
Abstract
The RadA/Sms protein facilitates DNA repair in Escherichia coli cells damaged by UV radiation, X-rays, and chemical agents. However, the precise mechanism by which RadA/Sms aids DNA repair is unknown. Here we report the production of monoclonal antibodies (MAbs) specific for RadA/Sms for use in biochemical and physiological investigations. Histidine-tagged RadA/Sms (RadA-6xHis) was overproduced in E. coli BL21 cells transformed with the radA/sms coding region in plasmid pRSET A and purified by nickel affinity chromatography. Splenocytes from female BALB/c mice hyperimmunized with the purified protein were fused to SP2/0-Ag14 myeloma cells, and the resultant hybridomas were selected in HAT medium. MAbs were detected in hybridoma culture supernatants by indirect ELISA and Western blot analysis against purified RadA-6xHis. MAbs from four cell lines were further evaluated by Western blotting against peptide maps generated by endoproteinase Glu-C digestion of RadA-6xHis. Each of the four MAbs recognized a unique epitope on the fusion protein. Two of the MAbs (6F5 and 2A2) also detected wild-type (tagless) RadA/Sms produced from the pJS003 plasmid in E. coli K-12 cells. We anticipate that these antibodies will prove useful for the detection, isolation, and functional analysis of RadA/Sms.
Introduction
In Escherichia coli, the gene coding for the RadA/Sms protein was originally called radA and was first described in 1982 by Diver and colleagues.(1) Ten years later, Neuwald and colleagues(4) described an E. coli DNA repair gene that they called sms to denote the fact that insertional inactivation of sms led to enhanced
Although it is clear that RadA/Sms is important in DNA repair, its precise function is not well defined.(8) In this report, we describe the production of monoclonal antibodies (MAbs) to histidine-tagged RadA/Sms (RadA-6xHis) and show that these antibodies can detect both the tagged and wild-type (tagless) forms of RadA in E. coli lysates. We anticipate that these antibodies will be useful tools during investigations of RadA/Sms structure, function, and gene induction.
Materials and Methods
E. coli strains, plasmids, and culture conditions
The E. coli strains and plasmids used in this study are shown in Table 1. Strain SR2876 is deficient in the chromosomal radA gene, but overproduces RadA protein from the radA gene carried on plasmid pJS003 (derived from plasmid pBR322). Strain SR4097 produces normal levels of RadA from the chromosomal copy of the radA gene and overproduces a RadA-6xHis fusion protein from the radA gene carried on plasmid pVK1009 (derived from pRSET A; Invitrogen, Carlsbad, CA). E. coli strains were grown in Luria-Bertani (LB) broth(10) with ampicillin added to 50 μg/mL when plasmids were present.
Genetic nomenclature is that used by Berlyn and colleagues.(29) Plasmid pVK1009 (radA-6xhis) carries a gene fusion adding 108 nt (36 amino acids including a 6xHis-tag) in frame to the N-terminus of the radA gene. AmpR and TetR indicate ampicillin and tetracycline resistance, respectively.
Unpublished data (courtesy of J. Seo, S.J. Pullen, and N.J. Sargentini, A.T. Still University (Kirksville, MO).
CGSC, Coli Genetic Stock Center at Yale University (New Haven, CT).
Overexpression and purification of E. coli RadA-6xHis
The radA open reading frame was PCR amplified using genomic DNA from E. coli strain SR932 as the template, a 5′ sense primer (P1) containing a BamHI site (underlined) (5′-
To further purify the protein to single-band homogeneity, gel slices containing unfixed and unstained RadA-6xHis that co-migrated with a 50 kDa pre-stained protein standard (Precision Plus dual-color markers, Bio-Rad, Hercules, CA) were excised from SDS-PAGE gels and minced into small pieces. The protein was passively eluted into distilled water by overnight agitation on a rotary mixer at 4°C. The eluate containing purified RadA-6xHis was lyophilized to dryness, resuspended in PBS, and stored in working aliquots at −80°C. This preparation of RadA-6xHis was used for the intravenous booster immunization of mice prior to hybridoma production, for screening hybridoma culture supernatants by ELISA and Western blotting, and for determining MAb epitope specificities in peptide mapping experiments.
