
Abstract
Pyruvate carboxylase [EC 6.4.1.1] plays an important anaplerotic role in many species by catalyzing the carboxylation of pyruvate to oxaloacetate. To extend our understanding about the structure and function of pyruvate carboxylase (PC), a series of monoclonal antibodies were raised against sheep liver PC and those displaying inhibitory activity were further characterized. The binding epitopes of two monoclonal antibodies that displayed strong inhibitory activity were mapped. Six overlapping fragments of the human enzyme were expressed as thioredoxin fusion proteins in Escherichia coli and subjected to Western blot analysis. Both monoclonal antibodies (MAbs) recognized fragments encompassing the enzyme's C-terminal region, known to contain the structured biotin domain. Through deletion analysis, this domain was determined to be a minimal size of 80 amino acids. Further deletions that disrupted the conformation of the domain abolished antibody binding, indicating these antibodies recognized discontinuous epitopes. To further define the critical residues required for antibody recognition, a model of the domain was produced and an alanine scan performed on selected surface-exposed residues. Our results show that residues encompassing the biotin attachment site, but not biotin itself, are critical for the binding of both antibodies. These data provide a mechanism to explain the inhibitory activity of the antibodies.
Introduction
Pyruvate carboxylase [EC 6.4.1.1] plays an important anaplerotic role in a wide range of species by catalyzing the carboxylation of pyruvate to oxaloacetate (see review(2)). In mammals, this reaction is of importance for gluconeogenesis in the kidney and liver, for lipogenesis in adipocytes, for biosynthesis of the excitatory neurotransmitters in astrocytes, and for glucose-induced insulin secretion by pancreatic beta cells. Accordingly, pyruvate carboxylase (PC) has attracted interest as a possible target for the treatment of obesity and type 2 diabetes.(4,5) The active form of PC is an α4 tetramer in eukaryotes and most bacteria.(3) Each ∼130 kDa subunit is a single polypeptide chain containing the N-terminal BC domain, central TC domain, and C-terminal BCC domain. Until recently, the macromolecular assembly of PC was unclear. Now electron microscopy studies,(6) together with complete X-ray crystal structures of PC from Rhizobium etli(7,8) and Staphylococcus aureus,(9) and a partial structure of human PC (minus the BC domain),(10) provide unparalleled understanding about the enzyme's architecture and reaction mechanism. The tetramer assembles as a dimer of dimers. Catalysis proceeds through a previously unrealized mechanism whereby the BCC domain oscillates between the BC site on its own monomer and the TC site on a partner subunit (i.e., not on the same chain). The active sites within the BC and TC domains are located 65–75 Å apart, suggesting that the whole BCC domain moves in a swinging-arm mechanism between the two partial reaction sites. This large conformational change is an essential feature of catalysis that helps PC to avoid the loss of the activated carboxyl anion to diffusion or unwanted side reactions. Interestingly, a structure with all four BCC domains fully resolved has yet to be reported, suggesting that conformational flexibility associated with activity is problematic for crystallization of the protein.(7,9,10) However, a complex of the biotin-BCC domain with the TC domain has been observed in several X-ray structures of S. aureus PC, allowing further molecular analysis of the transcarboxylation reaction.(9,10) There has only been a single account of the biotin-BCC in complex with the BC domain.(8)
The aim of the current study was to develop monoclonal anti-PC antibodies that inhibit enzyme activity. Such agents serve as valuable tools to further delineate protein structure and function relationships. Here a panel of monoclonal antibodies were raised against sheep liver PC and characterized against PC isozymes and relevant biotin-dependent enzymes. Two antibodies were identified that were both inhibitory and possessed broad immunoreactivity. The epitopes for both antibodies were mapped to distinct sites on the BCC domain of human PC. The significance of our findings in light of the recent crystal structures is discussed.
