
Abstract
Protein tyrosine phosphatase 1B (PTP1B), a member of the protein tyrosine phosphatase (PTP) family, plays a crucial role in metabolic signaling, with insulin and leptin signaling being well studied. New evidence indicates that PTP1B is also involved in cancer. In the present study, we report on the establishment of a monoclonal antibody specific for catalytic domain of PTP1B (PTP1Bc) generated through the hybridoma method. The monoclonal antibody is measured to have a titer of 4.1×106 against PTP1Bc in indirect ELISA. Western blot and immunofluorescent analyses indicated that this antibody can specifically combine native PTP1B in MDA-MB-231 and MDA-MB-453 cells. This monoclonal antibody against PTP1Bc can help enhance the understanding of PTP1B-related physiological and pathological mechanisms and may act as a therapeutic agent for diabetes, obesity, and cancer in the future.
Introduction
PTP1B is comprised of 435 amino acids with a molecular mass of 50 kDa and is expressed in a variety of organ tissues, such as liver, muscle, and adipose. PTP1B contains an N-terminal catalytic domain (1–300 residues), a proline-rich domain, and a C-terminal ER targeting domain. The ER targeting domain anchors the whole molecule into the cytoplasmic face of the endoplasmic reticulum (ER),(7) while the proline-rich domain was found to function properly in substrate binding and PTP1B activity in vitro.(8) The catalytic domain is recognized to be the active site involved in the removal of phophotyrosine residues on PTP1B substrates, such as insulin receptor (IR)(9) and endothelial growth factor receptor (EGFR).(10)
PTP1B has long been recognized to be a key modulator of insulin and leptin signaling. In insulin signaling, PTP1B-mediated dephosphorylation of IR inhibits IR signaling cascade and contributes to insulin resistance.(9) In leptin signaling, PTP1B targets a downstream element, the tyrosine kinase JAK2 (Janus kinase 2), and depholphorylates the kinase to influence the leptin signaling.(2) New evidence indicates that PTP1B is also involved in cancer, most notably breast(5) and ovarian cancer.(6) Recent findings suggested that PTP1Bc can act as a tumor suppressor and tumor promoter, depending on the substrate involved and the cellular context. For these reasons, PTP1B has attracted particular attention as a potential therapeutic target for diabetes, obesity, and cancer.
The purpose of this study was to establish a monoclonal antibody against PTP1Bc for use in PTP1B detection and for therapy relating to diabetes, obesity, and cancer.
Materials and Methods
Materials
RPMI 1640 and fetal bovine serum were purchased from Gibco BRL (Grand Island, NY). HAT medium and PEG solution were purchased from Sigma (St. Louis, MO). Ni2+ Sepharose column and rProtein A Sepharose column were purchased from GE Healthcare (Uppsala, Sweden). All cell lines and BALB/c mice were from the Cancer Research Center and Experimental Animal Center of Xiamen University of China, respectively.
Expression of PTP1Bc fusion protein
The recombinant vector of pET-22b(+)/PTP1Bc, constructed in our previous study,(11) was transformed into Escherichia coli BL21 (DE3) and cultured under isopropyl-beta-D-thiogalactopyranoside (IPTG) induction for 4 h at 37°C. The culture was centrifuged at 5000 g for 30 min at 4°C, and the bacteria were collected and lysed with lysozyme solution (20 mmol/L Tris-HCl [pH 8.0], 0.5 mol/L NaCl, 1 mmol/L EDTA, 1 mg/mL lysozyme) for 8 h at 4°C. Repetitive sonication was carried out to help DE3 dissolve and the inclusion body containing the PTP1Bc fusion protein was collected by centrifugation at 10,000 g for 30 min at 4°C. The collection was washed three times with washing buffer (20 mmol/L Tris/HCl [pH 8.0], 0.5 mol/L NaCl, 2mol/L urea, 20 mL/L Triton X-100) and solubilized by magnetic stirring in denaturation buffer (20 mmol/L Tris-HCl [pH 8.0], 8 mol/L urea, 1 mmol/L β-mercaptoethanol, 20 mL/L Triton X-100) overnight at 4°C. After centrifugation at 12,000 g for 30 min at 4°C, the supernatants were purified by Ni2+ Sepharose column. The purified PTP1Bc was dialyzed in renaturation buffer (0.4 mol/L Tris, 2.5 mmol/L PEG 4000, 133.3 mmol/L glycine, 0.4 mol/L L-arginine, 10 mL/L glycerol, in PBS) with decreasing concentration of urea. The protein concentration was determined by modified Bradford protein assay and the purity of the protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie Blue staining.