SDS-PAGE
SDS-PAGE was performed as described by Laemmli(11) in a discontinuous minigel system (Bio-Rad) using 12% or 15% resolving gels overlayed with 4% stacking gels. Protein loads were determined by the Bradford method(12) using a commercial Coomassie Blue dye reagent concentrate (Bio-Rad). Proteins resolved by SDS-PAGE were visualized by staining in 0.1% w/v Coomassie Brilliant Blue R-250 in 40% methanol/10% acetic acid, or were electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes for immunoblotting as described by Towbin and colleagues.(13)
Immunization of mice and monoclonal antibody production
Beginning at 6 weeks of age, four female BALB/c mice were immunized with nickel affinity purified RadA-6xHis protein in three intraperitoneal injections over the course of 5 months. Each injection contained 6.9–15 μg of RadA-6xHis in a total volume of 130 μL. Diluents for the first through third immunizations, respectively, were 50% Freund's complete adjuvant (MP Biomedicals, Solon, OH), 50% Freund's incomplete adjuvant (MP Biomedicals), and PBS without adjuvant. Titers of sera obtained by tail bleed 10 days after the third intraperitoneal immunization were determined by Western blotting(13) against nickel affinity purified RadA-6xHis. Three days prior to hybridoma production, the two mice with the highest serum antibody titers received intravenous booster immunizations containing 20 μg of RadA-6xHis diluted in 100 μL of PBS. Splenocytes from the mice were fused to Sp2/0-Ag14 myeloma cells as described by Van Deusen(14) and selected in HAT medium prepared from Dulbecco's modified Eagle's medium supplemented with 15% horse serum (Sigma-Aldrich, St. Louis, MO).
Beginning 2 weeks after fusion, hybridoma supernates were screened by indirect ELISA(15) against RadA-6xHis, as described below. Cell lines of interest were switched to HT medium on day 21 and cloned by limiting dilution(16) prior to storage in liquid nitrogen. To facilitate recovery of MAbs from culture supernatants, cells were gradually adapted to growth in Ex-Cell 610-HSF serum- and protein-free medium (Sigma-Aldrich). The medium was exchanged for PBS and the MAbs were concentrated by ultrafiltration through 30 kDa cut-off membranes (Millipore, Billerica, MA). MAb heavy chains were isotyped by ELISA using goat anti-mouse class- and subclass-specific primary antibodies (Sigma-Aldrich), followed by rabbit anti-goat IgG-alkaline phosphatase secondary antibody (Bio-Rad). Wells were developed with p-nitrophenylphosphate substrate (Pierce Chemical, Rockford, IL) at room temperature and evaluated by visual examination.
ELISA procedure for screening hybridoma supernatants
RadA-6xHis (300 ng/100 μL/well) purified by passive elution from polyacrylamide gels was diluted in PBS and adsorbed onto 96-well ELISA plates (BD Biosciences, Franklin Lakes, NJ) by incubation at 37°C for 1 h on an orbital shaker set at 100 rpm. The antigen-coated plates were blocked by a 10-min incubation in PBS containing 20% non-fat dry milk (NFDM, 200 μL/well) at room temperature. Blocked plates were incubated first with undiluted hybridoma supernatant (100 μL/well, 2 h, 37°C), and then with alkaline phosphatase-conjugated goat anti-mouse polyvalent immunoglobulins (IgG, IgM, IgA; Sigma-Aldrich) diluted 1:1000 in PBS containing 0.05% Tween-20 (PBST) and 0.5% BSA (100 μL/well, 1 h, room temperature). Plates were washed between steps with PBST. Wells were developed with p-nitrophenylphosphate substrate (100 μL/well) at room temperature, and absorbances were read at 405 nm (A405) in a Labsystems Multiskan MCC/340 ELISA reader (Fisher Scientific, St. Louis, MO). Hybridoma supernatants with A405 values greater than five times that of Sp2/0-Ag14 supernatants were considered reactive to RadA-6xHis.