Materials and Methods
Generation and purification of monoclonal antibodies
Sheep liver PC was prepared as previously described(11) before further purification on a monomeric avidin-sepharose column to a final specific activity of 22 U/mg. This material was diluted in Freund's complete adjuvant with emulsifier to a specific activity of 8–10 U/mg before subcutaneous injection into female BALB/c mice (140 μg per injection). Subsequent immunizations with 60 μg and 100 μg of PC were performed at 3 and 16 weeks. Four days after the final injection the mice spleens were harvested for fusion with NS-1 myeloma cells,(12) and cell suspensions were plated into four 96-well plates. To screen for clones expressing PC binding antibodies, medium from each well was tested using an ELISA(13) where each well of the microtiter plate was coated with 0.5 μg of sheep liver PC. On days 9–11, those wells containing the most useful antibodies were expanded and aliquots of the hybridoma cells stored at −8°C. Purification of the antibodies from cultured hybridoma supernatant was performed using Protein A–Sepharose (Amersham Biosciences, Uppsala, Sweden) following the manufacturer's instructions, and sera type was determined as previously described.(14)
Quantitation of PC activity
PC activity was determined using two alternative methods. For rapid screening of antibodies with inhibitory activity, cultured hybridoma supernatant (65 μL) was incubated for 1 h at 30°C with 0.5 mU of sheep liver PC. The reaction was initiated by addition of the reaction mix (100 mM Tris-HCl [pH 8.4], 5 mM MgCl2, 2.5 mM ATP, 10 mM sodium pyruvate, 10 mM NaH[ 14 C]O3 [5–15 cpm/μmol] and 330 μM acetyl-CoA) and allowed to proceed for 15 min at 30°C before termination of the reaction with HCl to a final concentration of 330 mM. After 20 min, 50 μL of the acidified products were spotted onto 2 cm squares of Whatman 3M paper and dried at 105°C before using liquid scintillation counting to measure 14 C-oxaloacetate. For quantitation of the inhibitory activity of the antibodies, PC was assayed as previously described.(15)
Truncation and mutational analysis of human PC to delineate antibody-binding sites
To broadly define the epitopes for MAbs 12 and 42, a series of six overlapping fragments spanning the entire coding region of hPC were generated by PCR. These fragments were designated BC (residues 1-488), F1 (residues 475-718), F2 (residues 711-954), F3 (residues 947-1178), TB1 (residues 947-1077), and TB2 (residues 1048-1178). The DNA encoding these fragments was amplified using the oligonucleotides outlined in Supplementary Table 1. EcoR1 and Xho1 endonuclease restriction sites were incorporated into the 5′ and 3′ ends of the PCR products, respectively, to facilitate cloning into the pET-32a (+) vector (Novagen, Darmstadt, Germany). For finer mapping of the antibody binding sites, a series of DNA fragments encoding N-terminally and C-terminally truncated forms of the 107 amino acid biotin domain (TB2) were amplified by PCR and similarly cloned as described above. Finally, site-directed mutagenesis was performed upon selected residues with the Quick-change site-directed mutagenesis kit (Stratagene, Santa Clara, CA) using plasmid pBlue (hPC-107) as the template. This plasmid was constructed by subcloning the EcoR1 and Xho1 treated DNA fragment encoding hPC-107 into pBluescriptII KS(+) digested with the same restriction enzymes. After confirmation by DNA sequencing, the modified DNA was reintroduced into the pET-32a vector. Recombinant proteins were expressed as a fusion to thioredoxin in E. coli BL21 following induction with 0.1 mM IPTG for 2 h. Whole cell lysates were prepared as previously described(16) for analysis either by SDS-PAGE or Western blot. The blots were probed either with MAb 12 (1:2500 dilution) or MAb 42 (1:5000 dilution), followed by an anti-sheep antibody conjugated with alkaline phosphatase. The immunoreactive products were visualized as previously described(16) and bands quantitated using ImageJ software (NIH, Bethesda, MD). For in vivo biotinylation assays, the blots were probed with avidin conjugated to alkaline phosphatase.
Preparation and labeling of Fab
Purified monoclonal antibody was digested with papain in the presence of the reducing agent cysteine, employing the method described previously(17) with minor modifications. The reactions were carried out in a total volume of 2 mL containing equal volumes of purified IgG (4 mg/mL) in PBS and 1 mL of 0.2 mg/mL papain (Boehringer, Mannheim, Germany) in digestion buffer (PBS [pH 7.5], 0.02 M cysteine, and 0.02 M EDTA) providing a protease-antibody ratio of 1:20 (w/w). The digestion was performed for 7 h at 37°C before termination with 30 mM iodoacetamide. The digested antibody was then dialyzed against PBS at 4°C overnight before separation of Fab from the Fc fragment by protein A-Sepharose chromatography column (1 mL Hitrap affinity column, Pharmacia Biotech, Piscataway, NJ). For Europium labeling, Fab 42 was labeled using the Delfia labeling kit (Wallac, Finland), according to the manufacturer's instructions.
