
Abstract
Patients with HIV-1 immune-related thrombocytopenia (HIV-1-ITP) have a unique antibody (Ab) against platelet GPIIIa49-66, which is capable of inducing oxidative platelet fragmentation in the absence of complement activation. By screening a human phage antibody library with the GPIIIa49-66 peptide as bait, we have developed several humanized phage Abs, which act similarly to the parental Ab. However, the presence of a stop codon in the heavy chain of the obtained phage clones limits their expression in soluble recombinant form. To circumvent this problem, we mutated the stop codon inside clone 11 that exhibits the highest binding activity to platelet GPIIIa49-66, resulting in a soluble scFv format (named A11) in Escherichia coli Rosseta. In in vitro binding assay, A11 exhibited similar binding specificity to parental Ab at various concentrations. Moreover, A11 is able to induce oxidative platelet fragmentation by preferentially binding to activated versus resting platelets. These findings provide a proof-of-principle for the development of a novel approach to inhibit arterial thrombosis by generating a selective scFv for the lysis of platelet-rich thrombi.
Introduction
By screening a human phage display library with the GPIIIa49-66 peptide as bait, we developed several phage-derived Abs against GPIIIa49-66 that exhibit comparable antigen-binding capacity.(5) In the present study, we have developed a scFv format (named A11) based on a previously reported phage antibody (clone 11). We demonstrate that 1. A11 could induce comparable oxidative platelet fragmentation to their parental anti-GPIIIa49-66 Ab. 2. It preferentially binds to activated platelets versus resting platelets, as well as induces activated platelet fragmentation at a low A11 concentration.
Materials and Methods
Materials
All reagents were obtained from Sigma (St. Louis, MO) unless otherwise designated. Plasmid pET-29a was from Novagen (Madison, WI). Peptides were synthesized by Bio-Synthesis (Lewisville, TX). GPIIIa−/− mice with C57/BL6 background and C57/BL6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME).
Generation of therapeutic scFv-A11 format
The Tomlinson J phage Library (MRC Gene Service, Cambridge, United Kingdom) was used to screen against a biotin-conjugated GPIIIa49-66 peptide. Specific clones enriched for anti-GPIIIa49-66 Abs were generated and clone 11 was selected for the highest binding activity, as described previously.(6) The stop codon in the heavy chain of clone 11 was mutated by a site-directed mutagenesis kit as described in the manual. After mutation, the cDNA of pIT2-A11 phagemid was used as a template for PCR with the sense primer 5′-CCATGGCCGAGGTGCAGCTG-3′ and the antisense primer 5′-CGGGCCGCACTCTTTGGTCC-3′. The restriction site NocI was introduced by the sense primer and the restriction site NotI was introduced by the antisense primer. The resulting 726-bp NocI-NotI fragment was digested by NocI and NotI and inserted into pET-29a to generate pET29a-A11. The ligated plasmid pET29a-A11 was then transformed into E. coli BL21(DE3) PlysS cells. Cells transforming with the pET29a-A11 expression vector were cultured in 1 L 2YT medium containing carbenicillin (50 μg/mL) and chloramphenicol (34 μg/mL) with shaking at 37°C until the optical density (600 nm) was 0.4−0.6. Production was induced by the addition of 1 mM IPTG, and the cells incubated at 37°C for 4 h with shaking. Refolding and purification of the scFv Ab were performed at 4°C as previously described.(6)
ELISA assay
For the binding activity assay, plastic microtiter plates (Corning, Lowell, MA) were coated with GPIIIa49-66 peptide (20 μg/mL) in 50 mM sodium bicarbonate buffer (pH 9.6) or washed platelets (1×106/well) in phosphate-buffered saline (PBS), and then blocked with blocking buffer (3% bovine serum albumin [BSA] in PBS with 0.1% Tween-20) for 2 h at room temperature (RT). Soluble A11 with histidine tag was added to the plates and incubated for 1 h at RT. A11 was detected with primary mouse anti-His tag antibodies followed by secondary goat anti-mouse horseradish peroxidase-conjugated IgG. After washing thoroughly, ABTS (Pierce Chemical, Rockford, IL) was added for color development, and the absorbance was determined at 405 nm. BSA and GPIIIa49-66 scrambled peptide act as negative control under the same condition. To characterize the binding affinity of scFv-A11 and parental IgG, the Abs were added at concentrations from 0.01 nM to 1000 nM and incubated on the plate for 1 h at RT. The other procedures were performed as described above.