Western blot analysis
Hybridoma supernatants were tested by Western blot analysis against RadA-6xHis purified by elution from polyacrylamide gels, and against an E. coli strain that produced tagless RadA from plasmid pJS003 (strain SR2876). A sample of purified RadA-6xHis (12 μg) or E. coli lysate (37 μg) was solubilized in electrophoresis sample buffer (62.5 mM Tris [pH 6.8], 2% SDS, 5% 2-mercaptoethanol [ME], 20% glycerol, 0.1% bromophenol blue) and loaded into a 6-cm wide well of a 12% polyacrylamide minigel. The protein was subjected to electrophoresis and electrotransferred to PVDF as described by Towbin and colleagues.(13) Membranes were blocked by a 1 h incubation in Tris-buffered saline (TBS; 20 mM Tris, 150 mM NaCl [pH 7.5]) containing 5% NFDM. Strips cut from the membranes were incubated for 90 min with hybridoma supernatants diluted 1:2 in TBS containing 0.05% Tween-20 (TTBS) and 1% NFDM. Strips were washed in TTBS and then incubated for 90 min in 1:1000 alkaline phosphatase-conjugated goat anti-mouse polyvalent immunoglobulins. After further washes, strips were developed with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate substrate (NBT/BCIP, Bio-Rad). Positive control strips were probed with 1:10,000 polyclonal mouse serum as the primary antibody. Negative control primary antibodies included 1:10,000 normal mouse serum, 1:2 spent Sp2/0-Ag14 myeloma medium, and PBST.
To further analyze the specificity of the MAbs, the antibodies were Western blotted against two additional His-tagged proteins, nitric oxide synthase-6xHis (NOS-6xHis) (unpublished, V.K. Singh, A.T. Still University) and methionine sulfoxide reductase B-6xHis (MsrB-6xHis).(17) Both are staphylococcal proteins overproduced from plasmids derived from pRSET A and carried in E. coli BL21 cells. The proteins were solubilized in electrophoresis sample buffer and applied to 3-cm-wide wells of a minigel at ∼5 μg/lane. After electrophoresis and transfer to PVDF, strips containing each protein were Western blotted with the MAbs as described above. Positive control strips were probed with HisDetector nickel-alkaline phosphatase conjugate (Ni2+-AP) diluted 1:20,000 in Detector Block Solution (Kirkegaard & Perry Laboratories, Gaithersburg, MD). MAbs that reacted with all three His-tagged proteins were scored as specific for the His tag rather than the protein to which the tag was attached. MAbs that reacted only with RadA-6xHis, but not with tagless RadA or the His-tagged staphylococcal proteins, were scored as specific for an epitope comprising the junction between the C-terminus of the His tag and the N-terminus of RadA protein.
Mapping of MAb epitope specificities on RadA-6xHis
A peptide map of RadA-6xHis was generated by partial digestion of the purified protein with staphylococcal V8 protease (endoproteinase Glu-C, Sigma-Aldrich). RadA-6xHis (30 μg) purified by passive elution from polyacrylamide gels was mixed with 1.5 μg of V8 protease in PBS (pH 7.4) and incubated in a 37°C waterbath. Aliquots containing 10 μg of partially digested RadA-6xHis were removed after 10 min, 60 min, and 120 min incubation times. The digests were mixed with 4X electrophoresis sample buffer (0.25 M Tris [pH 6.8], 40% glycerol, 8% SDS, 8% 2-ME), boiled 5 min to stop digestion, and immediately frozen at −80°C until analyzed by SDS-PAGE and Western blotting. An undigested control was prepared by solubilizing 10 μg of RadA-6xHis directly in sample buffer without enzyme co-incubation. Each sample was applied to a 3-cm-wide well of a 15% polyacrylamide minigel. After electrophoresis and transfer of peptides to PVDF, strips cut from each section were stained with Coomassie Blue R-250 or probed with individual MAbs. Incubation of the strips in MAb solutions (2 μg/mL) proceeded overnight at 4°C, and was followed by a 2 h incubation at room temperature in 1:4000 goat anti-mouse IgG-alkaline phosphatase conjugate (Sigma-Aldrich). As before, strips were washed between steps with TTBS and developed in NBT/BCIP substrate.
Results
Purification of RadA-6xHis from strain SR4097 lysates
RadA-6xHis was enriched from strain SR4097 cell lysates by nickel affinity chromatography. As shown in Figure 1, the major product eluted from the affinity column (lane 2) migrated with the same mobility as the 50 kDa marker (lane 1), consistent with the calculated mass of RadA-6xHis (53.6 kDa). To minimize contaminating proteins, RadA-6xHis was passively eluted from polyacrylamide gel slices, yielding a product of single-band purity, as assessed by both Coomassie staining (lane 3) and blotting with Ni2+-AP (lane 4). This highly purified product was used as an intravenous booster immunogen in spleen donor mice, and as an antigen to screen MAbs by ELISA, Western blotting (Figs. 2, 3), and epitope mapping (Fig. 4).
