
Abstract
PreS1 is a hypothetical candidate domain of L protein for hepatitis B virus (HBV) to adhere to and invade host hepatic cells. This report deals with the expression and purification of recombinant adw2 subtype of the preS1 peptide of hepatitis B virus surface antigen in Escherichia coli. The DNA fragments of the full-length or N/C terminal sequence of preS1 synthesized by PCR were inserted into the prokaryotic expression vector pGST-MOLUC, respectively. Reconstitute plasmids (named pGST-preS1, pGST-preS1N, and pGST-preS1C) were confirmed by sequencing analysis and transferred into Escherichia coli BL21(DE3). Recombinant full-length and N/C terminal of preS1 with GST tag were expressed at high levels in soluble form after induction with IPTG. The recombinant proteins were purified by a single-step affinity chromatography method. The immune reactivity of recombinant preS1 was confirmed by Western blot and virus capture assay. Furthermore, when the purified recombinant protein was used to immunize rabbit, the specific antibody titer can reach 10−7. Thus, our successful expression system and achievement of purified recombinant preS1 protein and its polyclonal antibody lay the foundation for better understanding of the mechanism of HBV PreS1 protein in virus endocytosis and are helpful in seeking the PreS1-related protein.
Introduction
In this study, we constructed the recombinant GST-preS1 prokaryotic expression plasmid. The fusion protein was highly expressed in soluble form and was purified by glutathione sepharose 4B affinity chromatography. Furthermore, we studied the immunogenicity and immunoreactivity of the purified fusion protein and achieved its polyclonal antibody. The successful expression and purification of fusion preS1 and preparation of its polyclonal antibody may help to study the character and function of preS1.
Materials and Methods
PCR amplification
To construct the prokaryotic expression plasmid containing the preS1 gene, the DNA sequence of preS1 was synthesized by PCR. A plasmid of pEcob6 containing full-length of subtype adw2 of HBV was used as the template. To synthesize the full-length of preS1, the primers were P1 (5′-ATGAAGCTTAATGGGAGGTTGGTCATC-3′) and P3 (5′-ATCTCTAGAGGCCTGAGGATGACT-3′). To synthesize the N terminal 1-183 bp DNA fragment, another downstream primer P4 (5′-TCGTCTAGATCCCACTCCTACCTG-3′) was used with the same upstream primer P1. To synthesize the C terminal 148-357 bp fragment, another upstream primer P2 (5′-ATCAAGCTTAGACCACTGGCCAGCA-3′) was used with the same downstream primer. All oligonucleotides were synthesized by Invitrogen (Shanghai, China). Fifty μL PCR mixtures contained 100 ng template plasmid, 20 pmol/μL each primer, 200 μM dNTP, standard pfu polymerase buffer, and 2.5 U pfu polymerase (Takara, Dalian, Liaoning, China). PCR amplification began with an initial denaturation step at 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 62°C for 30 sec, and 72°C for 30 sec, with a final elongation step at 72°C for 10 min. The PCR products were resolved in 1.5% agarose gel stained with ethidium bromide.
Construction of PreS1 expression plasmids
PCR product was cloned into the E.Coli expression vector pGST-MOLUC (kindly provided by TC He, University of Chicago, Chicago, IL). The purified PCR product and vector plasmid were digested with HindIII and XbaI restriction enzyme. The excised preS1 DNA fragment was ligated into the equivalent sites of vector with an insert ratio of 10:1. The ligation mixture was used to transform the competent E. coli strain JM109. The ampicillin resistant colonies were isolated, identified by PCR analysis of plasmid. The inserted DNA fragment was finally confirmed by DNA sequencing. The recombined plasmids were named pGST-PreS1, pGST-preS1N, and pGST-preS1C.