Competitive binding assay
A competitive binding assay was performed to determine if MAb 12 and MAb 42 recognized overlapping epitopes. One hundred nanograms of streptavidin (Sigma, St. Louis, MO) in 0.1 M borate buffer (pH 9.5) were coated onto the surface of microtiter plates (Lumitrac 600 white 96-well plate, Greiner, Germany) for 2 days at 4°C. The plates were washed three times with TBS-Tween (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween-20) before blocking with 1% bovine serum albumin in TBS for 2 h at 37°C. The C-terminal 107 amino acids of human PC, encompassing the BCC domain, was overexpressed in E. coli BL21 for use in the binding assay. One microgram of the whole cell lysate containing the BCC was bound to the streptavidin-coated plates for 2 h at 37°C. After washing the wells three times with wash buffer (TBS-Tween buffer containing 0.8 μg/mL diethylene triamine pentaacetic acid) to ensure that only covalently bound Europium remained associated with the Fab fragment, the antibody competition was performed in a 100 μL volume containing 50 μL of Europium-labeled Fab 42 (8448 cps/125 ng of Fab 42) and various concentrations of unlabeled Fab 42 or 12. The reaction was incubated overnight at 4°C before the plates were washed five times in wash buffer and three times in water. The amount of Europium-labeled Fab bound to the plate surface was quantitated using time resolved fluorescence, as described previously.(18)
Results
Characterization of inhibitory properties of anti PC antibodies
Our aim here was to identify high affinity monoclonal antibodies that could inhibit PC from a broad range of species. These would serve as valuable tools to delineate protein structure and function relationships for this important metabolic enzyme. Thus, a series of monoclonal antibodies directed against sheep liver PC (SLPC) were generated and screened for their ability to both bind and inhibit PC activity. Initial screening of the antibodies was performed using cultured hybridoma supernatants. Antibody binding was measured using an ELISA with immobilized SLPC. From the screen two classes of antibodies were identified. In class I, 12 monoclonal cell lines were obtained that produced anti-PC antibodies with no inhibitory activity. These were not pursued further. In contrast, five class II clones expressed monoclonal antibodies with inhibitory activity. Antibodies were subsequently purified using protein A affinity chromatography allowing further characterization of the inhibitory properties of the immunoglobulins and epitope mapping. Of particular interest were two IgG1 monoclonal antibodies, namely MAb 12 and MAb 42. Only these antibodies were shown to (1) have high titers against a panel of PCs in an ELISA (Supplementary Table 2); (2) have inhibitory activity against all PCs tested including SLPC and recombinant human PC isolated from HEK 293T cells(15) (Fig. 1); and (3) did not recognize sheep liver propionyl CoA carboxylase (Supplementary Table 2).

Inhibitory activities of MAb 12 and MAb 42. The activity of sheep liver or human PC was performed as described in the Materials and Methods. Enzyme (20 mU) was incubated for 1 h at room temperature with 5 μg of either MAb 12 or MAb 42 before quantitation of remaining PC activity. The results shown here are the mean of triplicate samples±SEM. Each assay was analyzed relative to the no-antibody control; ***=p<0.01.
To further explore the mechanism of binding, a competitive ELISA was performed using either avidin or an anti-biotin antibody. Both avidin and the anti-biotin antibody effectively competed against both monoclonal antibodies for binding to the SLPC antigen (Table 1). However, biotin itself was not the antigenic determinant as incubation of the antibodies with a greater than 1000-fold excess of free co-factor did not result in a loss of inhibitory activity (Supplementary Fig. 1). These data placed the epitope for both antibodies within the active site of SLPC, most probably near the biotin attachment site in the BCC domain. Having identified the active site as a key component of the epitope, the effect of other substrates was investigated. SLPC was pre-incubated for 1 h together with antibody and an excess of MgCl2, MgATP, or pyruvate. SLPC activity was subsequently measured. Neither MgCl2 alone nor pyruvate reversed the inhibitory properties of either antibody (Table 2 and Supplementary Table 3). Likewise, MgATP had no effect on the activity of MAb 12 (Table 2). In contrast MgATP totally abolished the inhibitory activity of MAb 42 (Table 2), suggesting interference at the site of the first partial reaction in the BC domain. Together these data demonstrate that MAb 12 and MAb 42 bind to epitope(s) that are essential for catalysis in all PCs. However, the two antibodies possess distinctly different properties.