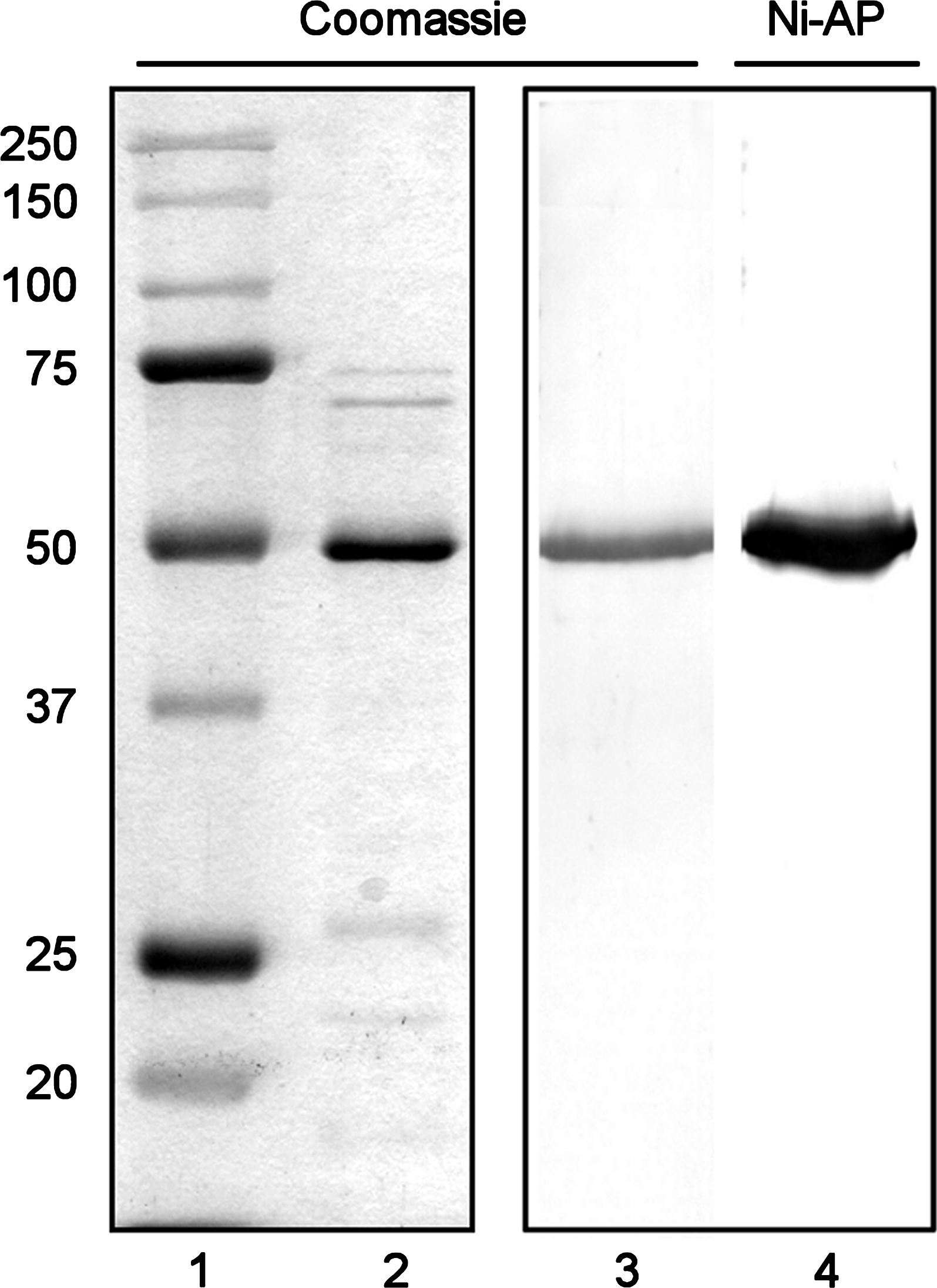
Purity of the RadA-6xHis fusion protein obtained by affinity chromatography and gel elution. The RadA-6xHis fusion protein was purified from E. coli SR4097 cells by nickel affinity chromatography (0.5 μg, lane 2) or by passive elution from polyacrylamide gel slices (0.2 μg, lanes 3 and 4) and resolved by SDS-PAGE through 12% polyacrylamide gels. Lanes 1–3 were stained with Coomassie Blue R-250. Lane 4 was transferred to PVDF and probed with Ni2+-AP conjugate to detect the His tag. Lane 1 contains 5 μL of Precision Plus dual-color pre-stained standards (Bio-Rad). Masses are expressed in kDa.