Expression of recombinant GST-preS1
For protein expression, the competent E. coli BL21(DE3) was transformed with pGST-preS1, pGST-preS1N, and pGST-preS1C, respectively. The transformed BL21(DE3) strain was inoculated into 100 mL fresh LB medium containing 100 g/mL ampicillin and incubated at 37°C overnight. The culture was seeded into fresh LB medium containing 100 g/mL ampicillin by a ratio of 1:50 and incubated at 37°C to reach an OD600 of 0.4∼0.6; then a final concentration of 1 mmol/L isopropylthio-D-galactoside (IPTG) was added into the culture and incubated for an additional 6 h. An aliquot of 1 mL was taken from each induced and non-induced control cell after incubation, centrifuged at 12,000 rpm (5100 g) at 4°C for 2 min to get cell pellet, to which 100 μL gel loading buffer (4×stacking gel buffer, 4% SDS, 20% glycerol, 0.2% bromophenol blue, 0.2 M DTT) was added. The pellet was vortexed and lysed in boiling water bath for 5 min, and then another centrifugation was done at 12,000 rpm (5100 g) for 2 min. Samples were stored at −80°C. For analysis of expression of protein, 10 μL of supernatant were loaded onto a 15% discontinuous SDS-PAGE. The gel was stained with Coomassie brilliant blue R-250, then destained with destaining solution (methanol–glacial acetic acid, 3.5:1), and assayed by thin layer scanning using BandScan 4.5 software (Glyko, Hayward, CA).
Solubility analysis of the expression product
The induced cell pellet was collected by centrifugation and resuspended in 100 μL PBS and lysed by sonication. The sonicate was centrifuged at 14,000 g for 15 min at 4°C. The supernatant was collected and the precipitate was resuspended by an equal volume of PBS. Ten μL of supernatant and precipitate resuspending solution were loaded onto a 15% discontinuous SDS-PAGE to analyze the soluble character of the expression products.
Western blot analysis
To examine the immunoreactivity of the fusion protein GST-preS1, Western blot analysis was carried out. Total cell lysate from induced BL21/preS1 was resolved by 15% SDS-PAGE and transferred by electroblotting on to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). Mouse monoclonal antibodies against preS1 (1:1000 v/v, 125E11, Alpha, Shanghai, China) and GST (1:200 v/v, Sigma-Aldrich, St. Louis, MO) were used as primary antibodies. A horseradish peroxidase (HRP)-conjugated second antibody (1:5000 v/v, Zhongshan, Beijing, China) against mouse IgG was used to detect the bound antigens with Super SignalWest Pico Chemiluminescent substrate kits (Pierce, Chicago, IL) according to the manufacturer's instructions.
Purification of recombinant GST-preS1
The GST-preS1 fusion protein was purified by glutathione sepharose 4B affinity chromatography according to the manufacturer's protocol. Briefly, cells from 400 mL induced BL21 containing preS1/preS1N/preS1C were harvested by centrifugation at 12,000 rpm (5100 g) for 10 min at 4°C and resuspended in 20 mL PBS (pH 7.4) with 0.1 mmol/L PMSF and 10 g/L TritonX-100 before sonication in an ice bath. The sonicated cell lysate was centrifuged at 14,000 g for 20 min at 4°C. The collected supernatant was incubated with 400 μL glutathione sepharose 4B pre-equilibrated with PBS at room temperature for 60 min. The mixture was transferred to affinity column and washed by PBS three times. The bound protein was eluted with 5 mL elution buffer (10 mmol/L reduced glutathione, 50 mmol/L Tris-HCL [pH 8.0]). The eluted fractions were run on 15% SDS-PAGE to identify the enriched GST-preS1/preS1N/preS1C fusion proteins. Protein concentration was determined according to Bradford's method.