Flow cytometry
For detection of scFv binding to activated platelets, the murine platelets were activated by addition of 20 μM ADP (Helena Lab, Beaumont, TX), and then incubated for 20 min at RT with increasing concentrations of scFv Ab. Secondary antibody staining was performed with FITC-labeled anti-His(6)-tag MAb (Dianova, Hamburg, Germany). The binding to the platelets was determined by flow cytometry using a FACScan (BD Biosciences, Mountain View, CA).
Platelet particle assay
Gel-filtered human platelets were isolated from ethylenediaminetetraacetic acid-anticoagulated blood and labeled with anti-CD61-fluorescein isothiocyanate (FITC) as previously described.(1) Fluorescent-labeled platelets/particles were measured by flow cytometry using a FACScan (BD Biosciences). Gates were adjusted for platelets by exclusion of other blood cells.
Results
Generation of soluble scFv module
Previous studies demonstrated that phage Abs against platelet integrin GPIIIa49-66 could induce oxidative platelet fragmentation as parental Ab.(5) Phage antibody is the fusion format of scFv fragment and filamentous phage coat protein 3 (g3p) (Fig. 1A), which renders it suboptimal for potential clinical use. A soluble scFv form that is amenable for production and purification is therefore required. As illustrated by Figure 1B, all of the selected clones, including clones 11, 41, 43, 49, and 54, contain stop codon (TAG) in their heavy chain region, which limits the generation of scFv module. To circumvent this hurdle, we chose clone 11, which exhibits the highest binding activity,(5) mutated the inside stop codon (TAG) into AAG (lys) (named A11), and cloned the resultant mutant to plasmid pET-29a for expression in E. coli Rosetta. Upon induction with 1 mM IPTG, this clone produced A11 protein with a poly histidine affinity tag (His-tag) that facilitates the purification by Ni-NTA affinity column after protein refolding (Fig. 2A). The homogeneity of the purified antibody was evaluated by 12% SDS-PAGE and Commassie blue staining, visualized as a 29 kDa band (Fig. 2B). The scFv form has additional unparalleled advantages for clinical utility,(7–9) with their recombinant feature allowing for further genetic engineering such as affinity maturation and construction of fusion molecules for high clot-target avidity.(10–13)
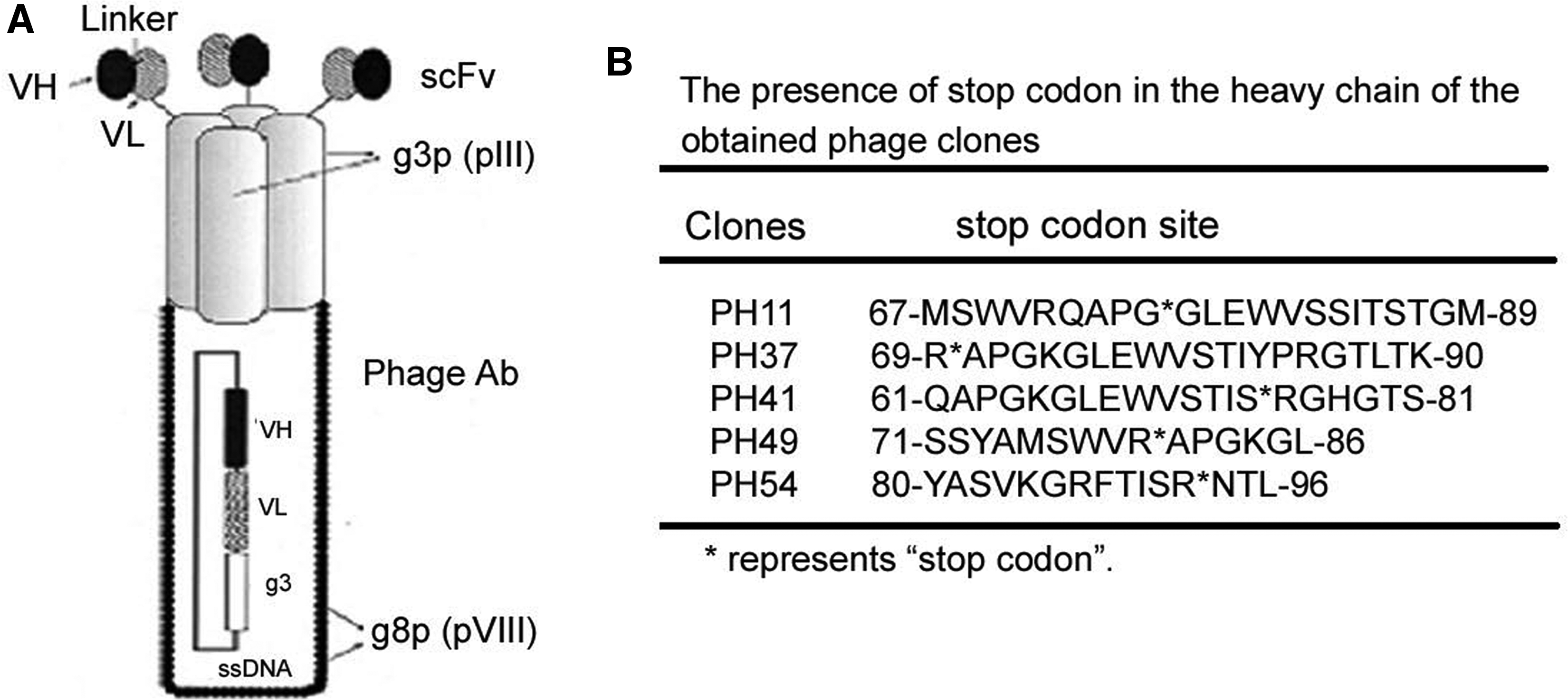
Phage Abs from the Tomlinson J phage Library. (
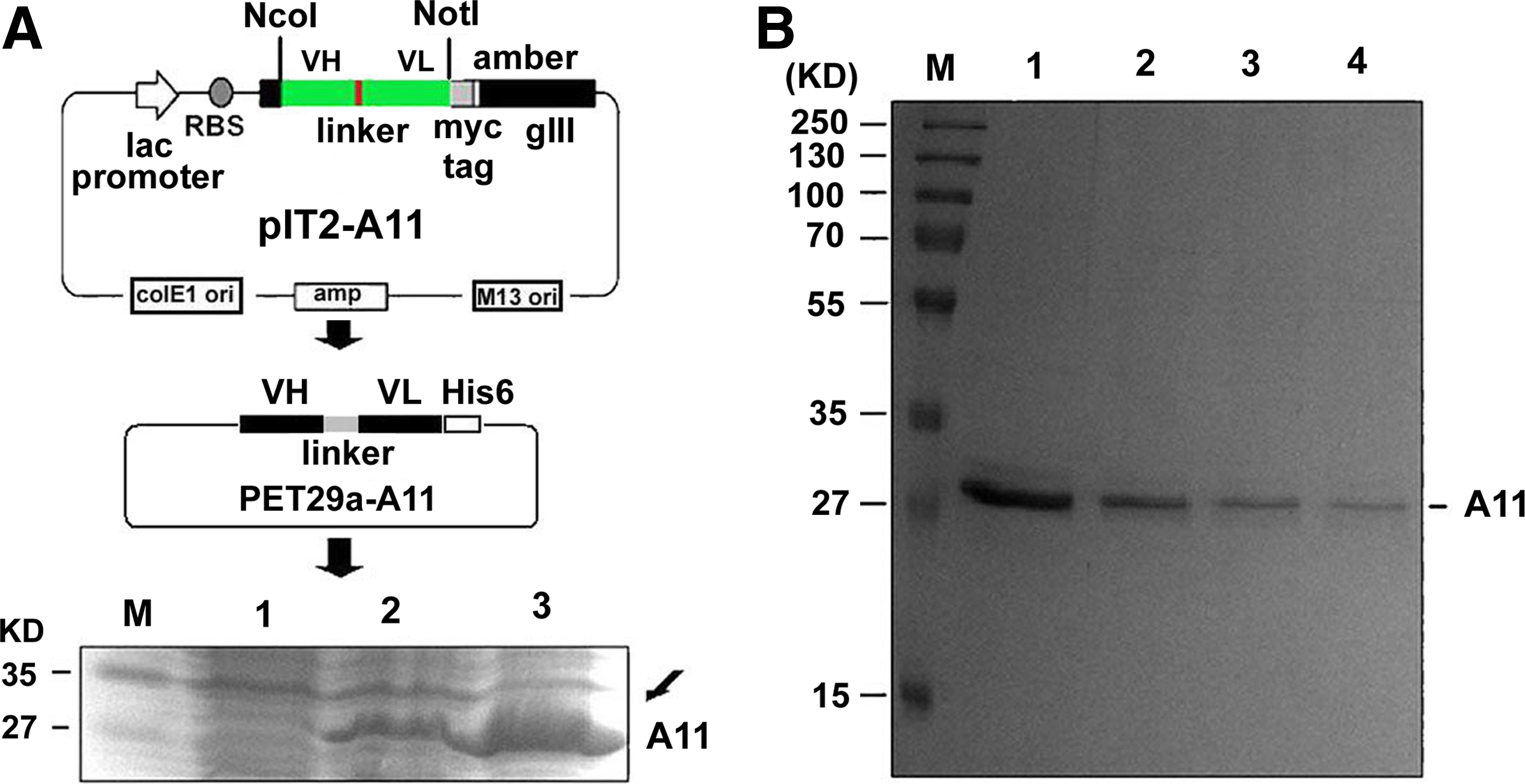
Generation of therapeutic scFv format (named A11). (
Binding activity of A11 to platelet integrin GPIIIa49-66