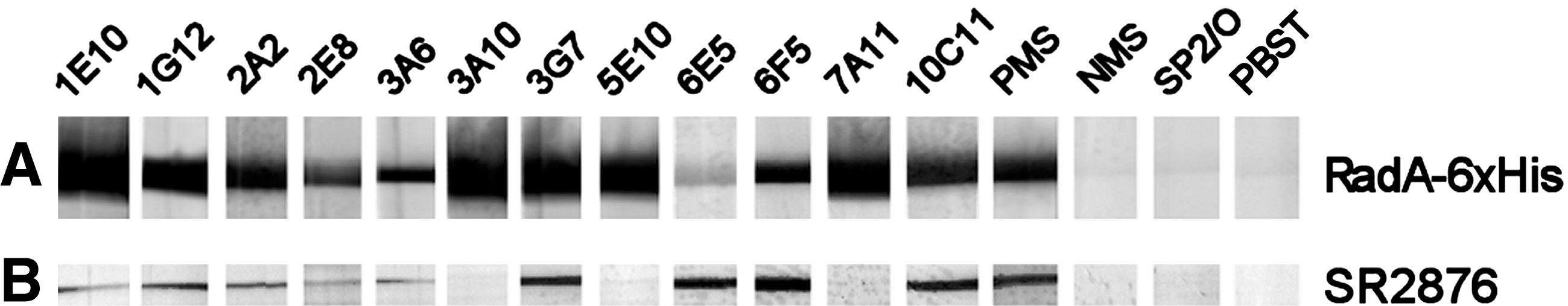
Western blot analysis of hybridoma supernatants. Hybridoma supernatants were Western blotted against (
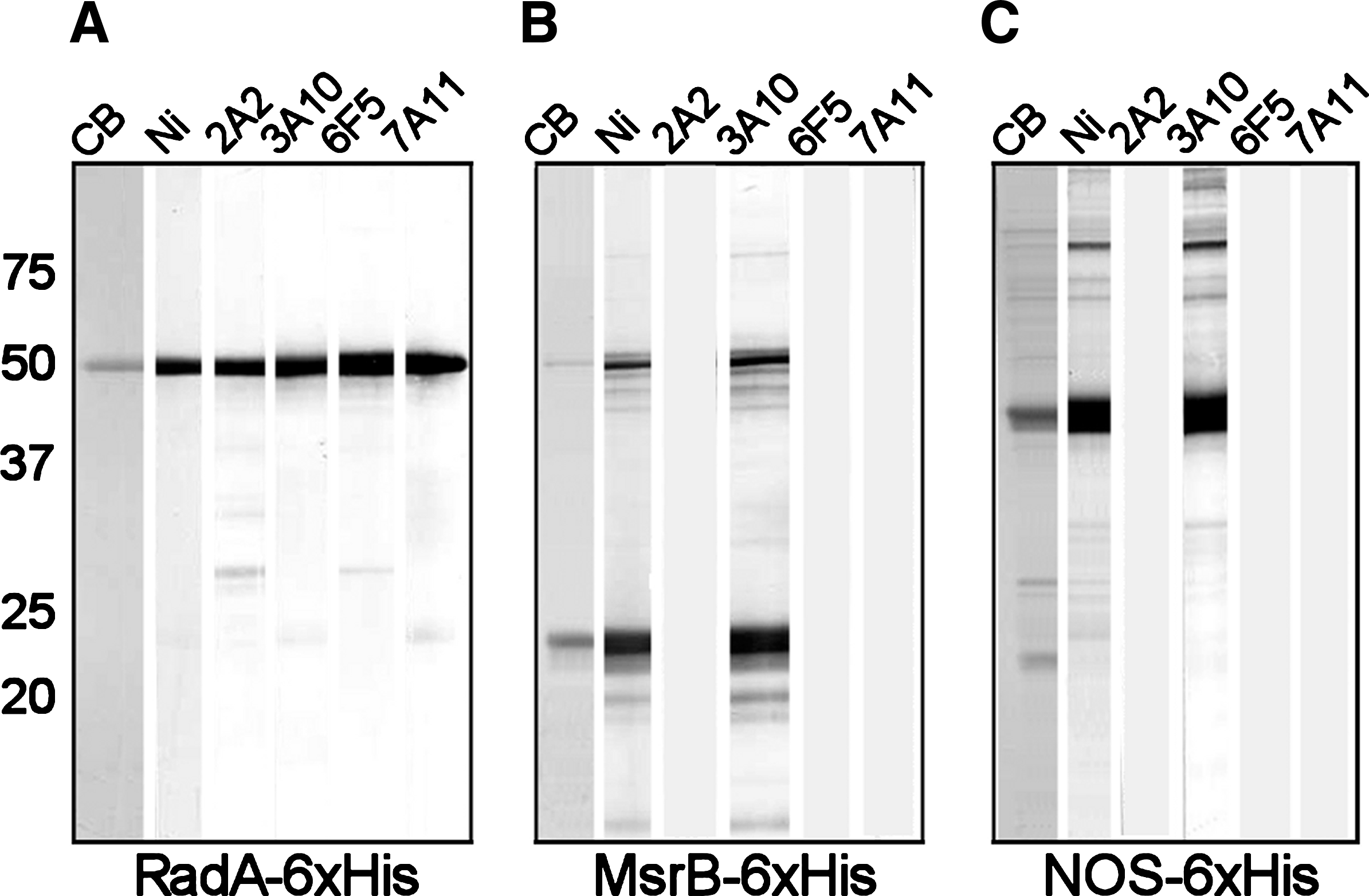
Western blot analysis of His-tagged proteins probed with monoclonal antibodies generated to RadA-6xHis. Each 3-cm wide lane of a 12% gel was loaded with ∼5 μg of a His-tagged protein produced from a modified pRSET A plasmid carried in BL21 cells. The proteins included (

Epitope mapping of monoclonal antibody specificities. Each 3-cm-wide lane of a 15% gel was loaded with 10 μg of RadA-6xHis that was (
Detection of MAbs in hybridoma supernatants
In ELISA screening assays, 101 hybridoma supernatants yielded A405 values at least 5-fold higher than Sp2/0-Ag14 negative controls against purified RadA-6xHis (data not shown). Twelve hybridoma lines with the highest and most consistent ELISA reactivities were screened by Western blot analysis (Fig. 2) against purified RadA-6xHis; all of these recognized the fusion protein at ∼53 kDa (Fig. 2A). The MAbs were further screened by Western blot for reactivity to tagless RadA produced by E. coli strain SR2876 (Fig. 2B). Nine lines were reactive to tagless RadA, while three lines (MAbs 3A10, 