Generation of anti-preS1
To develop an anti-serum specific to preS1, the purified GST-preS1 was used to immunize a New Zeland rabbit. The GST-preS1 was dissolved in PBS to a final concentration of 1 mg/mL. The initial injection was 1 mL GST-preS1 solution mixed with an equal volume of complete Freund's adjuvant at the site of underskin around the spine and arm of the rabbit. All subsequent injections (boosts) consisted of 1 mL GST-preS1 solution and 1 mL incomplete Freund's adjuvant every 2 weeks. After three rounds of immunization, the serum was collected and stored at −20°C.
ELISA
To test the specific antibody titer of collected serum, ELISA was carried out. In brief, 96-well microtiter ELISA plates were coated with 10 mg/L purified GST-preS1. Serially diluted rabbit anti-preS1 serum was added to the plate and incubated at 37°C for 60 min. Anti-rabbit IgG conjugated with horseradish peroxidase (HRP) (1:8000 v/v, Sigma-Aldrich) were sequentially added and incubated at 37°C for another 60 min. After washing with PBS buffer, 100 μL of substrate solution containing TMB (Sigma-Aldrich) was added to each well in the dark to produce a color change. Optical density (OD) was measured at 450 nm in an ELISA reader. All measurements were performed in triplicate.
Virus capture assay
The rabbit anti-serum was diluted to 10 mg/mL with carbonate sodium buffer (4.3 g/L NaHCO3, 5.3 g/L Na2CO3 [pH 9.4]) to coat the 96-well microtiter ELISA plates. HBV positive serum diluted with PBS to a titer of 106 copy of HBV was added into each well and incubated at room temperature for 60 min. A mouse anti-HBs IgG conjugated with HRP was added to detect the captured HBV particles. After incubation at room temperature for 60 min and washing with PBS, substrate solution containing TMB was added and the optical density was measured at 450 nm. To investigate the inhibition effect of preS1, various doses of purified GST-preS1/preS1N/preS1C were added into the diluted HBV positive serum, and the HBV titer was detected by ELISA, similar to the above. In this case, purified GST was used as negative control.
Results
Construction of prokaryotic expression vectors pGST-preS1, pGST-preS1N, and pGST-preS1C
To construct the recombinant plasmid expressing preS1, a prokaryotic expression plasmid pGST-MOLUC was used. DNA fragments of full-length preS1, N terminal preS1N, and C terminal preS1C were obtained by PCR with the primer P1/P3, P1/P4, and P2/P3, respectively. The fragments were inserted downstream of the T7 promoter in pGST-MOLUC. The recombinant plasmids were identified by PCR, showing the expected 357 bp, 183 bp, and 210 bp DNA fragments, respectively (Fig. 1). The inserted DNA sequences were further confirmed by DNA sequencing assay.