Immunization of mice
Eight BALB/c mice (female, 6–8 weeks old) were chosen and each was subcutaneously injected with 50 μg purified PTP1Bc (in 0.4 mL PBS) fully emulsified with 0.4 mL Freund's complete adjuvant. Two weeks later, each mouse was boosted with 50 μg PTP1Bc in Freund's incomplete adjuvant. The booster injection was repeated every 2 weeks for three injection times. Serum from the tail vena was monitored for their antibody titers against PTP1Bc by indirect ELISA. Mice with sustained antibody titers above 1×104 were selected and intravenously injected with 25 μg PTP1Bc without Freund's adjuvant 3 days before cell fusion.
Establishment of hybridomas
Spleen cells from the selected mice were fused with myeloma cells (SP2/0 cell line). The fusion was at a cell ratio of 1:5 (spleen to myeloma) in the presence of 50% polyethylene glycol (PEG) according to Kohler and Milstein.(12) The fusion cells were cultured in HAT medium in 96-well plates. All cell colonies were selected and their supernatants were detected for antibody titers against PTP1Bc. Cell colonies with high titer were chosen and cloned by the limiting dilution method(12,13) three times to establish hybridoma cell lines secreting monoclonal antibody (MAb). The four hybridoma cell lines with the highest titers were selected for further investigation.
Indirect ELISA was performed as follows: 10 μg/mL purified PTP1Bc in coating buffer (0.05 M bicarbonate, pH 9.6) was coated in the 96-well plates overnight at 4°C. The plates were blocked with 5% fat-free milk (200 μL/well) at 37°C for 2 h and washed with PBS-T (0.05% Tween-20 in PBS) three times. The supernatants of serum or hybridoma cell culture were incubated in the plates for 1 h at 37°C. After washing, goat anti-mouse IgG-HRP was added and incubated for 1 h at 37°C. O-phenylenediamine (OPD) was added to develop color and the optical density (OD) was measured at 490 nm by a microplate reader (model 680, Bio-Rad, Tokyo, Japan).
MAb production
One hybridoma cell line detected to secrete antibody with the highest titers was massively cultured for hybridoma injection. Twenty BALB/c mice (female, 6–8 weeks old) were intraperitoneally injected with sterile paraffin oil (0.5 mL per mouse) 7 days before the hybridoma injection. Each mouse was injected with 2×105–106 hybridoma cells. Seven to 10 days later, ascites were collected and centrifuged at 10,000 g for 30 min to obtain the supernatant. The supernatant of the ascites was further purified by rProtein A Sepharose column according to the manufacturer's protocol. The purity and concentration of the purified MAb (designated as Garbu MAb) was analyzed by SDS-PAGE and Bradford protein assay, respectively.
Titer analysis
For the titer analyses, supernatant of the MAb ascites, the Garbu MAb, and a negative control antibody (mouse IGg1, monoclonal anti-β-actin antibody, Sigma) were tested by indirect ELISA as described above. Data were analyzed and graphed with OriginPro 8.1 software (OriginLab, Northampton, MA).
MAb isotyping
The class and subclass of the Garbu MAb were identified by a mouse monoclonal antibody isotyping reagent (Sigma) according to the manufacturer's directions.
Western blot analysis
Both MDA-MB-231 cells and MDA-MB-453 cells (about 1×107) were lysed in 1 mL RIPA solution (containing 1% Triton X-100, 1% deoxycholate, and 0.1% SDS, Beyotime, Jiangsu, China) for 30 min at 4°C and centrifuged at 12,000 g for 30 min at 4°C. Supernatants of these lysates and purified PTP1Bc were loaded to the SDS-polyacrylamide gels and electrophoretically transferred to PVDF membranes by Mini-Protean (Bio-Rad, Hercules, CA). Garbu MAb diluted with PBS at 1:1000 was used as the primary antibodies to incubate with the membranes overnight at 4°C. After being washed with PBST three times, the membranes were incubated with goat anti-mouse IgG-HRP conjugate (1:3000 dilution) for 1 h at 37°C. Proteins of interest were detected by enhanced ChemiLuminescence (ECL, Sangon, Shanghai, China) in a Kodak Image Station 4000R (Carestream Health, Rochester, NY).