5E10, and 7A11) failed to detect the tagless protein. Based on these results, four MAbs were chosen for additional analysis: MAbs 2A2 and 6F5 recognized both purified RadA-6xHis and tagless RadA in SR2876 lysates, while MAbs 3A10 and 7A11 recognized only RadA-6xHis. Isotyping analysis showed that MAbs 3A10, 6F5, and 7A11 are IgG1 antibodies, while MAb 2A2 is an IgG2b antibody (data not shown).
The antibodies were further tested by Western blot analysis against two additional His-tagged proteins, MsrB-6xHis and NOS-6xHis, both containing the same N-terminal 36 amino acid tagged as RadA-6xHis (Fig. 3). MAbs 2A2, 6F5, and 7A11 were reactive to RadA-6xHis (Fig. 3A), but not to MsrB-6xHis (Fig. 3B) or to NOS-6xHis (Fig. 3C). MAb 3A10, however, was reactive to all three 6xHis-tagged proteins and mirrored Ni2+-AP reactivity. From these data, we inferred that MAbs 2A2 and 6F5 recognize epitopes on the RadA protein, while MAb 3A10 is specific for an epitope confined to the 36 amino acid tag. MAb 7A11, which recognizes RadA-6xHis but does not bind to tagless RadA or to either of the 6xHis-tagged staphylococcal proteins, apparently recognizes an epitope that is formed by the junction of the His tag and RadA protein.