The specificity of A11 against GPIIIa49-66 peptide was determined by ELISA. It was demonstrated that A11 binds to GPIIIa49-66, in comparison to scrambled GPIIIa49-66 peptide and BSA control (Fig. 3A). Notably, A11 did not bind to intact mouse platelet that is deficient of β3 integrin (β3-/-), indicating its specific interaction with β3 intact platelets (Fig. 3B). As demonstrated by Figure 3C, A11 possesses a comparable binding specificity to the parental Ab at tested concentrations, whereas the control scFv (13CG2) exhibited little affinity to the antigen. Since activated platelets display more GPIIb-IIIa reactive receptors on the surface, we reasoned that A11 might bind more avidly with activated platelets. Figure 3D demonstrates A11 preferably bound to ADP activated platelets in a concentration-dependent manner, which is ∼1.8 fold more potent than its interaction with resting platelets.
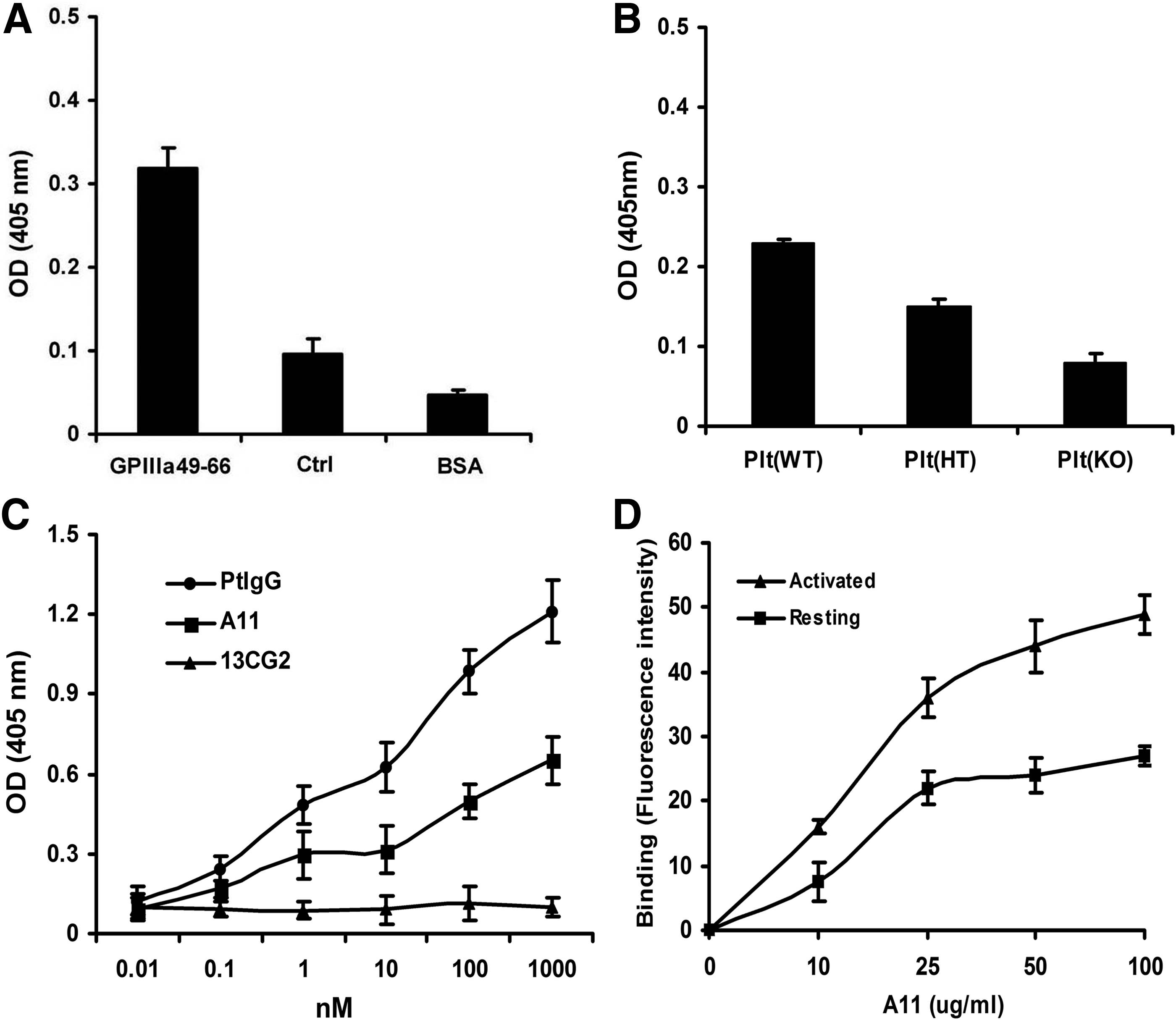
Binding of A11 to platelet integrin GPIIIa49-66. (
Effect of A11 on oxidative platelet fragmentation
Since A11 format demonstrated identical binding specificity to platelet β3 as patient GPIIIa49-66 Ab (parental Ab), we postulated that it could similarly induce oxidative fragmentation as patient Ab. To analyze the potential effect of A11 on platelet fragmentation, we performed flow cytometric