A competitive ELISA was performed using SLPC as immobilized antigen. Mouse anti-SLPC antibodies were then incubated in the well with a challenge agent, either avidin or goat anti-biotin antibody (αBiotin). Following binding for 1 h at room temperature, wells were washed and amount of bound anti-SLPC antibody was determined using either anti-mouse or anti-goat antibody, as appropriate, conjugated to horseradish peroxidase. Values given are mean absorbance at 450nm±SEM (n=3). Determination of these values included use of control wells for which the contribution of the challenge antibody or serum to the absorbance of the competition assay was ascertained.
SLPC activity was measured as described in Methods and Materials in the presence of either MAb 12 or 42. The assay was also performed with either 7 mM MgCl2 or 7 mM MgCl2 and 2.5 mM ATP (sodium salt). Values given are mean SLPC activity expressed as mU/mL±SEM (n=3). The percentage of PC activity is shown in parentheses, in the presence or absence of MgCl2 or MgATP, and is derived from the value in which no antibody was present for that particular treatment.
Epitope mapping of inhibitory antibodies 12 and 42
To more precisely define the epitope, mapping of the antibody binding sites was first performed using six overlapping fragments spanning the entire coding region of human PC. It was decided to perform the mapping on human PC as both antibodies reacted strongly with it, and we had previously reported cloning and recombinant expression of the human enzyme.(15,19) Additionally, structural data for the human enzyme was available in the literature.(10) Each fragment was recombinantly expressed in E. coli BL21 as a fusion to thioredoxin, then analyzed by Western blot. As shown in Figure 2, both MAbs 12 and 42 cross-reacted with fragments F3 (residues 947-1178) and TB2 (residues 1047-1178), but not with BC (residues 1-488), F1 (residues 475-718), F2 (residues 711-954), and TB1 (residues 947-1077). This localized the epitopes for both MAbs 12 and 42 within the C-terminal 131 amino acids of hPC containing the BCC domain (residues 1047-1178).
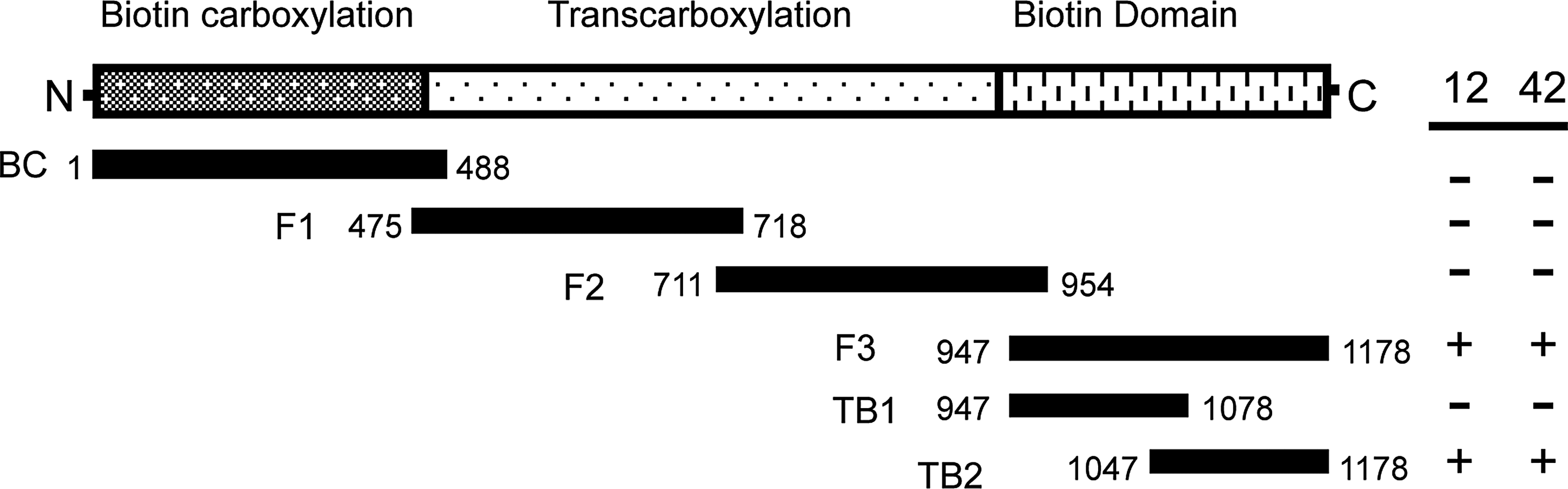
Localization of the epitopes for MAb 12 and MAb 42. The primary structure of human PC is diagrammatically represented with the enzymes' three functional domains indicated above. Six overlapping fragments made to encompass the full-length protein are shown below. The table to the right indicates if fragments were detected (+) or not detected (-) by Western blot analysis using MAbs 12 and 42.