Immunofluorescent analysis
MDA-MB-453 cells were cultured on glass cover slides overnight at 37°C and fixed with 4% paraformaldehyde at room temperature for 30 min. Non-specific binding was blocked with blocking solution (containing 1% BSA, 5% goat serum, 0.2% NaN3) at room temperature for 30 min. The slides were then washed with PBS three times and incubated with the Garbu MAb (1:1000 dilution) at 37°C for 1 h. After washing, the cells were mounted with fluoresceinisothiocyanate (FITC)-conjugated goat anti-mouse antibody (Boster Bio-Engineering, Wuhan, China) and re-stained with Hoechst 33258 (Beyotime Biotechnology, Nantong, China). The fluorescence images were captured by Olympus IX51 Inverted Fluorescence Microscope (Olympus, Melville, NY).
Results
Expression and purification of PTP1Bc fusion protein
PTP1Bc fusion protein (∼40 kDa) was expressed in DE3 with 0.3 mM IPTG induction for 4 h at 37°C and purified by Ni2+ Sepharose column. Results of lanes 1 and 2 (Fig. 1) showed that PTP1Bc fusion protein was not expressed significantly without IPTG induction and highly expressed under 0.3 mM IPTG induction. Lanes 3 and 4 showed PTP1Bc fusion protein was mainly located in the DE3 inclusion bodies. Three samples of purified PTP1B fusion protein (lanes 5–7, Fig. 1) were measured by Bradford protein assay to be 0.68 mg/mL, 0.44 mg/mL, and 0.22 mg/mL, respectively. The purified PTP1Bc migrated at molecular masses of ∼40 kDa, which fits the calculated molecular weight of ∼39.3 kDa (128 kDa per amino acid ×307 amino acids).
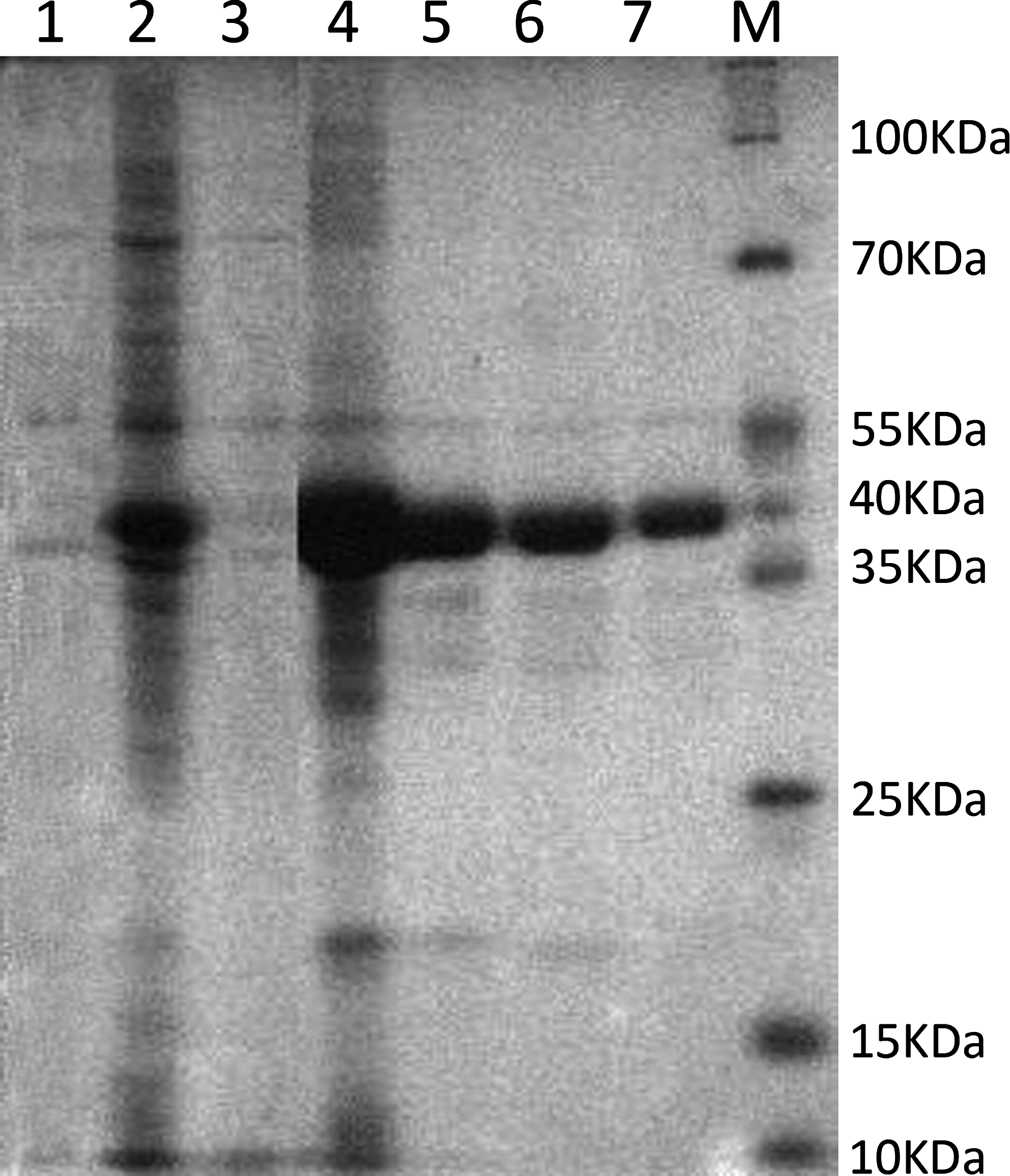
SDS-PAGE analysis for the expression and purification of PTP1Bc in DE3. M, protein marker; lane 1, DE3 culture without IPTG induction; lane 2, DE3 culture after 0.3mM IPTG induction; lane 3, supernatant of DE3 culture with 0.3 mM IPTG induction; lane 4, precipitate of DE3 culture with 0.3 mM IPTG induction; lanes 5–7, purified PTP1Bc in different concentrations.
Generation of hybridomas secreting MAb against PTP1Bc
At 10–12 days after spleen cells and myeloma cells fusion, supernatants of growing colonies were detected for their ability to secrete specific antibodies by indirect ELISA. Four growing colonies that were detected to secrete MAb with high titers (designated