analysis of platelets labeled with anti-GPIIb-FITC. Upon A11 treatment, there was a significantly shifted fluorescence intensity, which was accompanied by an altered cellular distribution from the right upper quadrant (RUQ) to the lower end of the left upper quadrant (LUQ) (Fig. 4). These findings indicate platelet fragmentation and diminished fluorescence intensity in the gate, presumably via loss of membrane GPIIb fluorescence as well as the production of smaller particles. A11 retained the efficacy in inducing platelet fragmentation (Fig. 5). It was shown that the fragmentation induced by A11 was inhibited by peroxide inhibitor catalase and diphenylene iodonium (DPI) (Fig. 6), as previously reported for anti-GPIIIa49-66 Ab, indicating the requirement of oxidation reaction for platelet fragmentation.
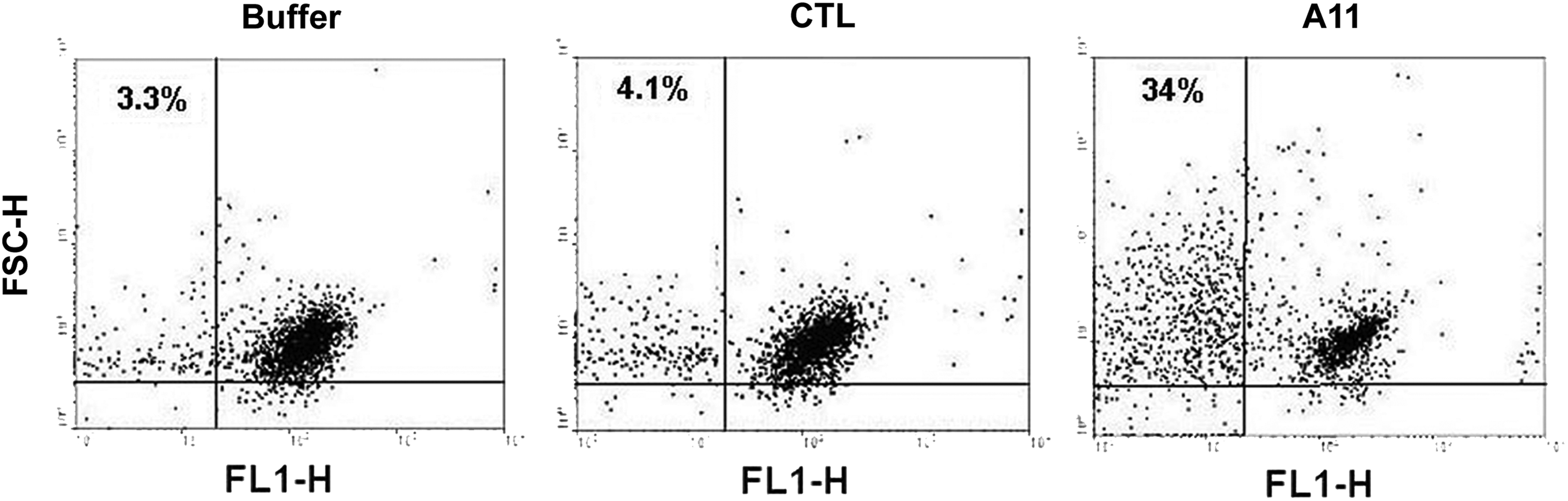
Flow cytometry of platelet in in vitro particle formation. Gel-filtered platelets were labeled with anti-GPIIb-FITC monoclonal Ab, washed, and then treated with various agents: buffer alone (

Comparison of A11 versus parental anti-GPIIIa49-66-induced platelet particle formation. A11 or parental anti-GPIIIa49-66 IgG was incubated with gel-filtered platelets at varying concentrations of Ab and analyzed by flow cytometry as described above.
