
Abstract
Chimeric antigen receptor T cell (CAR-T) therapy has achieved great success and progress for treatment of hematological malignancy, but it still cannot overcome the obstacles in solid tumors. The hostile tumor microenvironment (TME), such as dense extracellular matrix, hypoxia, low pH, and tumor-derived metabolites, largely impedes CAR-T function. Oncolytic virus, as a form of immunotherapy, provides a way to antagonize the TME and improve the efficacy of CAR-T cells in solid tumors. In this study, the chemokine CXCL9 and interleukin 15 (IL15) genes were genetically integrated into adenoviral vector to construct oncolytic adenovirus (OAV) Ad-CXCL9-IL15, which could infect tumor cells to express and secrete CXCL9 and IL15. Ad-CXCL9-IL15 showed potent antitumor activity in xenografted prostate cancer model and augmented the tumor infiltration of CD45+CD3+ T and CD8+ T cells in immunocompetent mice. Moreover, Ad-CXCL9-IL15 treatment decreased Treg cells in tumor mass and increased CD44+CD62L+ T cells in spleen. Indicating that Ad-CXCL9-IL15 modified the TME and augmented antitumor immune responses in vivo. Furthermore, administration of Ad-CXCL9-IL15 dramatically promoted infiltration and survival of B7H3-targeting CAR-T cells, improved the therapeutic efficacy, and prolonged the survival time of prostate cancer-bearing mice. Therefore, cytokine-armored OAV Ad-CXCL9-IL15 could be used as a bioenhancer to modify TME and boost immunotherapy for solid tumors.
INTRODUCTION
In recent years, cancer immunotherapies have demonstrated remarkable clinical benefits in advanced cancers due to their ability to induce durable responses compared with conventional chemotherapy. 1 However, their efficacy remains limited in a significant proportion of patients, largely attributed to the immunosuppressive tumor microenvironment (TME). The TME is not merely a passive bystander but an active orchestrator of immune evasion. It creates physical and biochemical barriers that impair immune cell infiltration, promote T cell exhaustion, and foster tumor immune tolerance. 2,3 These mechanisms collectively dampen the activity of immunotherapies such as immune checkpoint inhibitors (ICIs) or adoptive T cell therapies, particularly in immunologically “cold” tumors characterized by low T cell infiltration and high stromal resistance. 4,5
To overcome TME-mediated immunosuppression, strategies that simultaneously target tumor cells and remodel the TME are critical. Oncolytic viruses (OVs), a class of genetically engineered or naturally selective viruses, have emerged as potent agents to address this dual challenge. 6 OVs selectively infect and lyse tumor cells, releasing tumor-associated antigens (TAAs) and damage-associated molecular patterns, which prime dendritic cells and activate tumor-specific T cells. 7 More importantly, OVs are uniquely equipped to reprogram the immunosuppressive TME. 8 –10 This TME-remodeling capacity positions OVs as ideal partners for combination immunotherapy. Talimogene laherparepvec, an U.S. Food and Drug Administration–approved herpes simplex virus type 1 (HSV-1)-based OV, not only lysed tumor cells but also enhanced intratumoral CD8+ T cell density and PD-L1 expression, synergizing with anti-PD-1 therapy to improve response rates in melanoma. 11 Hence, combination of OVs and immunotherapy is worthy of further exploration.
Chimeric antigen receptor T cell (CAR-T) is proved to be effective for refractory/relapsed B cell leukemia, lymphoma, multiple myeloma, and several autoimmune diseases in clinic. However, the therapeutic effect of CAR-T in treating solid tumors is limited. 12 The high heterogeneity of solid tumors provides them with an effective mechanism to evade CAR-T cells. 13 Solid tumors are often surrounded by dense physical barriers that effectively prevent T cell penetration. In addition to physical barriers, T cells also face highly immunosuppressed TME, which share cellular, molecular, and metabolic features that seriously lead to T cell depletion and dysfunction. So far, CAR-T cells themselves have not been able to overcome these obstacles. 14
As an immunological therapy, OVs change the expression profile of cytokines or chemokines in TME after tumor cells are infected, so that TME is transformed into an environment more conducive to immune cell activation and antitumor immune response is initiated. 15 Therefore, the combination of OVs and CAR-T cells can overcome the obstacle of abnormal function of CAR-T cells in TME and improve the antitumor efficacy. 16,17 Combination therapy with herpes simplex OV G47Δ and CAR-T targeting podoplanin has shown excellent efficacy in clinical glioma and melanoma models, extending overall survival by more than double compared with OVs or CAR-T alone. 18
Studies have shown that the overexpression of CXCL9 in solid tumors is related to CD8+ T cell infiltration. 19 As a CXCR3 ligand expressed by activated immune cells, CXCL9 plays a dual role in antitumor immunity: it not only mediates effector T cell recruitment into the TME but also sustains their intratumoral retention—both processes being critical for immunotherapy efficacy. 20 However, the therapeutic potential of CXCL9 may be limited by insufficient activation and persistence of tumor-infiltrating lymphocytes. IL15 demonstrates unique pharmacokinetic advantages with prolonged serum half-life compared with other cytokines, and its pleiotropic effects directly enhance the feasibility of clinical translation. 21 Mechanistically, IL15 promotes the proliferation and functional maintenance of both CD8+ T cells and natural killer (NK) cells 22 while simultaneously upregulating CXCR3 expression on these effector cells 23,24 —thereby amplifying their responsiveness to CXCL9-mediated chemotaxis. Importantly, emerging clinical evidence suggests that IL15 synergizes with checkpoint inhibitors by reversing T cell exhaustion, 25 which aligns with the reported role of CXCL9 in predicting ICI responsiveness. 19
Studies have shown that B7H3 (CD276) is overexpressed on the surface of many tumors, which is closely related to tumor growth, metastasis, recurrence, and poor prognosis. 26 B7H3 exhibits distinct advantages as a therapeutic target, and the current strategy is mostly to develop CAR-T therapy. 27 However, B7H3-targeted CAR-T therapy remains suboptimal due to the same challenges as usual CAR-T cells in solid tumors: the dense extracellular matrix and the immunosuppressive TME. In this article, we used oncolytic adenovirus (OAV) carrying CXCL9 and IL15 to cooperate with B7H3-CAR-T cells to treat solid tumors, aiming to improve the activity of CAR-T therapy in solid tumors. The OAV Ad-CXCL9-IL15 directly lysed tumor cells, released tumor antigens, and recruited CAR-T cells to the tumor site. Moreover, Ad-CXCL9-IL15 infected tumor cells and mediated the expression of chemokine CXCL9, which caused more CAR-T cells to infiltrate into tumor tissues. At the same time, the secreted IL15 can promote the proliferation and activation of CAR-T cells, releasing interferon-γ (IFN-γ) and other cytokines, improve the tumor immunosuppressive microenvironment, enhance the function of enriched immune cells, and improve the synergistic antitumor effect.