as D2, E5, C4, and F3) were chosen to subclone to 96-well plates. After three rounds of subcloning and detection, four hybridomas were established and the D2-31-18-11 hybridoma (designated as Garbu hybridoma) with highest titer by indirect ELISA was chosen to produce MAb ascites.
SDS-PAGE analysis of MAb ascites and purified MAb
MAb ascites and the Garbu MAb were loaded to a pre-cast gel for the SDS-PAGE analysis. Figure 2 shows two bands in lane 1 and lane 2 with molecular masses of ∼50 kDa and ∼25 kDa, representing the heavy chain and light chain of antibodies, respectively.
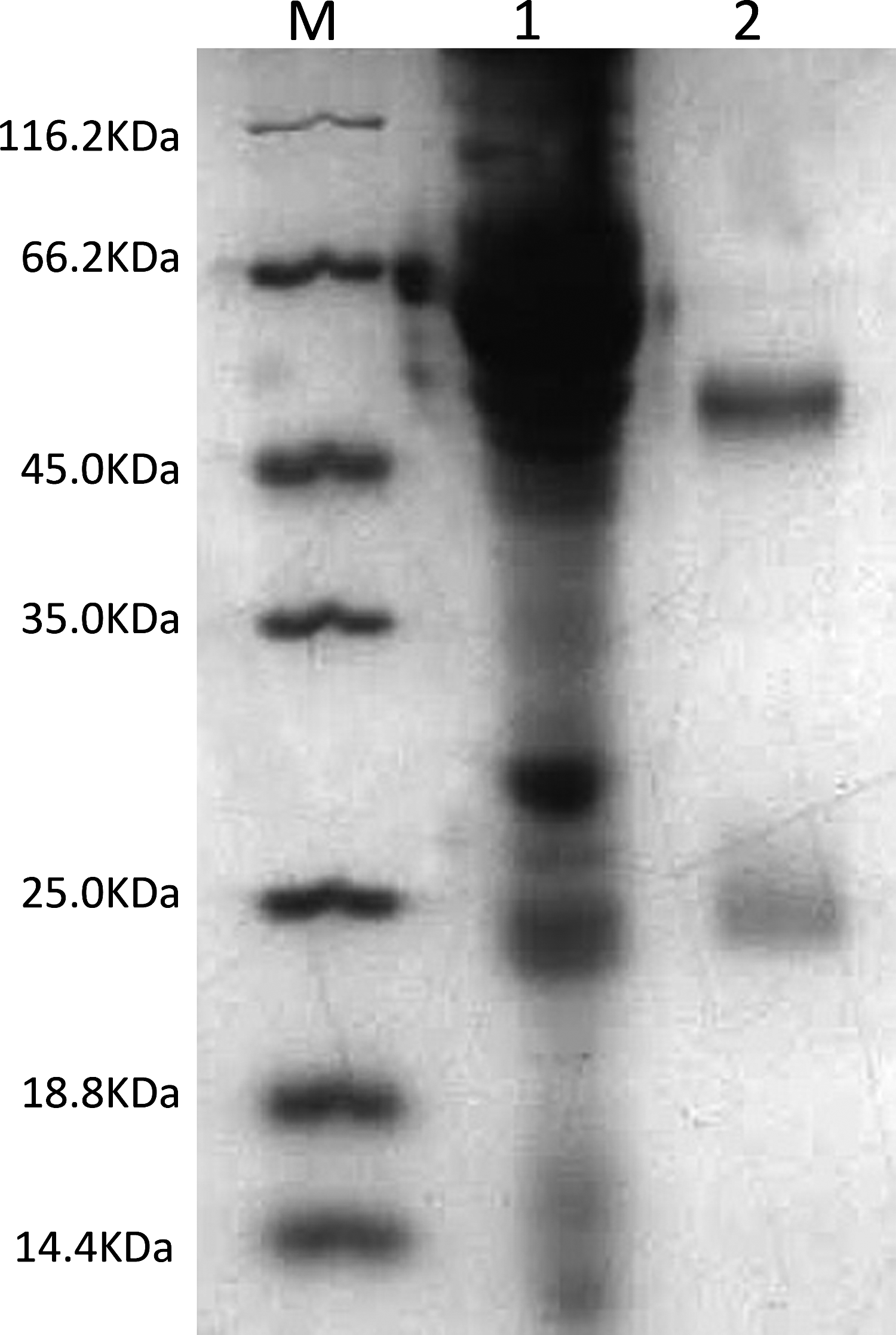
SDS-PAGE analysis of MAb ascites and Garbu MAb. M, protein marker; lane 1, MAb ascites; lane 2, Garbu MAb.
Isotype identification of the MAb
The results of the isotyping analysis for the Garbu MAb indicated that it was IgG1 isotype.
Titer analysis
Serial dilutions of MAb ascites and the Garbu MAb were measured in indirect ELISA for their titers against PTP1Bc. As shown in Figure 3, MAb ascites and Garbu MAb recognized the fusion protein PTP1Bc with titer of 4.1×106 and 2.6×105, respectively.