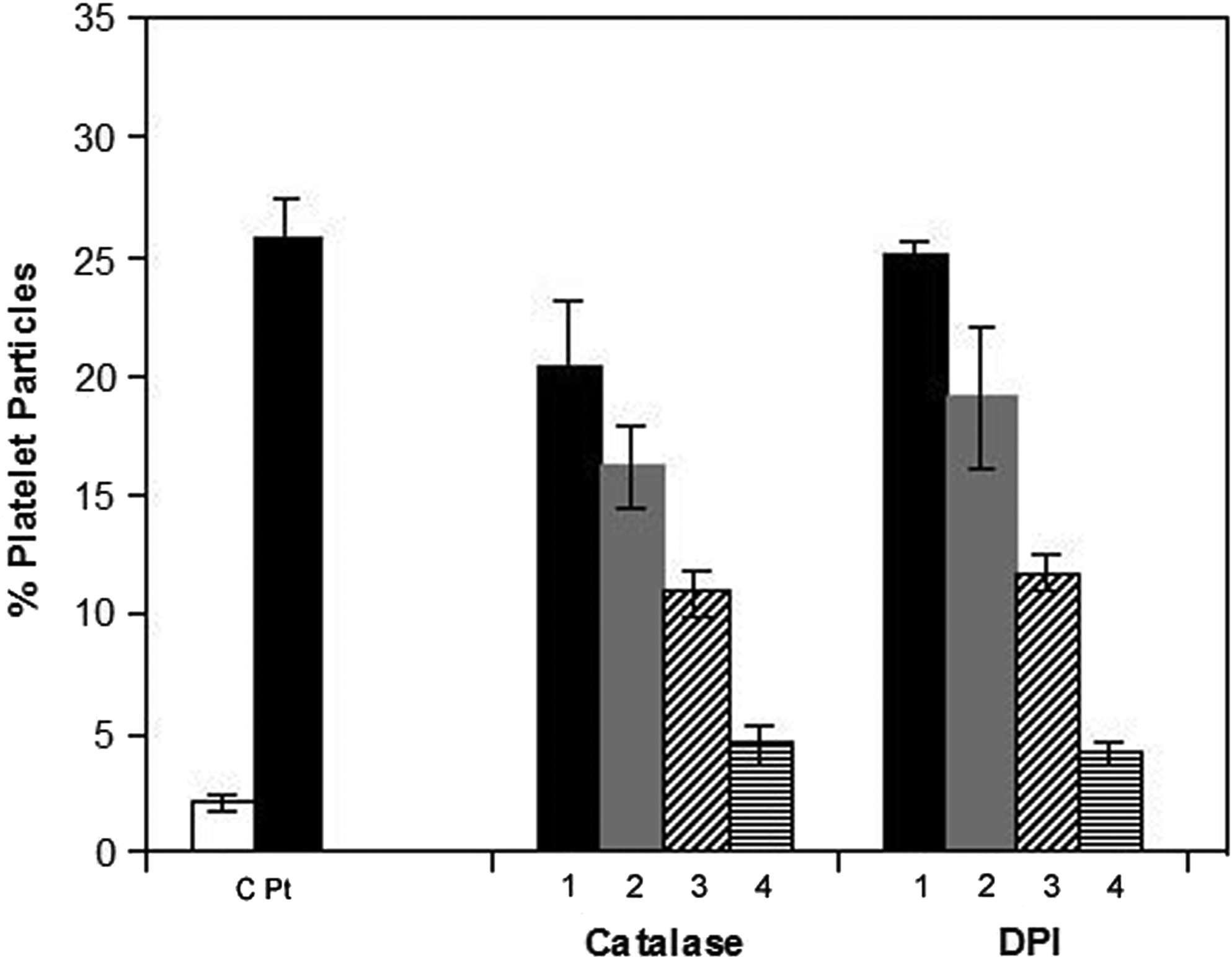
Effect of peroxide inhibitors catalase and diphenyleneiodonium (DPI) on platelet particle formation induced by A11. Gel-filtered platelets were pre-incubated with inhibitor or buffer for 15 min prior to the addition of A11, control scFv 13CG2 (
Effect of A11 on activated platelet fragmentation
Since A11 preferably binds to activated platelets, it is likely that A11 interacts more avidly with platelets preactivated by the thrombin receptor agonist TFLLRN. A11 has a preferential effect on activated platelets pretreated with the thrombin receptor agonist TFLLRN, particularly at low A11 concentration (∼2.6-fold greater sensitivity; Table 1).
Gel-filtered platelets (plts) were treated with the thrombin PAR-1 agonist TFLLRN (100 μM) for 30 min at 37°C and then followed with various A11 concentrations (20–5 μg/mL) for 4 h. Note increased sensitivity of activated platelets to A11 at low A11 concentration. CTL: irrelevant control scFv Ab (13CG2; 20 μg/mL).
Discussion
Single-chain variable fragment of antibody (scFv Ab) provides many advantages over monoclonal antibodies, especially for therapeutic purposes, including enhanced tissue penetration due to their small size and abrogated immunogenicity. Currently, phage surface display antibody technology has been successfully employed to generate antibody that specifically blocks the activated form of GPIIb/IIIa.(14–16) In contrast to clinically used anti-platelet agents, none of which are conformation specific, scFvs specifically against activated platelets do not induce conformational changes in GPIIb/IIIa or outside-in signaling, thereby avoiding unwanted global activation of platelets.(15) The exclusive binding property of activated platelet-specific scFvs is mediated by RXD motifs in the heavy chain complementary-determining region (CDR) 3 of the antibodies.(16) While these blockers selectively recognize activated platelets, they permit necessary physiological functions of platelets, including cell adhesion and spreading on immobilized fibrinogen.(16) Therefore, the activation-specific GPIIb/IIIa blockade via human single-chain antibodies represents a promising novel strategy for antiplatelet therapy with higher benefit-to-risk ratio.