MATERIALS AND METHODS
Cells and viruses
Mouse prostate cancer cell RM-1, human prostate cancer cell Du145, and human embryonic kidney-293 (HEK293) cells were cultured in Dulbecco’s modified Eagle’s medium. Human prostate cancer cell PC-3 and LnCap were cultured in RPMI 1640 medium. All media were supplemented with 10% fetal bovine serum, 100 U/mL penicillin (P), and 100 mg/mL streptomycin (S). Cells were cultured in an incubator containing 5% carbon dioxide at 37°C.
The open reading frames expressing CXCL9 alone and CXCL9 and IL15 simultaneously were cloned into adenovirus vector pZD55 to construct adenovirus shuttle plasmids pZD55-CXCL9 and pZD55-CXCL9-IL15. pZD55-CXCL9 and pZD55-CXCL9-IL15 were co-transfected into HEK293 cells with adenovirus skeleton plasmid pPE3, which is a 5-serotype adenovirus vector, and the OAVs Ad-CXCL9 and Ad-CXCL9-IL15 were reconstituted. After amplification and purification, the titers of Ad-CXCL9 and Ad-CXCL9-IL15 were determined by half tissue culture infective dose method.
Isolation, culture, and transduction of primary T cells
Peripheral blood mononuclear cells were collected from healthy blood donors and activated with human T cell activator CD3/CD28 (Gibco; Life Technologies). T cells were cultured in X-vivo 15 (Lonza) supplemented with 1% human serum albumin, 1% P/S, and 200 U IL-2. The supernatant was changed every other day. After 48 h of activation, T cells were infected with the B7H3-specific CAR retroviral constructed by our laboratory and centrifuged in a centrifuge (1500 g, 2 h). Three days after infection, the percentage of B7H3-CAR-T cells was determined by flow cytometry.
Western blot
Total protein was extracted from the cells. A 10% SDS-PAGE gel was prepared, 100 μg of total protein was loaded, and the protein was transferred to PVDF membrane. After that, the PVDF membrane was incubated in blocking buffer (5% skim milk prepared in TBS with 0.1% Tween 20) for 2 h. Primary antibodies against E1A (Merk Millipore) or GAPDH (Santa Cruz) were added and incubated overnight at 4°C. The samples were washed three times in TBS with 0.1% Tween 20 for 10 min each time, incubated with secondary antibody (Proteintech) for 1 h, and exposed using a Bio-Rad chemiluminescence instrument.
Enzyme-linked immunosorbent assay
Ad-CXCL9 and Ad-CXCL9-IL15 were infected with prostate cancer cells at a multiplicity of infection (MOI) of 20, respectively. After 24 h, the cell medium was changed to serum-free medium, and the supernatant was collected after another 24 h of culture. The expression of CXCL9 and IL15 was detected by enzyme-linked immunosorbent assay (ELISA) kit (Proteintech, KE00102, KE00165) according to the manufacturer method.
Transwell assay
The tumor cells were seeded in six-well plates, and the virus was added to the cells at MOI = 20. After 24 h, the cell medium was changed to serum-free medium, and the culture continued for 24 h, and the supernatant was collected. The 5-μm transwell (Corning Life Science) plates were used to add the collected supernatant to the lower chamber of the transwell, and 100 μL CAR-T cells with a density of 1 × 107/mL were added to the upper chamber. After 1 h, CAR-T cells in the lower chamber were counted.
xCELLigence
Tumor cell killing by OAV or CAR-T cells was measured using the xCELLigence RTCA system instrument (ACEA Biosciences). Tumor cells were seeded in E-plate wells with 10,000 cells per well. After 12 h, the cells were completely adherent, and OAV or CAR-T cells with different MOI or effect-target ratio were added. For experiments with the combination of OAV and CAR-T cells, both were added to the wells at the corresponding time points.
Flow cytometry analysis
To detect the expression of B7H3 in the cell lines used, the cells were incubated with antihuman B7H3 (PE) antibody. PBMC or CAR-T cells obtained from tumor tissue were stained with the following antibodies: human CD45 (APC-Cy7) and human CD3 (APC). Peripheral blood or intratumoral cells of wild-type mice were stained with the following antibodies: mouse CD3 (PerCP, PE), mouse CD8 (APC), mouse CD45 (PE-Cy7), mouse CD4 (PE), mouse CD25 (APC-Cy7), mouse FOXP3 (PE), and mouse NK (AF488NK1.1). Spleen cells were stained: mouse CD44 (APC) and mouse CD62L (PE). All antibodies were purchased from BioLegend. All samples were examined using a BD FACS Canto II flow cytometer. Data were analyzed with FlowJo (Tree Star Inc.).
CFSE-labeled cells
To detect the effect of OAV on the proliferation of B7H3-CAR-T cells, B7H3-CAR-T cells were first labeled with CFSE at a final concentration of 10 μM, and then Ad, Ad-CXCL9, and Ad-CXCL9-IL15 were added to B7H3-CAR-T cells at 50 MOI. B7H3-CAR-T cells were collected at 24 h, 48 h, 72 h, 96 h, and 120 h, respectively, and the cell proliferation was detected by flow cytometry.