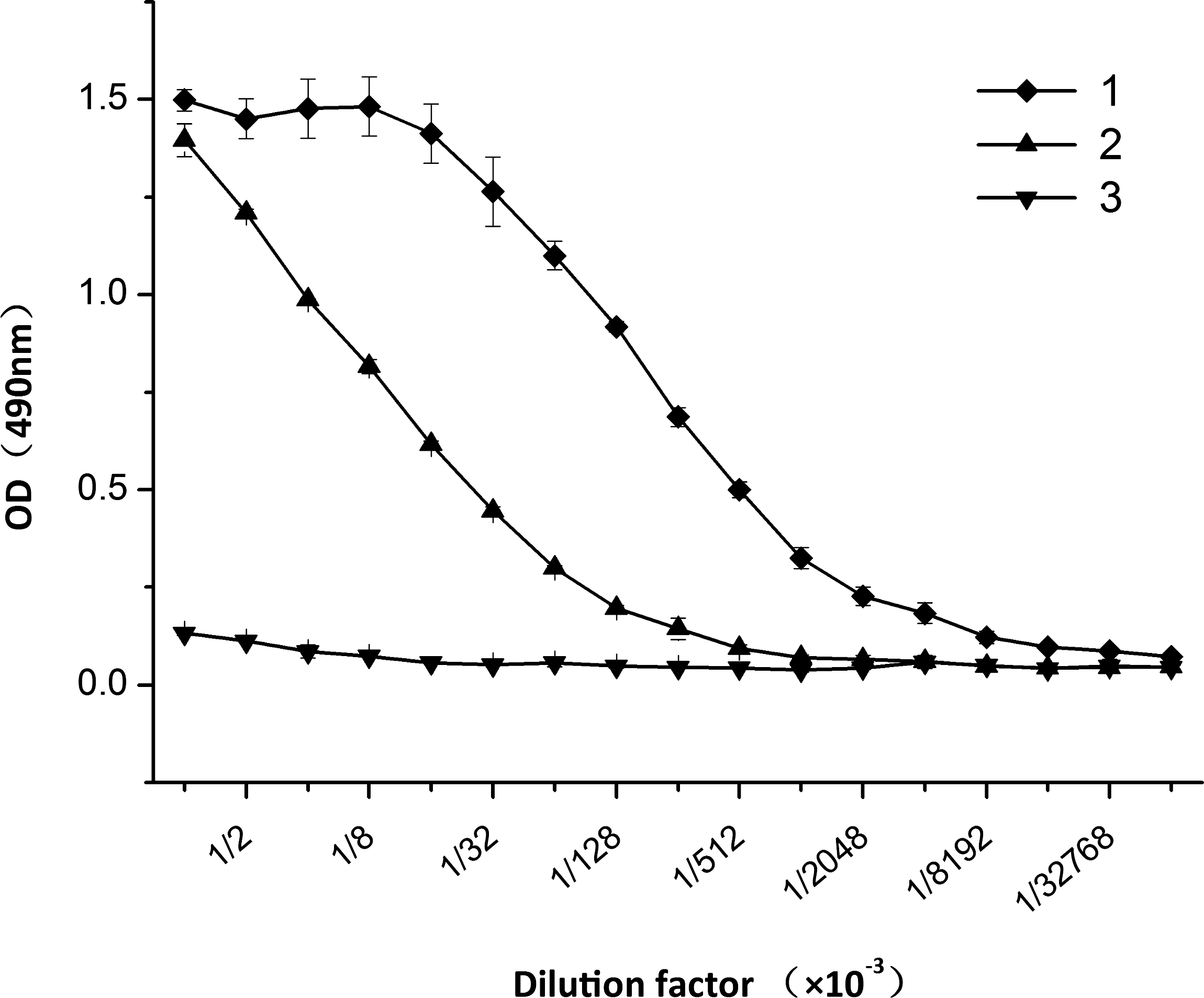
Titration analysis of MAb ascites and Garbu MAb. 1, MAb ascites; 2, Garbu MAb; 3, mouse IgG1 as control. The coating concentration of PTP1Bc was 10 μg/mL. Each point represents the OD values with the mean±standard deviation from six determinations (n=6).
Western blot analysis
Figure 4 shows that the Garbu MAb detected the PTP1Bc fusion protein at a molecular weight of ∼40 kDa (lane 3), as well as the native protein in the extracts of MDA-MB-231 (lane 1) and MDA-MB-453 (lane 2) at molecular weight of ∼50 kDa.