In the present study, we have generated a novel humanized scFv Ab that could induce oxidative platelet fragmentation. The advantage of A11 over the previously described antiplatelet Abs includes: (1) selective induction of fragmentation in activated versus resting platelets; (2) a human-origin Ab that is unlikely to induce hypersensitivity as a foreign antigen; (3) absence of an Fc domain that precludes complement activation and the phagocytic/cytokine response; and (4) smaller size scFv that renders an easier clot penetration than the full-length Ab.
The selectivity of A11 for activated platelets, however, is relatively suboptimal. The 2.5-fold difference between the fragmentation of activated platelets versus resting platelets cannot completely prevent the possible fragmentation of non-activated circulating platelets. Therefore, to avoid the possible side effects, this newly generated scFv Ab should be fused with other anti-thrombotic agents such as plasminogen activators to maximize its therapeutic window of efficacy, while being administered at a relatively lower dose.(11–13)
In the future, this proof of principle needs to be further evaluated in at least one animal thrombotic model by monitoring the platelet counts upon injection of the scFv Ab. Nevertheless, the current data have established the concept of developing a different approach to combat arterial thrombosis by generating an scFv for the lysis of platelet thrombus.
Footnotes
Acknowledgments
This article is dedicated to the memory of Dr. Simon Karpatkin who initiated this project and tragically passed away on August 21, 2009. This work was supported by NIH grants DAO-04315 (S.K.), and in part by grant ECNU (77202203) in China.
Author Disclosure Statement
The authors have no financial interests to disclose.