The antibody binding site was further defined through additional truncation analysis. Here Western blot analysis was performed in conjunction with an in vivo biotinylation assay employing avidin blot analysis.(20,21) As it has been established that biotin protein ligase specifically attaches biotin only onto a structured BCC domain,(20,22,23) protein biotinylation can be used as a probe to confirm correct folding. Deletion of as few as 10 amino acids from the C-terminus resulted in complete abrogation of all antibody binding and protein biotinylation (Fig. 3). Similarly, N-terminal truncations beyond amino acid 1098 produced polypeptides that were neither immunoreactive nor biotinylated. Together these results suggest that both MAb 12 and MAb 42 are conformational antibodies capable of interacting only with a structured BCC domain. A fragment of 80 amino acids, contained within residues 1098-1178 of human PC, was identified as the minimal substrate for antibody binding and enzymatic biotinylation.

Truncation analysis of human PC. A series of fragments of human PC were expressed in E. coli as fusions to thioredoxin and analyzed for immunoreactivity by Western blot analysis probed with MAbs 12 and 42. The black bars indicate the relative lengths of the fragments, whereas the grey bars represent thioredoxin. Numbers above the bars correspond to the amino acids in human PC. The table to the right indicates if fragments were detected (+) or not detected (-) by Western blot. Biotinylation of each construct by bacterial biotin protein ligase was also assessed by avidin blot, with the results shown in the column identified as “Bio” at right.
Verification of the BCC domain as the antigen was obtained with a competitive binding assay. Here holo-BCC domain was immobilized onto the surface of a streptavidin-coated microtiter plate through the tethered biotin moiety. Both antibodies were able to bind to the BCC domain in this form, thereby confirming the earlier observation that biotin itself was not the epitope. The competition assay was performed using the Fab fragments from both antibodies, and Fab 42 was labeled with Europium to permit quantitation of bound material by time-resolved fluorescence. Both Fab 12 and Fab 42 competed against labeled Fab 42 with IC50 values of 230 and 75 ng, respectively (Fig. 4), again indicating that the MAbs bound to the BCC domains but at subtly discrete epitopes. Both antibodies failed to bind a 60 amino acid truncated variant of the antigen (residues 10680-1178). As this fragment was also not biotinylated, this datum is consistent with the finding that the anti-PC antibodies bind to structured epitopes.