RNA extraction, reverse transcription, and real-time quantitative polymerase chain reaction
Total RNA from cells or tumor tissues was extracted with Trizol reagent (Invitrogen), and complementary DNA samples were prepared with HiScript reverse transcriptase kit (Vazyme Biotech). Three duplicate wells of each sample were set as template, and specific primers were used. The data were calculated by 2-δδCt method to calculate the fold change of target gene expression.
Animal experiments
All animal experiments were performed according to the protocol approved by the Animal Ethics Committee of Xuzhou Medical University. The experiment was divided into 8 groups with 10 mice in each group. Human prostate cancer cells PC-3 (5 × 105 cells per mouse) were injected subcutaneously into the right back of male NCG mice (4 weeks old, purchased from Jiangsu Jicui Pharmaceutical Biotechnology Co., Ltd.), and tumor growth and mouse body weight were monitored twice a week. Tumor volume was calculated as volume = length × width 2 × 0.5. When the tumor volume reached about 50–100 mm3, the OAV Ad, Ad-CXCL9, and Ad-CXCL9-IL15 were injected into the tumor, once every other day, for a total of three times. Each mouse was injected with a total of 1 × 109 plaque-forming units (pfu) of virus, and the same volume of normal saline was injected into the tumor of the control mice. For the combination treatment group or the single CAR-T cell treatment group, B7H3-CAR-T cells (2 × 106 cells/mouse) were injected into the mice via tail vein on the second day after the second OAV treatment. Blood samples were collected from the tail vein on days 7, 14, or 21 after CAR-T cell infusion to detect the content of B7H3-CAR-T cells in the peripheral blood of mice, and blood samples were collected from three mice in each group.
To detect the content of B7H3-CAR-T cells in tumor tissue, one mouse in each group was sacrificed 7 days and 14 days after intravenous injection of B7H3-CAR-T cells, and the tumor tissue was taken to make single cell suspension to detection CAR-T cell content. The tumor tissue was also used to extract RNA and detect the expression of cytokines in the tumor. Mice with tumors volume larger than 2000 mm3 were euthanized. To test the effect of OAV Ad-CXCL9-IL15 on tumor growth of tumor-bearing immunocompetent mice, a subcutaneous tumor model of RM-1 C57BL/6 mice (purchased from Jiangsu Jicui Pharmaceutical Biotechnology Co., Ltd., male, 5 weeks old) was constructed. When the tumor volume reached about 50–100 mm3. The OAV Ad, Ad-CXCL9, and Ad-CXCL9-IL15 were injected into the tumor once every other day for a total of three times. Each mouse was injected with 1 × 109 pfu of virus, and the same volume of normal saline was injected into the tumor of the control mice. Changes in tumor volume and immune cells in vivo were monitored regularly.
Liver and kidney function tests
The mouse blood of each treatment group was collected from the eyeball, and the supernatant was centrifuged at 4°C, 3000 RPM for 15 min after blood collection, and the parameters of liver and kidney function (alanine aminotransferase [ALT], aspartate aminotransferase [AST], creatinine [CRE] and urea nitrogen [BUN]) were measured by an automatic biochemical analyzer.
Immunohistochemical staining analysis
Tumor tissue was fixed in 10% formalin, embedded in paraffin, and cut into sections with a thickness of 3 μm. Tumor tissue sections were incubated with antihuman CD3 antibody and stained with hematoxylin.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8. Data are presented as mean ± standard deviation. Statistical comparisons between groups were performed using t-tests to calculate p-values. Survival curves were analyzed by Kaplan–Meier analysis, and p < 0.05 was considered statistically significant.
RESULTS
OAV Ad-CXCL9-IL15 can effectively infect prostate cancer cells
The genes of chemokine CXCL9 and cytokine IL15 were cloned into the adenoviral vector pZD55 with deletion of E1B55KD, named Ad-CXCL9-IL15 (Ad5-ZD55-CXCL9-IL15). The control adenoviruses Ad (Ad5-ZD55) and Ad-CXCL9 (Ad5-ZD55-CXCL9) were also constructed. OAVs Ad (Ad5-ZD55), Ad-CXCL9, and Ad-CXCL9-IL15 infected human prostate cancer cells PC-3, Du145, and LnCap at MOI = 20, respectively. Quantitative polymerase chain reaction (Q-PCR) results showed that the expression of adenovirus E1A and Hexon could be detected in these cells but there was no expression in the blank cells (Fig. 1A). Western blot results showed that E1A protein was effectively expressed in tumor cells infected with different Ads, while E1A protein was not detected in nonreplicating adenovirus Ad-CA13 and blank cells, which was consistent with the messenger RNA expression level (Fig. 1B). ELISA results showed that compared with the corresponding control group, high level of CXCL9 was produced and efficiently released from Ad-CXCL9 and Ad-CXCL9-IL15–infected prostate cancer cells (Fig. 1C). Ad-CXCL9-IL15–infected prostate cancer cells could also effectively express IL15 (Fig. 1D). These results indicate that the OAV Ad-CXCL9-IL15 can effectively infect prostate cancer cells and mediate the expression of corresponding factors.