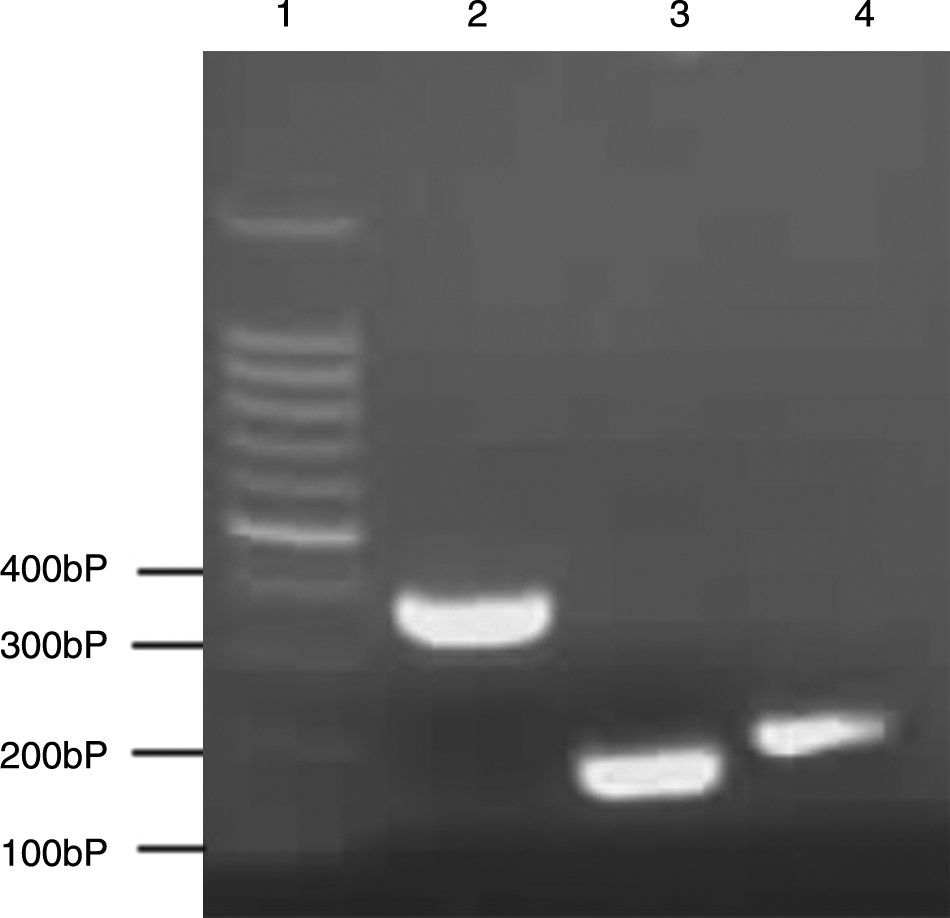
PCR identification of recombinant plasmids. The PCR products were analyzed by agarose gel electrophoresis. The ampicillin resistant colonies were isolated and used as PCR templates. Lane 1, DNA marker; lane 2, recombinant plasmid pGST-preS1; lane 3, recombinant plasmid pGST-preS1N; lane 4, pGST-preS1C.
Expression and purification of GST-preS1
To obtain the fusion proteins of GST-preS1/preS1N/preS1C, the engineering strains BL21/pGST-preS1/preS1N/preS1C were induced by adding IPTG to a concentration of 1 mM in the medium. SDS-PAGE analysis of the supernatant of the induced cell lysates demonstrated that the recombinant proteins were inducibly expressed (Fig. 2). The molecular weights of induced fusion proteins of GST-PreS1, GST-PreS1N, and GST-PreS1C were approximately 39, 31, and 32 kDa, respectively, as expected. The inducible protein accounted for about 8% of the total proteins as assayed by densitometric scanning. The solubility analysis of the three induced proteins showed that more than 80% of the induced proteins existed in soluble forms. To obtain purified fusion proteins, glutathione sepharose 4B affinity chromatography was carried out according to the manufacturer's protocol. Approximately 20 mg proteins were harvested from 400 mL culture, with a purity level above 90% (Fig. 3).

SDS-PAGE analysis of preS1 fusion proteins expressed in E. coli. Lane 1, SeeBlue®Plus2 Pre-Stained Standard Marker; lane 2, 10 μL total proteins of E. coli BL21 after induction; lanes 3 and 4, 10 μL total proteins of E. coli BL21/pGST-MOLUC before and after induction; lanes 5 and 6, 10 μL total proteins of E. coli BL21/pGST-preS1 before and after induction; lanes 7 and 8, 10 μL total proteins of E. coli BL21/pGST-preS1N before and after induction; lanes 9 and 10, 10 μL total proteins of E. coli BL21/pGST-preS1C before and after induction.

Western blot analysis of expressed protein. (
Immunogenicity of GST-preS1
To test the immunoreactivity of GST-preS1, Western blot analysis was carried out. All three fusion proteins were recognized by anti-GST (Fig. 4A). GST-preS1 and GST-preS1N reacted with antibody against preS1. GST-preS1C did not react with the antibody because it did not contain the antigenic determinant sequence 21-47 aa, which was recognized by the antibody 125E11 (Fig. 4B). The results of Western blot analysis demonstrated that the fusion proteins had good immunoreactivities.