Competition assay of Fab12 and Fab42. Streptavidin, coated directly onto microtiter plates, was used to capture TB2-107n (solid symbols) for use in a competitive binding assay. A non-biotinylated peptide, TB2-60n (open symbols), served as a negative control to demonstrate that protein immobilization was specifically via the biotin moiety. A fixed amount (125 ng) of labeled Fab42 was added to each well along with varying concentrations of unlabeled Fab12 (squares) or Fab42 (circles). After overnight incubation at 4°C, the amount of bound label was quantitated using time-resolved fluorescence. Data shown are the mean of three experiments±SEM.
Mutational analysis of antibody binding
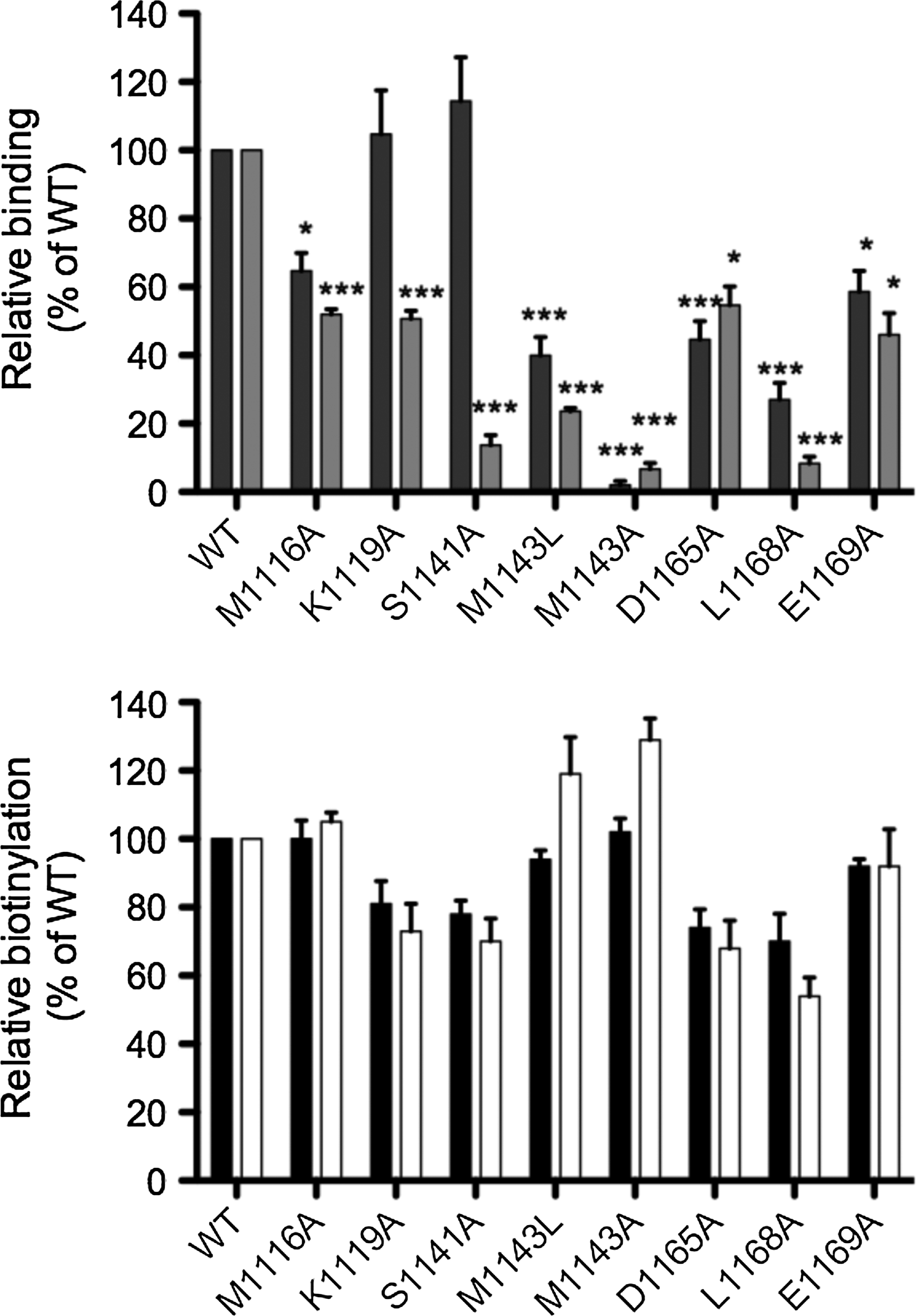
We have previously characterized BCC domains using mutagenesis studies and structural biology.(20,22,24) Here we employed these findings, in conjunction with other available structural data, to design a mutational series to probe the antibody binding sites in finer detail. Specifically, amino acid residues in the vicinity of the biotin attachment site (K1144) were selected for alanine substitution. A total of eight single amino acid substitutions were engineered in the BCC domain of human PC: M1116A, K1119A, S1141A, M1143L, M1143A, D1165A, L1168A, and E1169A. Each mutant was expressed in E. coli BL21 and cell lysates were analyzed by Western blot probed with either MAb 12 or 42 (Fig. 5A) or streptavidin to confirm the structural integrity of the biotin domain (Fig. 5B). The results from the mutagenesis study were mapped onto the human PC structure using coordinates from PDB 3BG3(10) (Fig. 6). Both antibodies failed to recognize the M1143A mutant. The more conservative leucine substitution was less severe, but still reduced binding for both antibodies to 30–40% of wild type. For MAb 12, K1191A and S1141A had no effect, whereas L1168A (27%), M1116A (64%), D1165A (44%), and E1169A (58%) showed reduced binding (Fig. 5A). These data suggest that Leu1168 and Met1143 provide most of the binding energy required for recognition by MAb 12 with Met1116, Asp1165, and Glu1169 making additional contributions to the epitope (Fig. 6B). In comparison, the four mutants M1116A, K1119A, D1165A, and E1169A all reduced binding of MAb 42 by at least 50% (Fig. 5A). As observed with MAb 12, L1168A (8%) also severely reduced binding for MAb 42. The most notable differences between the two antibodies were the interactions with K1119A and S1141A. Both mutants had no effect on the binding of MAb 12, but significantly affected interaction with MAb 42 (13% of wild type, p<0.001). These data suggest that S1141, M1143, and L1168 are critical to the binding of MAb 42 and form part of an extended epitope also encompassing M1116, K1119, D1165, and E1169 (Fig. 6C). Molecular modeling of the epitope on the isolated biotin domain structure revealed the solvent-exposed antibody binding surface near the biotinylation site. The epitopes were also mapped onto the intact S. aureus PC tetramer (PDB 3BG5(10)) where three out of the four BCC domains are defined (Supplementary Fig. 2). It was clear from this analysis that an antibody complexed to a BCC domain would have a dramatic inhibitory effect upon the conformational changes required for catalysis.