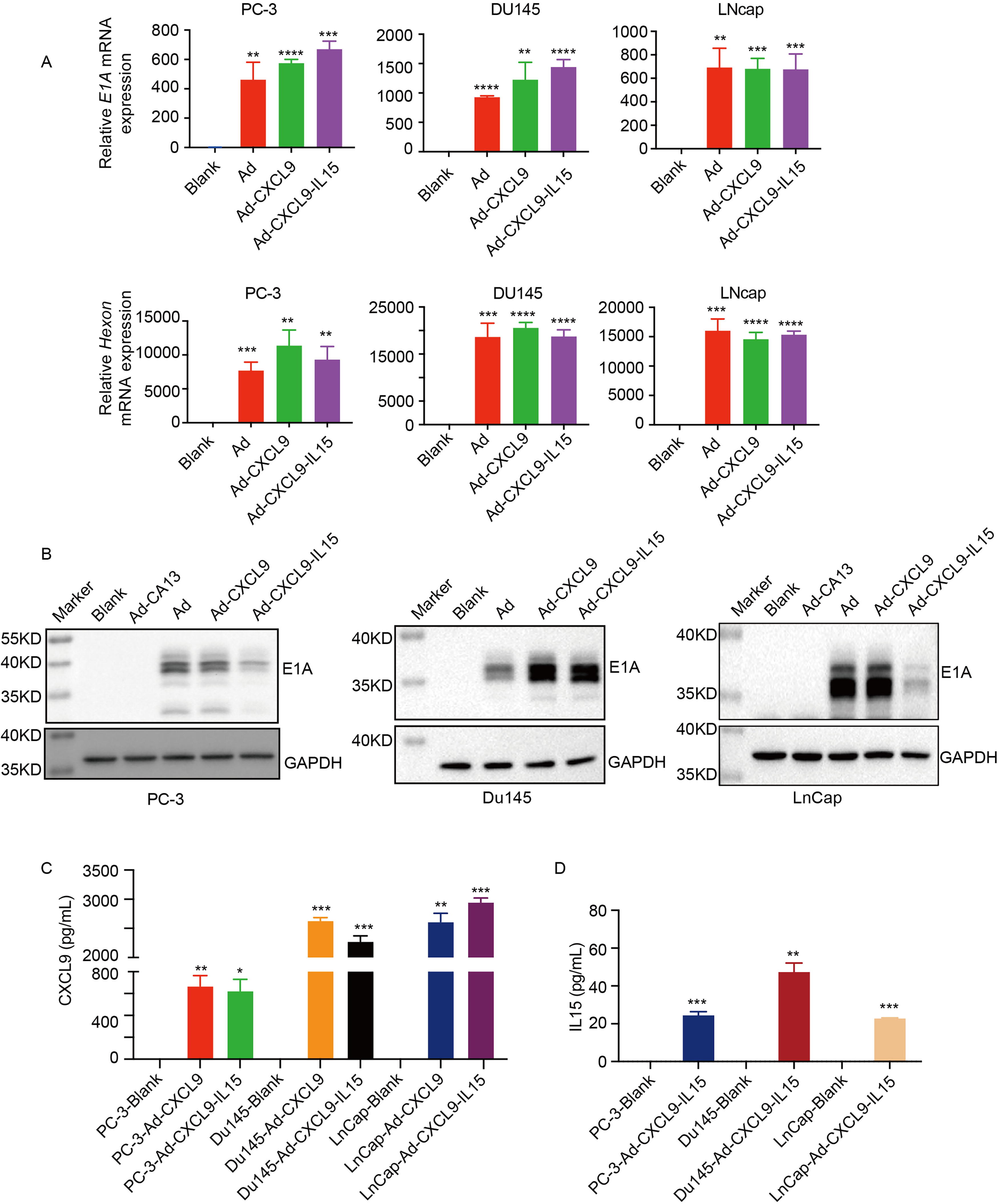
Oncolytic adenovirus Ad-CXCL9-IL15 infected human prostate cancer cells efficiently.
OAV Ad-CXCL9-IL15 killed prostate cancer cells
Real-time cell analyzer (RTCA) was used to detect the killing effect of oncolytic adenovirus Ad, Ad-CXCL9, and Ad-CXCL9-IL15 on prostate cancer. The results showed that compared with blank cells, Ad, Ad-CXCL9, and Ad-CXCL9-IL15 had killing effect on prostate cancer cells at MOI = 20. With the extension of action time, the killing effect was enhanced (Fig. 2A). To test the function of the factors expressed after tumor cells were infected with OAV, the supernatants of PC-3 cells were collected after infection with Ad, Ad-CXCL9, and Ad-CXCL9-IL15, and the chemotactic effect of different cell supernatants on T cells was detected by transwell. The supernatant from PC-3 cells infected with Ad-CXCL9 and Ad-CXCL9-IL15 could attract more T cells to the lower chamber, which was similar to the positive control group with recombinant CXCL9, while the supernatant of empty virus Ad group could not attract T cells (Fig. 2B). Compared with the control group, the supernatant from Ad-CXCL9-IL15 treatment group promoted the proliferation of T cells (Fig. 2C) and induced T cells to express higher levels of IFN-γ (Fig. 2D). Western blot results showed that T cells expressed higher levels of p-STAT5 protein after co-culturing with the cell supernatant of PC-3 cells infected with Ad-CXCL9-IL15 (Fig. 2E). These results indicate that Ad-CXCL9-IL15 can effectively kill prostate cancer cells, and the secreted CXCL9 and IL15 factors are biologically active.

Oncolytic adenovirus Ad-CXCL9-IL15 inhibited human prostate cancer cells growth in vitro.