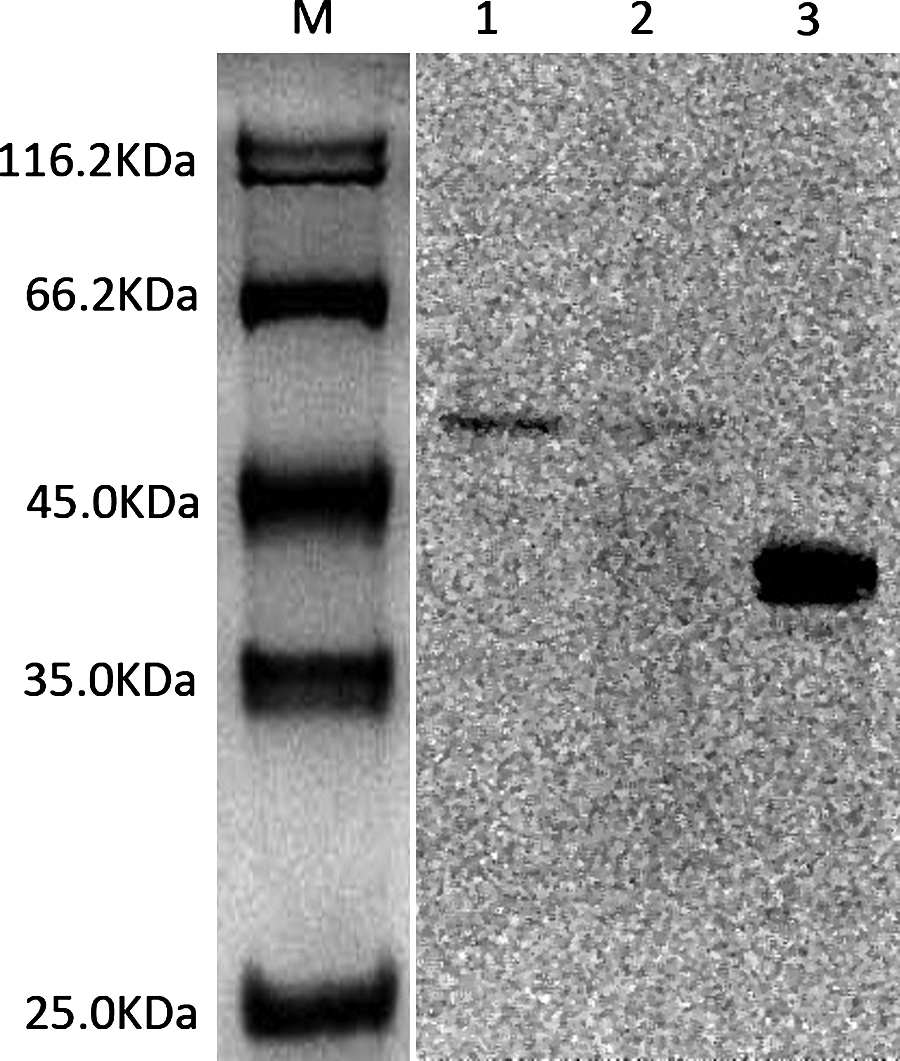
Western blot analysis with Garbu MAb. M, protein marker; lane 1, extracts of MDA-MB-231 cells; lane 2, extracts of MDA-MB-453 cells; lane 3, purified PTP1Bc fusion protein.
Immunofluorescent analysis
The binding specificity of the Garbu MAb with the MDA-MB-231 and MDA-MB-453 cells was characterized by immunofluorescence analysis. Results shown in Figure 5 revealed a bright signal around the nucleus of the cells, which was identical to the location of PTP1B in cells (PTP1B was expressed on the cytoplasmic face of the endoplasmic reticulum).
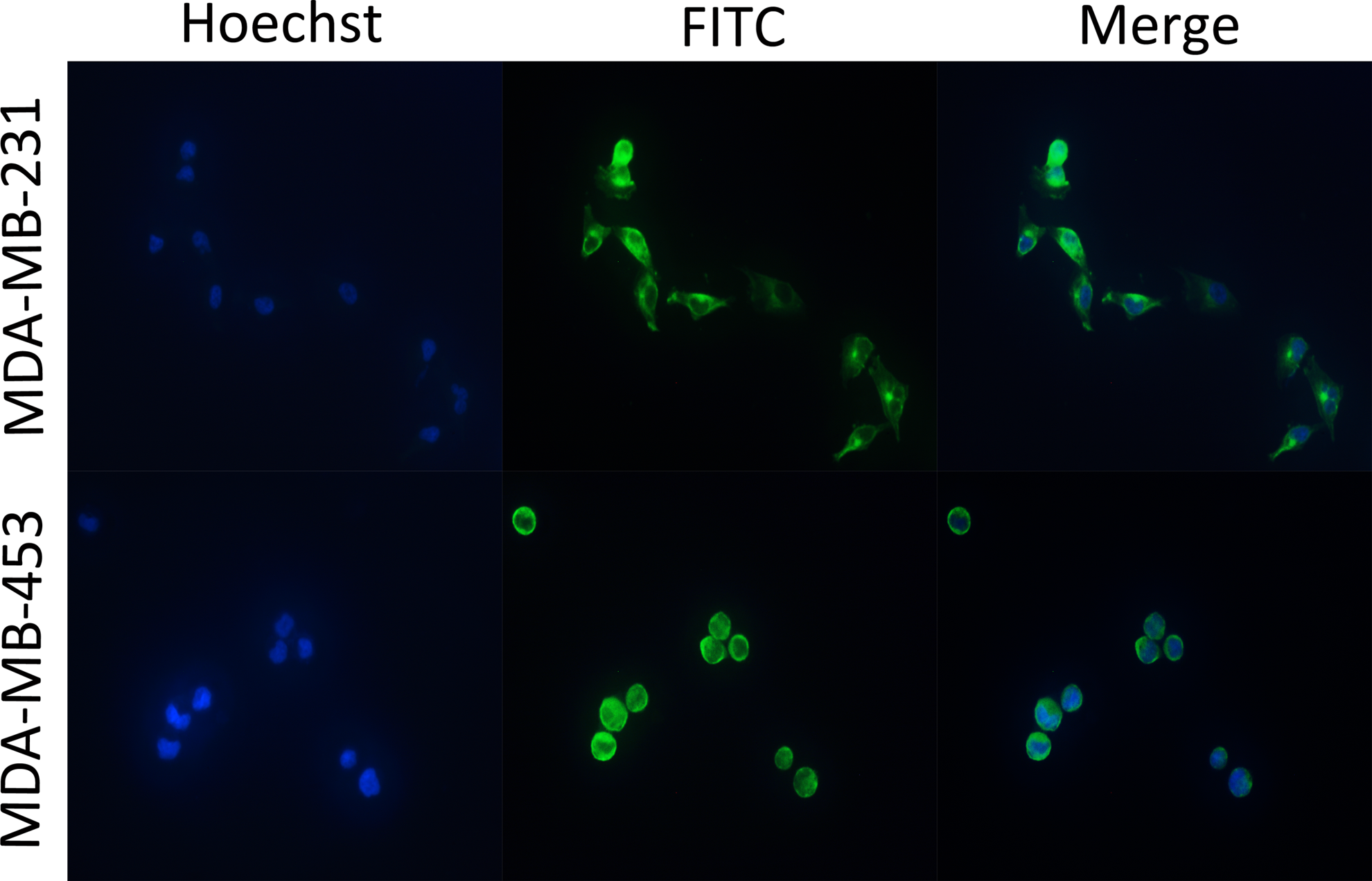
Immunofluorescent analysis of MDA-MB-231 and MDA-MB-453 cells with Garbu MAb.
Discussion
PTP1B, a member of the super family of protein tyrosine phosphatases, dephosphorylates its substrates to activate them and correspondingly mediates their downstream signaling.(1) This enzyme has been known to play a crucial role in metabolism and is reported to be associated with cancer, particularly breast cancer(5) and ovarian cancer.(6)
As the catalytic domain of PTP1B is the functional site of dephosphorylation, we chose this domain as the target of MAbs and used the PTP1Bc fusion protein as immunogen. Although anti-PTP1Bc has been reported, it is often desirable to obtain the hybridoma cells secreting specific MAb for several reasons: first, the hybridoma allows massive MAb production by hybridoma cell injection of the mice; second, the hybridoma RNA provides the gene fragments of the heavy chains and light chains to generate engineered antibody with favorable properties (for example smaller recombinant antibodies and engineered variants); third, based on our work, monoclonal antibodies against different epitopes of PTP1Bc would be generated for use in the further understanding of the role of PTP1B both structurally and functionally.
In our study, we successfully developed a stable hybridoma secreting MAb against PTP1Bc. Titers of MAb ascites (2.6×105) and the Garbu MAb (4.1×106) (Fig. 3) showed this MAb has high affinity against PTP1Bc. Both PTP1Bc fusion protein and native PTP1B in MDA-MB-231 and MDA-MB-453 cells can be specifically combined by the Garbu MAb in Western blot analysis. Immunofluorescent analysis confirmed that the Garbu MAb specifically recognized the native PTP1B in the MDA-MB-231 and MDA-MB-453 cells.
In conclusion, we established an MAb against PTP1Bc that may be useful in the detection of PTP1B and may be used to treat diabetes, obesity, and cancer in the future.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant nos. 30973485, 81172970) and the Science and Technology Foundation of Fujian Province of China (grant no. 2011R1039-1).
Author Disclosure Statement
The authors have no financial interests to disclose.