Mutational analysis hPC-107. Whole cell lysates expressing the hPC-107 mutant series were analyzed by Western blot (

Analysis of antibody binding sites. The structure of the human PC BCC domain, obtained using coordinates from PDB 3BG3,(10) is shown as a ribbon schematic (
All mutants were again assessed for correct folding using the in vivo biotinylation assay (Fig. 5B). This was performed at both 30°C and 37°C in order to observe whether any mutations induced temperature sensitive conformational changes to the molecule.(20) Importantly, none of the mutations severely affected the ability of the BCC domain to fold into the appropriate tertiary structure necessary for biotinylation. M1116A, M1143L, M1143A, and E1169A were essentially similar to wild-type protein at both temperatures, while K1119A and S1141A were slightly reduced to 80% (Fig. 5B). The most severe effects were observed with D1165A and L1168A where biotinylation was reduced to 70% at 30°C and 50–60% at 37°C relative to wild type (Fig. 5B), indicating the substitutions of these mutants had minimal effect on the overall structure of the domain.
Discussion
The aim of the present study was to produce monoclonal antibodies that inhibited the enzymatic reaction catalyzed by PC. We were successful in obtaining two conformation-dependent antibodies that localized to the C-terminal 80 amino acids of human PC encompassing the BCC domain. During its life cycle, the BCC domain participates in a number of heterologous protein-protein interactions. Firstly, it is enzymatically biotinylated by biotin protein ligase. Subsequently, the BCC domain participates in catalysis where it oscillates between the two partial reaction sites within PC, carrying carbon dioxide between carboxyphosphate in the BC active site and pyruvate in the TC site. Thus, one possible mechanism explaining the inhibitory properties of our antibodies is that they sterically interfere in the movement of biotin-BCC between BC and TC domains. Alternatively, the antibodies may hinder docking of the biotin moiety into the BC or TC domains. Both scenarios are consistent with electron microscopy studies(6) and X-ray crystal structures of PC that reveal conformation changes are essential for catalysis.(7–10) The BC and TC domains reside 65–75 Å apart, a distance that cannot be transversed by either biotin or the BCC domain alone. Hence, the biotin-BCC domain tethered to a flexible linker is employed to achieve the movement of the biotin group between the distant reaction sites. Furthermore, this swinging-arm mechanism is analogous to other multifunctional enzymes in which reactions proceed at distant partial reaction sites, such as fatty acid synthase(25) and pyruvate dehydrogenase.(26,27) An alternative mechanism explaining enzyme inhibition is that the epitope is biotin itself, as is observed with avidin inactivation of PC.(28) Here we argue this is unlikely as the inhibitory properties of MAb 12 or 42 could not be reversed by pre-equilibration of the antibodies with a vast excess of free biotin. Furthermore, when the BCC domain was coupled through the prosthetic group to the surface of a microtiter plate coated with streptavidin, Fab fragments from both antibodies retained the ability to bind the BCC domain. Interestingly, neither of the IgGs interacted with active PC when pre-complexed with avidin. This implies that regions of PC structure outside of the biotin domain also participate in sterically inhibiting access of the intact antibody, as would be expected from our electron microscopy study of avidin-PC complexes.(29) Once this structure was removed, the isolated biotin domain could simultaneously interact with both streptavidin and a Fab fragment of either monoclonal antibody.