Ad-CXCL9-IL15 inhibited the growth of subcutaneous transplanted mouse prostate cancer RM-1
To detect the effect of OAV Ad-CXCL9-IL15 on immune response in TME, immunocompetent mice were used for the experiment. Human adenovirus has been reported to be weak in replication in most mouse cells; therefore, the infection and replication of Ad-CXCL9-IL15 in the mouse prostate cancer cell line RM-1 were first determined. Q-PCR results showed that compared with blank cells, OAV Ad, Ad-CXCL9, and Ad-CXCL9-IL15 were able to express adenovirus E1A and Hexon after infecting RM-1 cells (Fig. 3A). ELISA results showed that Ad-CXCL9-IL15 produced CXCL9 and IL15 in infected RM-1 cells (Fig. 3B). Western blot results showed that E1A protein was effectively expressed in tumor cells infected with Ad, Ad-CXCL9, and Ad-CXCL9-IL15, while E1A protein was not detected in nonreplicating adenovirus Ad-CA13 and blank cells (Fig. 3C). RTCA assay showed that Ad-CXCL9-IL15 inhibited the growth of prostate cancer cell line RM-1 (Fig. 3D). The Ad-CXCL9-IL15 could act on mouse prostate cancer cell in vitro. In the in vivo experiment, the wild-type C57BL/6 mouse tumor bearing model was established by mouse prostate cancer cell RM-1. The mice were divided into 5 groups with 10 mice in each group. A total of 1 × 109 pfu of Ad-CA13, Ad, Ad-CXCL9, and Ad-CXCL9-IL15 were injected into the tumor, and the same volume of normal saline was injected as a control. The results showed that intratumoral injection of Ad-CXCL9 had a better tumor inhibition effect than PBS group, nonproliferative adenovirus Ad-CA13 group, or Ad group, and Ad-CXCL9-IL15 had the best antitumor activity (Fig. 3E–G). The tumor growth inhibition rate of RM-1 prostate cancer model was 63.52% (Fig. 3H). All groups of Ad, Ad-CXCL9, and Ad-CXCL9-IL15 prolonged the survival time of mice (Fig. 3I). The serum of mice was collected to detect the liver function indexes ALT, AST, and the kidney function indexes CRE, and BUN. As shown in Figure 3J, compared with the control group, all the OAVs treatment had no significant influence on the liver and kidneys of the mice. The above results suggested the used OAVs had a good safety in vivo.

Ad-CXCL9-IL15 inhibited subcutaneous xenograft tumor growth of murine prostate cancer RM-1.
OAV Ad-CXCL9-IL15 improved tumor immune microenvironment
On days 7 and 14 after OV injection, 3 mice in each group were selected for tail venous blood, and the proportions of CD45+CD3+ immune cells, CD8+ T cells, and CD4+ T cells in peripheral blood were detected by flow cytometry. The percentages of CD45+CD3+ T cells and CD8+ T cells in the peripheral blood of Ad-CXCL9 and Ad-CXCL9-IL15 treated mice were higher than those of other experimental groups. The percentages of CD45+CD3+ T cells and CD8+ T cells in the peripheral blood of Ad-CXCL9-IL15 group were higher than those of Ad-CXCL9 group (Fig. 4A–D). However, there was no significant difference in the proportion of CD4+ T cells among the groups (Fig. 4E).

Detection of immune cells in peripheral blood of RM-1 tumor-bearing mice.
On day 7 after treatment with OAV, two mice in each group were euthanized, and genomic DNA and protein were extracted from tumor tissue. As shown in Figure 5A, the expression of adenovirus E1A and Hexon could be detected in Ad, Ad-CXCL9, and Ad-CXCL9-IL15 groups. ELISA results showed that both CXCL9 and IL15 were efficiently secreted in the corresponding OAV treated group (Fig. 5B). Flow cytometry was used to detect the proportion of infiltrating immune cells in the tumor tissue of different groups. The results showed that the infiltration of CD45+CD3+ T cells and CD8+ T cells in the tumor increased after the injection of Ad-CXCL9-IL15. However, there was no significant difference in the proportion of CD4+ T cells among the groups. The absolute numbers of infiltrating CD45+CD3+ T cells and CD8+ T cells in the tumor were also increased by adding counting beads. Compared with Ad-CA13 treatment group, Ad-CXCL9-IL15 treatment group had the largest number of infiltrating immune cells, and the difference was statistically significant. Compared with the Ad-CXCL9 group, the infiltration of CD45+CD3+ T cells and CD8+ T cells in the Ad-CXCL9-IL15 treatment group was also increased, and the difference was statistically significant (Fig. 5C–E).

Detection of immune cells in tumor tissues of RM-1 tumor-bearing mice.
In addition, an increased percentage of intratumoral NK cells was detected, although there was no significant difference between Ad-CXCL9-IL15 and Ad groups (Fig. 6A). At the same time, the results showed that the number of infiltrating regulatory T cells (Treg) in the Ad-CXCL9-IL15 treatment group was significantly decreased compared with the other treatment groups (Fig. 6B), and the proportion of memory T cells in the spleen of the Ad-CXCL9-IL15 treatment group was increased compared with the other treatment groups (Fig. 6C). These results indicate that Ad-CXCL9-IL15 can infect tumor cells, express CXCL9 and IL15, improve tumor immunosuppressive microenvironment, induce antitumor immune response, and improve tumor treatment effect.

Regulation of the tumor microenvironment by Ad-CXCL9-IL15.
OAV Ad-CXCL9-IL15 combined with B7H3-CAR-T enhanced the killing effect on prostate cancer cells
In the wild-type mice, we verified that Ad-CXCL9-IL15 could activate the antitumor immune response. To further study the promotion effect of Ad-CXCL9-IL15 on CAR-T immune cell therapy, the experiment of Ad-CXCL9-IL15 combined with B7H3-CAR-T cell therapy for prostate cancer was designed. B7H3 is highly expressed in a variety of cancer cells and is closely related to cancer metastasis, recurrence, postoperative cancer progression, and death.[16] First, the expression level of B7H3 in prostate cancer cells was detected by flow cytometry, and the results showed that B7H3 was highly expressed in human prostate cancer cells PC-3 and Du145 (Fig. 7A). In this experiment, a retrovirus targeting B7H3 was constructed, including a single chain variable fragment targeting B7H3 CAR, the transmembrane region of CD8a and the intracellular signaling domain of CD28 and CD3ζ (Fig. 7B). To test the killing effect of B7H3-CAR-T cells on prostate cancer, the effect–target ratios of B7H3-CAR-T cells to prostate cancer cells were 1:10, 1:5, 1:1, and 5:1. RTCA results showed that with the increase of the effect–target ratio, the killing effect of B7H3-CAR-T on PC-3 and Du145 cells was more significant (Fig. 7C).