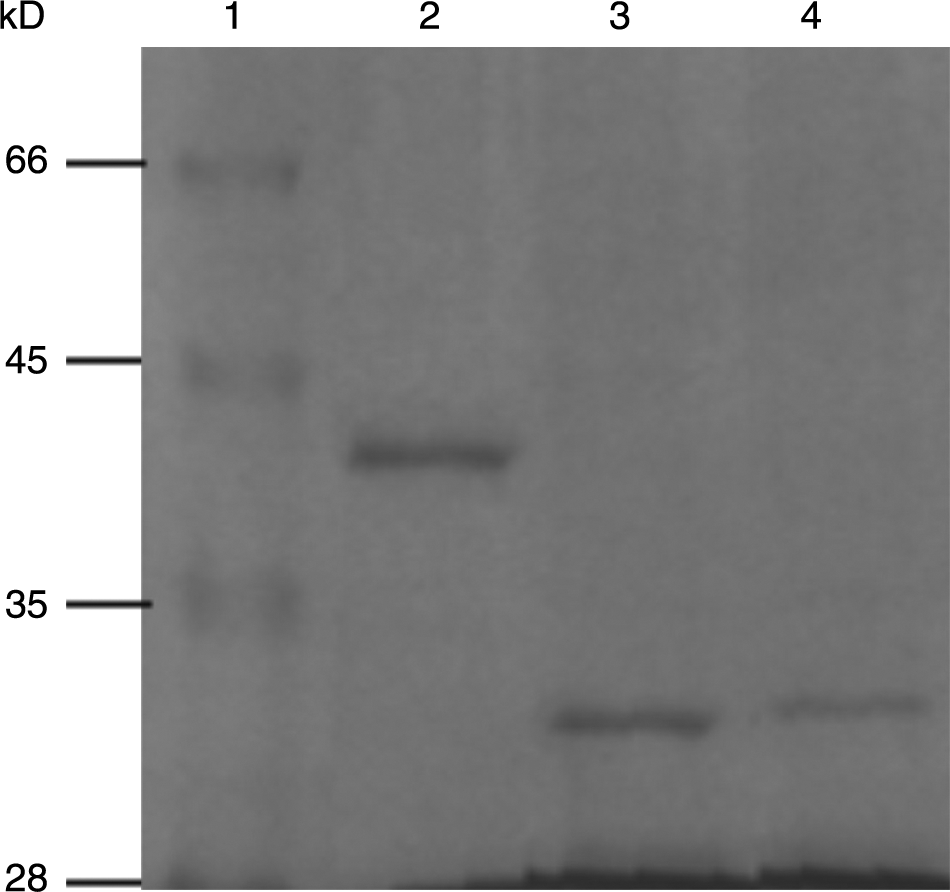
SDS-PAGE analysis of purified preS1 fusion proteins. The fusion proteins were purified by glutathione sepharose 4B affinity chromatography and 10 μL of eluted fraction was analyzed by 15% SDS-PAGE. Lane 1, middle molecular weight protein marker; lane 2, GST-preS1; lane 3, GST-preS1N; lane 4, GST-preS1C.
GST-preS1 stimulates antibody production
To develop a poly-antibody against GST-preS1, rabbits were immunized with the purified recombinant protein. After three rounds of immunization, a high titer (10−7) anti-serum against GST-preS1 was produced. Immunization of the rabbit with 1 mg of purified fusion protein was able to stimulate strong immunoreactivity, and the anti-preS1 was detected in the serum 2 weeks after the initial immunization. These results indicated that the purified protein had good immunogenicity.