Epitope mapping of RadA-6xHis
Purified RadA-6xHis partially digested with staphylococcal V8 protease was probed with the MAbs to compare the epitope specificities of the antibodies (Fig. 4). After 120 min of digestion, bands containing intact RadA-6xHis at 52.2 kDa and cleavage fragments at 35.4 and 23.9 kDa were stained with nearly equal intensities by Coomassie Blue (Fig. 4B, lanes 2, 8). Minor bands could also be seen at 49.4, 34.1, 28.3, 21.0, and 20.3 kDa by Coomassie staining. All four MAbs recognized intact RadA-6xHis and its 35.4 kDa cleavage fragment (Fig. 4B, lanes 4–7). Intact RadA-6xHis and fragments of 35.4, 21.0, and 20.3 kDa all reacted with the Ni2+-AP conjugate (Fig. 4B, lane 3), indicating that these polypeptides contained the N-terminus bearing the His tag. In keeping with the hypothesis that epitopes recognized by MAbs 3A10 and 7A11 are formed completely or in part by the tag, it is noteworthy that both of these MAbs recognized all fragments recognized by Ni2+-AP (Fig. 4B; compare lanes 3, 5, and 7). Additional fragments that migrated between the 25 kDa and 37 kDa markers were detected by MAbs 3A10 and 7A11 but not by Ni2+-AP; these apparently lack the histidine residues at positions 5–10 but contain most of the remaining residues in the 36 amino acid tag of the fusion protein. Besides the 35.4 kDa cleavage fragment, MAbs 2A2 and 6F5 both recognized a fragment of 28.3 kDa lacking the N-terminus (deduced by its non-reactivity with Ni2+-AP), but only MAb 6F5 recognized the 23.9 kDa fragment. MAb 6F5 also recognized a smaller N-terminal fragment (20.3 kDa) than MAb 2A2, suggesting that the 6F5 epitope is upstream of the 2A2 epitope on the RadA protein. The potential binding sites for the MAbs are shown schematically in Figure 5.
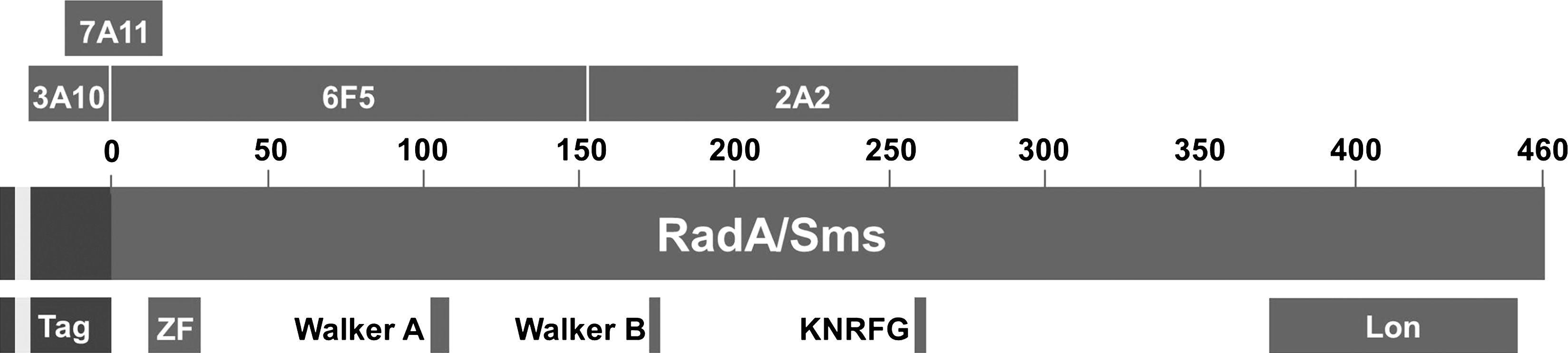
Potential binding sites for MAbs generated to RadA-6xHis. The potential binding sites for MAbs 2A2, 3A10, 6F5, and 7A11 on the RadA/Sms protein, determined by peptide mapping experiments described in Figure 4, are shown. The locations of the N-terminal tag (including 6xHis shaded in light gray), zinc finger (ZF) motif, Walker ATP-binding motifs, KNRFG nuclease-like region, and Lon protease-like region are indicated.