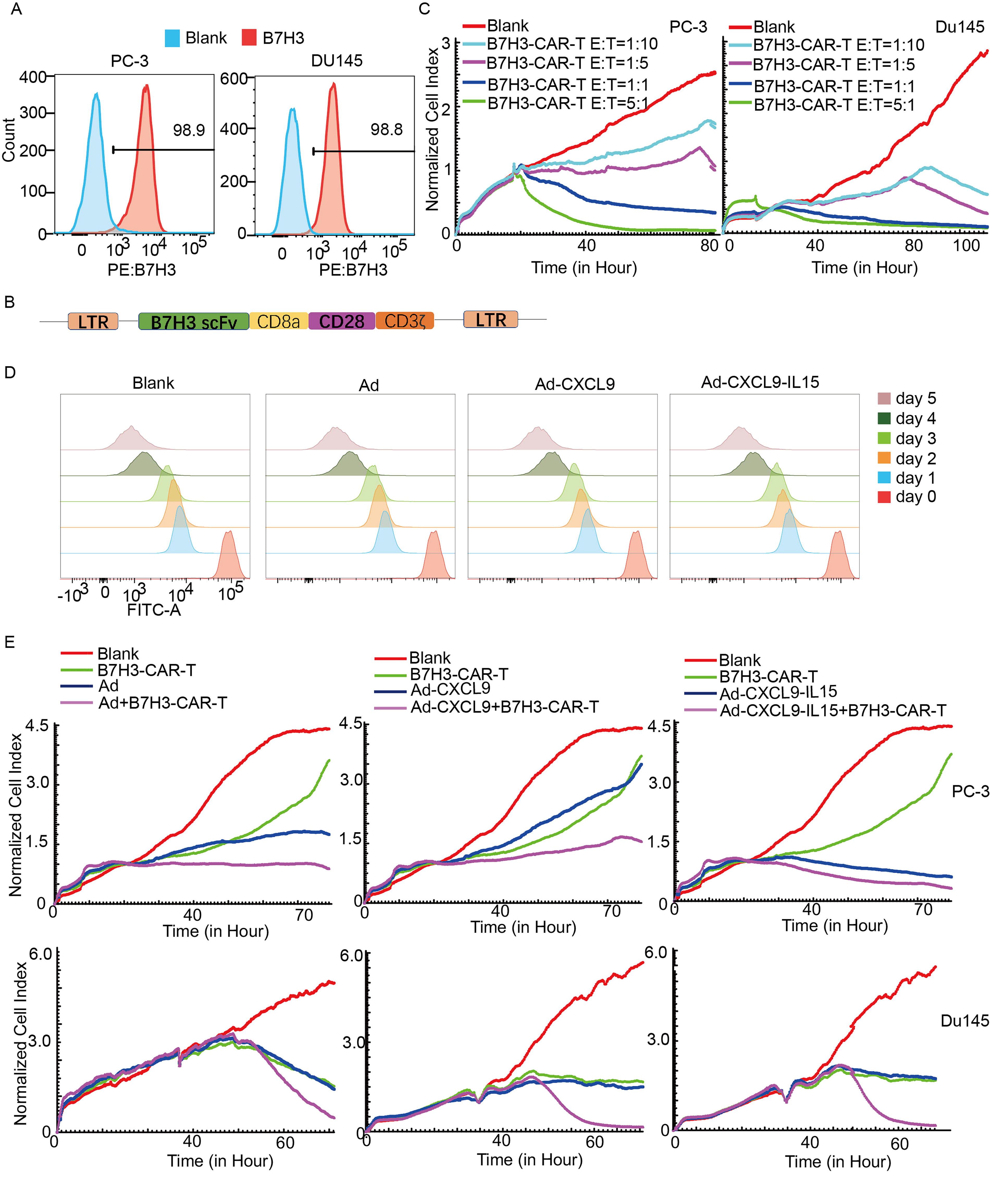
Oncolytic adenovirus Ad-CXCL9-IL15 combined with B7H3-CAR-T cells inhibited human prostate cancer cells growth in vitro.
To detect the antitumor effect of OAV combined with B7H3-CAR-T cells in vitro, the effect of each OAV on the proliferation of B7H3-CAR-T cells in vitro was first detected. CFSE-labeled B7H3-CAR-T cells were incubated with Ad, Ad-CXCL9, and Ad-CXCL9-IL15 at a high MOI (50) for 5 days. The proliferation of CAR-T cells was measured daily by flow cytometry. As shown in (Fig. 7D), the used Ads had no effect on the proliferation of B7H3-CAR-T cells, and the OAV combined with B7H3-CAR-T cells was feasible. PC-3 and Du145 cells were infected with Ad, Ad-CXCL9, and Ad-CXCL9-IL15 at MOI = 20, and then B7H3-CAR-T cells with low target ratio (E:T = 1:10, E T, effect–target ratios) were added. The results of RTCA assay showed that Ad-CXCL9-IL15 or B7H3-CAR-T alone could effectively inhibit the growth of prostate cancer cells compared with the control group, and the inhibitory effect of the combination of the two was enhanced with the extension of incubation time (Fig. 7E). These results indicate that Ad-CXCL9-IL15 combined with B7H3-CAR-T has a more significant killing effect on prostate cancer cells.
OAV Ad-CXCL9-IL15 and B7H3-CAR-T combination therapy had better antitumor effect than single therapy in vivo
To test the antitumor effects of Ad-CXCL9-IL15 and B7H3-CAR-T in vivo, a subcutaneous transplantation model of NCG mice was established using human prostate cancer PC-3 cells. When the tumor diameter grew to about 5 mm, NCG mice were randomly divided into eight groups: PBS, Ad, Ad-CXCL9, Ad-CXCL9-IL15, B7H3-CAR-T, Ad+B7H3-CAR-T, Ad-CXCL9+B7H3-CAR-T, and Ad-CXCL9-IL15+B7H3-CAR-T combined group (n = 7 each). OV was injected intraperitoneally once every other day for a total of three times, with a total dose of 1 × 109 pfu of virus per mouse. One day after the third injection of virus, 2 × 106 B7H3-CAR-T cells were infused into the tail vein, and the treatment regimen was as shown in Figure 8A. Tumor volume and body weight were measured regularly. The results showed that the tumor inhibition effect of Ad+B7H3-CAR-T, Ad-CXCL9+B7H3-CAR-T, and Ad-CXCL9-IL15+B7H3-CAR-T combined treatment group was better than that of the corresponding single treatment group. Among them, the Ad-CXCL9-IL15+B7H3-CAR-T group had the most significant antitumor effect, and the tumor volume was significantly reduced compared with the PBS control group (the tumor inhibition rate reached 70.84%) (Fig. 8B–D). In addition, the Ad-CXCL9-IL15+B7H3-CAR-T combination group significantly prolonged the survival of mice (Fig. 8E). During the experiment, 7 days after B7H3-CAR-T cell infusion, one mouse in each group was euthanized, and the tumor tissue was taken for genomic DNA extraction. The expression of adenovirus gene E1A and Hexon in the tumor was detected by Q-PCR. E1A and Hexon were expressed in tumor tissues in both the single-virus group and the combined treatment group (Fig. 8F).