PreS1 inhibit virus capture by anti-preS1
To investigate whether the produced anti-preS1 is able to bind to naive HBV particles, virus capture assays were performed. As shown in Figure 5, poly-antiserum against preS1 coating on the surface of a 96-well plate strongly bound to HBV particles in infection sera. The binding was inhibited by adding purified GST-PreS1, GST-preS1N, and GST-preS1C to the HBV sera. The inhibition effect was dose-dependent in that the binding virus particles reduced with an increasing amount of fusion proteins in the sera. The GST-preS1 was shown to have the strongest inhibition effect, followed by GST-preS1N, with the GST-preS1C having the weakest inhibition effect. The possible reason for this lies in the fact that the full length of preS1 had the most antigenic determinants, and the main epitopes, especially the neutralizing antigens, were located in the N terminal of preS1.
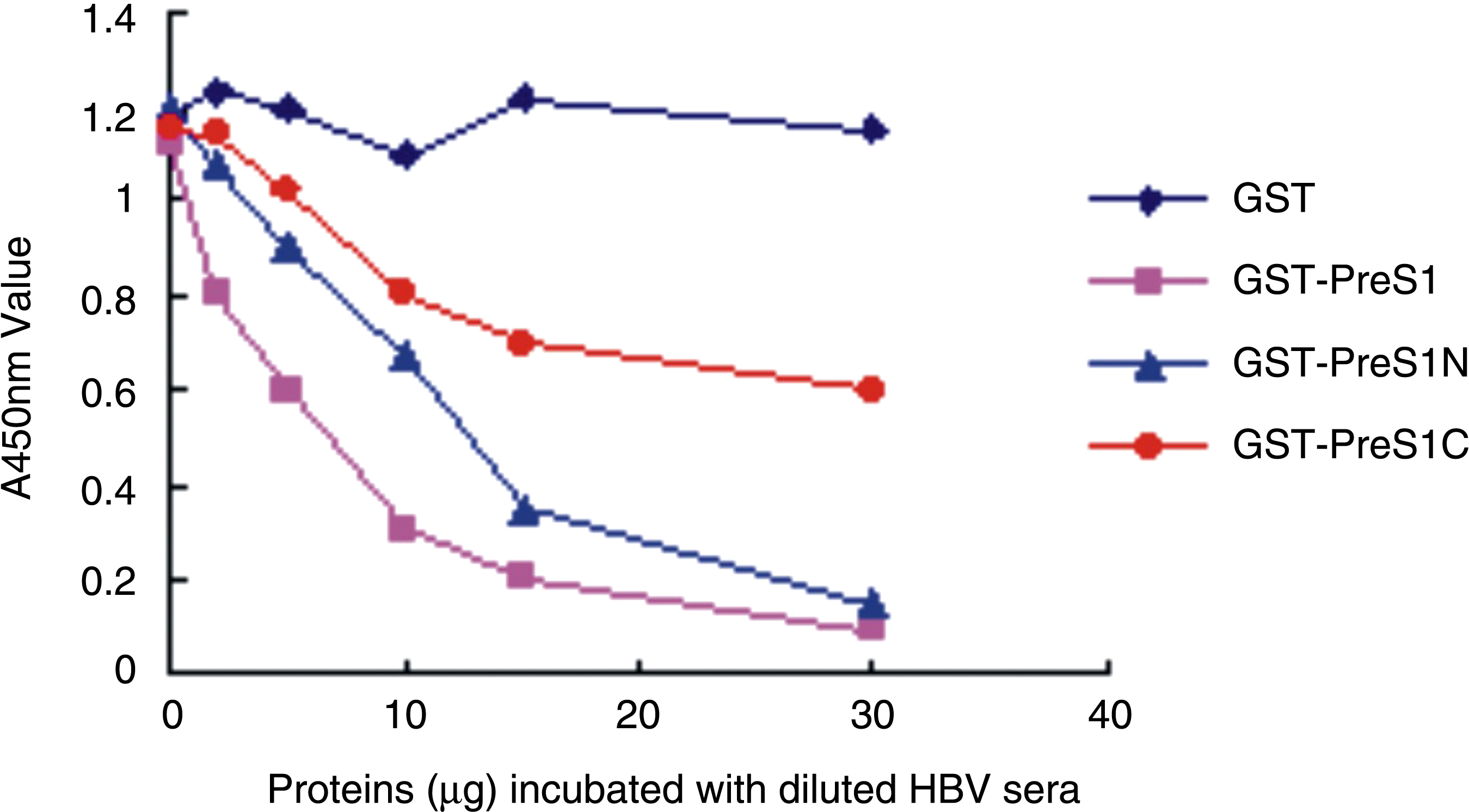
Virus capture assay. The concentration of HBV particle was measured by optical density at 450 nm for their positive relationship. GST was used as a negative control.
