLEDGMENTS
The authors thank Microbix Biosystems, Inc. for providing reagents for helper-dependent adenovirus construction and propagation. We thank the University of Washington Institute for Stem Cell and Regenerative Medicine, of which D.A.D. is a member, for its support of this project.
AUTHORS’ CONTRIBUTIONS
B.K.W.: Data curation, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing—original draft, and writing—review and editing. L.B.: Investigation, methodology, and writing—review and editing. G.S.-P.: Investigation, methodology, and writing—review and editing. N.S.: Data curation, formal analysis, project administration, investigation, methodology, and writing—review and editing. A.Z.R.: Investigation and writing—review and editing. N.S.L.: Investigation and writing—review and editing. L.L.: Investigation, methodology, and writing—review and editing. F.K.: Methodology, writing—review and editing, and project administration. D.A.D.: Conceptualization, methodology, writing—original draft, writing—review and editing, supervision, project administration, and funding acquisition.
DATA AVAILABILITY STATEMENT
The data that support the findings of these studies are available from the authors on reasonable request.
AUTHOR DISCLOSURE STATEMENT
The authors have no conflicts to disclose.
FUNDING INFORMATION
This work was supported by the
SUPPLEMENTARY MATERIAL
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.