Oncolytic adenovirus Ad-CXCL9-IL15 combined with B7H3-CAR-T cells inhibited human prostate cancer PC-3 cells growth in vivo.
ELISA was used to detect the expression of CXCL9 and IL15 in the tumor. The tumors of Ad-CXCL9, Ad-CXCL9-IL15, Ad-CXCL9+B7H3-CAR-T, and Ad-CXCL9-IL15+B7H3-CAR-T group expressed high levels of CXCL9. IL15 was expressed in Ad-CXCL9-IL15 and Ad-CXCL9-IL15+B7H3-CAR-T group (Fig. 8G). These results indicate that the OV Ad-CXCL9-IL15 can infect tumors and mediate the expression of target genes after intratumoral injection, and Ad-CXCL9-IL15 combined with B7H3-CAR-T cells has a better tumor inhibition effect than the mono therapy in vivo.
OAV Ad-CXCL9-IL15 effectively promoted the invasion and survival of B7H3-CAR T cells in tumors
To understand the potential mechanism of Ad-CXCL9-IL15 combined with B7H3-CAR-T cells to enhance antitumor activity, mice venous blood was collected on days 7, 14, and 21 after B7H3-CAR-T cell infusion, and CD3-APC and CD45-APC-Cy7 antibodies were labeled. The survival of B7H3-CAR-T cells in peripheral blood of mice was detected by flow cytometry. The results showed that the percentages of C45+CD3+ cells in peripheral blood of mice in B7H3-CAR-T, Ad+B7H3-CAR-T, Ad-CXCL9+B7H3-CAR-T, and Ad-CXCL9-IL15+B7H3-CAR-T groups on day 7 were 0.5%, 0.71%, 0.72%, and 1.11%, respectively (Fig. 9A). The results of counting beads showed that the number of CAR-T cells in the peripheral blood of Ad-CXCL9-IL15+B7H3-CAR-T group was the highest (706 cells/100 μL), which was statistically significant compared with that of B7H3-CAR-T group (124 cells/100 μL). However, there was no significant difference between Ad-CXCL9-IL15+B7H3-CAR-T and Ad-CXCL9+B7H3-CAR-T groups (day 7) (Fig. 9B). On day 14, the proportion of C45+CD3+ positive cells in the peripheral blood of mice increased, the most significant was in the Ad-CXCL9-IL15+CAIX-CAR-T group (4.76%). The number of Ad-CXCL9-IL15+B7H3-CAR-T cells infiltrated was the highest (1119 cells/100 μL), which was statistically significant compared with the control B7H3-CAR-T group (147.5 cells/100 μL). At the same time, the difference between Ad-CXCL9-IL15+B7H3-CAR-T and Ad-CXCL9+B7H3-CAR-T groups was statistically significant (day 14) (Fig. 9B). On day 21, the number of CAR-T cells in the combined groups decreased significantly, and the number of Ad-CXCL9-IL15+B7H3-CAR-T group (0.28%) was still higher than that of B7H3-CAR-T (0.025%). In addition, on the 7th and 14th day after CAR-T infusion, one mouse in each group was selected for euthanasia, and the tumor tissue was taken and ground. The content of CAR-T cells in the tumor was analyzed by flow cytometry, and the number of infiltrating CAR-T cells in the tumor was counted by counting beads. The results showed that the proportion and number of CAR-T cells in Ad-CXCL9+B7H3-CAR-T group were higher than those in PBS, B7H3-CAR-T, and Ad+B7H3-CAR-T groups. However, the Ad-CXCL9-IL15 combined with B7H3-CAR-T group had the highest proportion and number of CAR-T cells in tumor tissue, which was consistent with the proportion of CAR-T cells in peripheral blood (Fig. 9C and D). On day 39 after the experiment, the number of infiltrating CAR-T cells in the Ad-CXCL9-IL15+B7H3-CAR-T group was the highest (1.54 × 106 cells/100 mg), which was significantly higher than that in the B7H3-CAR-T group (26,509.7 cells/100 mg). At the same time, the difference between Ad-CXCL9-IL15+B7H3-CAR-T and Ad-CXCL9+B7H3-CAR-T groups was also statistically significant (Fig. 9D). Immunohistochemical results of tumor tissues showed that CD3 positive staining in tumor tissues of Ad-CXCL9-IL15+B7H3-CAR-T group was significantly increased compared with other groups (Fig. 9E). These results suggest that Ad-CXCL9-IL15 enhances the tumor therapeutic effect by enhancing the invasion and survival of B7H3-CAR-T in tumor tissues.

Ad-CXCL9-IL15 promotes B7H3-CAR-T cell infiltration in tumors.