Discussion
In this study we produced four hybridoma cell lines secreting monoclonal antibodies to different epitopes on RadA-6xHis. MAb 2A2 is an IgG2b antibody specific for a RadA epitope located between 20.3 kDa and 35.4 kDa from the N-terminus of the RadA-6xHis fusion protein. MAbs 3A10, 7A11, and 6F5 are IgG1 antibodies whose binding sites on RadA-6xHis are located upstream of the MAb 2A2 epitope. MAbs 3A10 and 7A11 recognize epitopes formed completely or in part by the 36 amino acid tag at the N-terminus of RadA-6xHis. MAb 6F5 recognizes an epitope located within 20.3 kDa of the N-terminus of RadA-6xHis that is not dependent on the tag for its conformation.
The locations of several putative functional regions of the RadA protein are known (Fig. 5), enabling predictions regarding the potential of each MAb to affect RadA activity through steric interference or by inducing conformational changes in the RadA protein. Keeping in mind that the 36 amino acid tag adds 4.1 kDa to the mass of RadA-6xHis, MAb 6F5 recognizes an epitope that lies within 16.2 kDa of the N-terminus of tagless RadA in the K-12 strain of E. coli (NCBI nr GI:1401238). This region contains a zinc finger with a CXXC-Xn-CXXC motif between residues 11-28, and a nucleotide-binding P-loop with the Walker A sequence motif GxxGxGK(T/S) spanning amino acids 102-109.(4) The importance of zinc fingers in the function of other DNA repair proteins has been demonstrated. For example, mutations in the zinc finger of the RecQ DNA helicase impede DNA binding and confer instability to the protein as a whole.(18) The importance of the zinc finger in RadA is not yet defined in terms of DNA binding. However, the original radA100 mutation is the result of substitution of cysteine by tyrosine at amino acid residue 28 and is believed to disrupt the zinc finger in RadA protein.(3) The importance of the zinc finger to RadA protein function is supported by the finding that the radA100 mutant shows the same diminished cell survival after treatment with DNA damaging agents as a ΔradA mutant.(3) Because MAb 6F5 is a large molecule (∼150 kDa), it may sterically interfere with zinc finger-DNA binding and provide mechanistic information on the importance of the RadA zinc finger in future protein-DNA interaction studies. MAbs 3A10 and 7A11, which recognize epitopes near the N-terminus of the fusion protein, might similarly impede interactions between DNA and the zinc finger of RadA-6xHis fusion protein.
The binding site for MAb 2A2 is located between 16.2 kDa and 31.3 kDa from the N-terminus of tagless RadA. This region contains the Walker B consensus sequence hhhhD/E between residues 173-177, where h represents a hydrophobic amino acid preceding an aspartic acid or glutamic acid.(19) The Walker A and B motifs are conserved among ATPases; they confer ATP binding and hydrolysis activities that provide energy for DNA binding, helicase activity, branch migration, and the assembly of polymeric protein ring structures (e.g., RuvB hexamers) necessary for DNA recombination and repair.(20–22) Downstream of the Walker motifs, the RadA/Sms consensus sequence KNRFG is located between amino acids 258-262 of tagless RadA, offering another potential binding/steric interference site for MAb 2A2. The KNRFG motif is similar to the KNRXG motif of the mycobacteriophage D29 gp65 protein. Mutation of KNRXG in gp65 abolishes its nuclease activity, which is specific for forked DNA structures.(23) The RadA/Sms protein also contains a region of homology to Lon, a serine protease that functions in the elimination of mutant and abnormal proteins and in the rapid turnover of certain short-lived regulatory proteins.(24) However, the Lon-like region (NCBI CDD: pfam05362), which spans amino acids 372-454 of tagless RadA, lies outside the portion of the protein recognized by the monoclonal antibodies characterized in this study.
Understanding DNA repair is critical to understanding carcinogenesis (reviewed by Lahtz and Pfeifer(25)). For example, mutations in the gene encoding p53 can explain half of all human cancers.(26) Studies in DNA repair genes in E. coli have provided, and they continue to provide, great insight into how DNA repair is accomplished and its roles in carcinogenesis and other human diseases (reviewed by Krwawicz and colleagues(27)). The MAbs described here will provide valuable tools for studying the poorly understood role of RadA/Sms protein in DNA repair, which should ultimately provide insights into carcinogenesis in humans.
Footnotes
Acknowledgment
This research was supported by the Master of Science in Biomedical Sciences program at A.T. Still University – Kirksville College of Osteopathic Medicine.
Author Disclosure Statement
The authors have no financial conflicts to disclose.