Discussion
Attachment and invasion of the virions to human hepatic cells are the initial step in HBV infection, followed by replication. The procedure of HBV invasion is still a puzzle to investigate. PreS1 is thought to play an important role in HBV attachment.(9) The 21-47 aa of preS1 is considered to be a specific binding site for hepatic receptors, consistent with the fact that the antibody against preS1 inhibits HBV attachment.(10–12) The human immunoglobulin A receptor, Interleukin-6, asialoglycoprotein receptor, apolipoprotein H, and so on were found to bind preS by different methods.(13–16) But there is still much controversy about these candidate receptors as to whether they indeed introduce the virion into the hepatic cell membrane for lack of convincing proof on infection models. None of the candidate receptors exogenously expressed in the hepatic cell line increased susceptibility for virus infection. Most of the candidate receptors were identified with a synthetic partial fragment of preS1. The experimental bias may result from the impairment of whole construction of the preS1 or the loss of important antigenic determinants. On the other hand, successful HBV infection may require multiple cellular co-factors for its multiple steps, including cell attachment, endocytosis, membrane fusion, and post-fusion.(3) Novel receptor or co-receptor candidates may be continuously discovered and help to understand the truth about HBV invasion. It was reported that HBV infection of the cell model may be inhibited by recombinant preS1, indicating that prokaryotic expression preS1 has a similar secondary structure and function with the natural protein.(17) In our study, the recombinant preS1 fused with GST tag was also able to inhibit the virus capture by specific antibody.
Previously approximately 350 million people were chronically infected with HBV, most of whom reside in Asia, Africa, and Latin America. Standard immunization programs were put in place, producing a reduced infection rate. However, conventional HBV vaccines with standard immunization schedules have a 5–10% non-responder rate, especially in adults.(18) Furthermore, HBV mutations decreased the protective efficiency of conventional vaccines.(19) Development of new, more efficient vaccines is needed for better infection control.(20) PreS1 has many epitopes, including B cell and T cell epitopes, to produce protective antibody, and have been shown to play an important role in immunogenicity against HBV.(21) PreS1 may be an ideal candidate for prophylactic and therapeutic vaccine design.(22) In our study, when the purified recombinant preS1 was used to immune rabbit, a specific antibody against preS1 was produced, indicating that the recombinant preS1 has strong immunogenicity. Furthermore, the specific antibody from immunized rabbit sera captured the HBV virions in virus capture assay and it would be inhibited by purified preS1. The results indicated that preS1 may have the potential to develop vaccines.
In conclusion, we have successfully expressed the preS1 of HBV with a tag of GST in E. coli and isolated large quantities of purified protein. The fusion protein has similar antigen determinants with natural preS1 and excellent antigenicity to produce specific antibody. The purified protein and polyclonal antibody can be used for identification and isolation of preS1 binding protein, which may function as a receptor for HBV, as well as for the development of a new recombinant HBV vaccine and diagnostic detection kits.
Footnotes
Acknowledgments
This work was supported in part by research grants from the National Natural Science Foundation of China (no. 30972582) and Chongqing Science and Technology Commission, China (2009BB5276).
Author Disclosure Statement
The authors have no financial interests to disclose.