DISCUSSION
The TME affects the infiltration and survival of effector immune cells, assists the immune escape of tumor cells. 28 Breaking the immunosuppressive microenvironment of tumor is conducive to improve the efficacy of immunotherapy. 29 OVs have also made significant progress in cancer treatment. 30 As a new adjuvant for immunotherapy, they provide a new treatment method for improving the efficacy of patients despite many challenges. 31 –33 CAR-T cell therapy has achieved remarkable efficacy in the treatment of hematological tumors. However, in solid tumors, there are problems such as poor invasion ability, short survival time, and dysfunction. To overcome these obstacles, OVs can be combined with immune cell therapy for antitumor studies, and the use of engineered OVs carrying therapeutic genes can further improve the therapeutic effect. 34,35
OVs as one promising antitumor method play an antitumor role through a variety of mechanisms. 36 Its selective lysis of tumor cells releases TAAs, triggers tumor-specific immune responses, and prevents tumor escape caused by antigen loss or antigen heterogeneity. 37 OVs can also release danger signals through immunogenic cell death, reverse tumor immunosuppression, and enable immune cells to proliferate and activate in TME. 38 OVs carry therapeutic genes that promote immune cell function, which can further enhance the effector capacity of immune cells. 39,40 The antitumor mechanism of OVs promotes the function of CAR-T cells to a certain extent, which greatly improves the antitumor effect of the combination of the two.
In this study, the OAV Ad-CXCL9-IL15 carrying chemokine CXCL9 and cytokine IL15 was constructed. The results showed that Ad-CXCL9-IL15 could infect prostate cancer cells and replicate in vitro, express adenoviral genes E1A and Hexon, and kill tumor cells. It could produce and secrete functional factors CXCL9 and IL15. The constructed CAR-T cells targeting B7H3 effectively killed prostate cancer cells in vitro. The antitumor effect of Ad-CXCL9-IL15 combined with B7H3-CAR-T cells was further verified in a subcutaneous tumor model of prostate cancer. The data showed that the combined treatment of Ad-CXCL9-IL15 and B7H3-CAR-T cells had a significant tumor growth inhibition effect than single treatment and prolonged the survival time of mice (Fig. 8). Ad-CXCL9-IL15 could enhance the infiltration and survival of B7H3-CAR-T cells in tumor mass, which suggested that CXCL9 and IL15 expressed by Ad-CXCL9-IL15 infection had a positive effect on B7H3-CAR-T cells promotion (Fig. 9). The efficiency of IL15 arming OV was also verified in the combination with EGFR-CAR NK cells in glioblastoma. 41
Oncolytic virotherapy was elucidated to exert the anticancer effects by multifaceted mechanisms. 42 To explore the mechanism by which Ad-CXCL9-IL15 enhanced the antitumor immune effect, a C57BL/6 immunocompetent mouse model of prostate cancer cell line RM-1 was established. The OAV Ad-CXCL9-IL15 infected tumors and expressed CXCL9 and IL15 in tumor tissues (Fig. 5). The proportion of CD8+ T cells and NK cells in tumor tissues increased, while the proportion of Treg cells decreased (Figs. 4–6), which improved the immunosuppressive microenvironment in tumor tissues. This might be related to Ad-CXCL9-IL15 mediating IL15 secretion. However, a separate OV expressing IL15 alone (Ad-IL15) was not constructed as a control group, as it would provide more direct mechanistic insights into the synergistic efficacy between IL15 and CXCL9. We fully acknowledge the scientific rigor of this approach and prioritize the construction and validation of Ad-IL15 in subsequent studies.
The OAV Ad-CXCL9-IL15 or B7H3-CAR-T cells alone treatment produced tumor inhibition but played a limited antitumor effect on prostate cancer in vivo. When combined with B7H3-CAR-T cells, Ad-CXCL9-IL15 acted as an immunotherapy adjuvant or booster to activate innate and specific immune responses and recruit immune cells (including CAR-T cells) to the tumor site by regulating the tumor immunosuppressive microenvironment. It has been pointed out that local intratumoral injection of OVs can cause distant effects, and distal untreated tumors can also be affected by antitumor effects mediated by immune responses. 43 –45 In this study, bilateral tumor model or metastatic tumor model should be established to further explore the distal effect of Ad-CXCL9-IL15. Moreover, here, we used cell lines and mouse models to test the antitumor activity of Ad-CXCL9-IL15 combined with B7H3-CAR-T. Due to lack of Extracellular matrix (ECM) deposition and different characteristics from parental tumors, these models might fail to recapitulate the highly reliable interactions between the tumor and its microenvironment. The continuously developing in vitro tumor models may use to be a good choice in prostate cancer. 46
In conclusion, the OAV Ad-CXCL9-IL15 carrying chemokine CXCL9 and cytokine IL15 constructed in this study can not only directly lyze tumor cells but also enhance the activity of CAR-T cells targeting B7H3 and activated the antitumor immune response. It modified TME through promoting the infiltration of CD8+ T cells and NK cells and decreasing immunosuppressive cells (Treg) in xenografted tumor in immunocompetent mice. It provided a new strategy for the treatment of solid tumors.
Footnotes
AUTHORS’ CONTRIBUTIONS
L.F.: Writing—original draft, visualization, validation, investigation, formal analysis, data curation, and methodology. X.W.: Writing—original draft, visualization, validation, formal analysis, and data curation. Y.Z.: Visualization and investigation. C.Z.: Writing—original draft, investigation, and methodology. X.L.: Writing—review and editing, visualization, and validation. W.L.: Writing—review and editing and formal analysis. Y.Z.: Writing—original draft, visualization, and investigation. N.S.: Data curation and software. J.Z.: Supervision, project administration, and funding acquisition. G.W.: Writing—review and editing, supervision, and project administration. All authors have read the journal’s authorship agreement, and the article has been reviewed and approved by all named authors.
AUTHOR DISCLOSURE
All authors have read the journal’s policy on disclosure of potential conflicts of interest. The authors declare no competing conflicts of interest.
FUNDING INFORMATION
This work was supported by grants from the National Key Research and Development Plan (2018YFA0900900), the National Natural Science Foundation of China (Grant No. 82273334), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190986), Social Development Key Project of Jiangsu Province (Grant Nos. BE2020641 and BE2020640), the Key R&D Program of Xuzhou (Grant No. KC20008), and the Qing Lan Project of Jiangsu Province.