The recent structural data of complete PC tetramers have highlighted the importance of the assembly of PC homodimer for catalysis. Furthermore, high-resolution X-ray crystal structures have also been reported for other biotin-dependent enzymes, including propionyl CoA carboxylase,(30) 3-methylcrotonyl-CoA carboxylase,(31) and urea carboxylase.(32) This has revealed surprising diversity in the assembly of these enzymes, given that they all share a conserved reaction mechanism that utilizes biotinylated BCC to transfer CO2. Like PC, urea carboxylase encodes the BC, TC, and BCC domains on a single polypeptide chain. In contrast to PC, however, urea carboxylase is catalytically active as a monomer, suggesting that the BCC must interact with the BC and TC subunits within the same molecule.(32) The architecture of propionyl CoA carboxylase and 3-methylcrotonyl CoA carboxylase are different again, with both adopting an α6β6 heterododecameric assembly composed of the BC and BCC domains on an α subunit and TC on a second β subunit.(30,31) The active holoenzyme complexes consist of a hexameric cylindrical core of β subunits decorated at the ends with the α subunits. As discussed above for PC, a flexible linker attached to the BCC domain facilitates the movement between the BC and TC domains. An interesting feature of all the available structures is the distance between the two partial reaction sites. For 3-methylcrotonyl-CoA carboxylase, the distance between BC and TC active sites is 80 Å, suggesting that the entire biotin-BCC domain must translocate during catalysis. For R. etli PC, the distance between the corresponding subsites is 65 Å when the enzyme is in complex with the allosteric activator ethyl CoA,(7) but 80 Å apart in its absence—a distant too great for the BCC domain to transverse in this example. Therefore, the role of the activator is to induce conformational changes within PC that reduces the distance between the two partial reaction sites. Uniquely, R. etli PC can only accommodate two equivalents of ethyl CoA in the tetramer.(7) Here the enzyme is assembled as a dimer of dimers, where one subunit can bind the activator while the other remains vacant. This provides a rare example of allosteric activation that is coupled with negative cooperativity. Employing conformational change as a regulator of enzymatic activity has also been proposed for the BC subunit of acetyl CoA carboxylase(33,34) and may be a conserved feature in other biotin-dependent enzymes. Here structural biology has played an essential role in understanding the intra- and intermolecular interactions that are central to catalysis for the biotin-dependent enzymes.
The two distinct monoclonal antibodies identified in the present study both exhibit their mechanism of inhibition through binding to the structured BCC domain. The BCC domain contains the single target lysine to which the biotin prosthetic group is attached through the enzymatic activity of biotin protein ligase. Previous studies have shown that biotin protein ligase recognizes the target residue within the context of a structured biotin domain.(20,22,23,35) Hence, enzymatic biotinylation can be readily employed as a probe to confirm structural integrity of the BCC domain.
It has long been known that biotin protein ligases from a wide variety of species can specifically biotinylate substrates from other species.(36) The protein-protein interaction has remained conserved through evolution due to the conservation of the BCC domain structure. The three-dimensional structures of many biotin-accepting domains have now been determined using both NMR and X-ray crystallography and show a remarkably conserved fold (see review(37)). This domain adopts a flattened β-barrel structure comprising two four stranded β-sheets, with the N-terminal and C-terminal residues close together at one end of the structure. At the other end of the molecule, the biotin accepting lysine resides on a highly exposed, tight hairpin loop between β-strands four and five. The biotinyl-lysine is found in the motif Met-Lys-Met, which is highly conserved in all biotin domains. The flanking methionine residues have been implicated in carboxylase and carboxyl-transferase reactions.(38,39) In the present report M1143, present on the C-terminal side of the target lysine, was implicated in the epitopes for both conformational antibodies as well as substrate recognition by biotin protein ligase.
Our mutagenic data suggest the two antibodies have distinct but overlapping binding sites on the BCC domain. Functional studies on other protein-protein complexes indicate that a small number of residues can confer tight binding affinity.(40) Mutational analysis of antibodies(41) or protein antigens(42) have shown that only 3 to 10 side chains account for most of the binding energy, although the structures of complexes between antibodies and protein antigens(43) show that typically between 14 and 21 residues are in contact. The predominant role of these side chains is to slow the dissociation of the complex.(44) Our result is consistent with these studies showing the alanine substitution of M1116, M1143, and E1169 can account for the reduction of the binding of MAb 12. For MAb 42, the alanine substitution of S1141 and M1143 most significantly accounts for reduction of binding to human PC, and K1119 and D1165 are likely to be modest contributors to MAb 42 binding. These residues cluster on one face of the BCC domain.
A consequence of having two different binding sites is that our two monoclonal antibodies also possess different inhibitory properties. Reversal of inhibition with MgATP was only observed with MAb 42. This suggests MAb 42, but not MAb 12, occludes the BCC-BC interaction at the site of the first partial reaction, presumably through the interface involving residues S1141, K1119, and M1143 on the BCC domain. Complexes involving the biotin-BCC-TC subunits have been crystallized for S. aureus PC,(9,10) propionyl CoA carboxylase,(30) urea carboxylase,(32) and 3-methylcrotonyl-CoA carboxylase.(31) However, there is only one crystallographic example of the biotin-BCC in complex with BC.(8) One possible explanation for this is that the BCC domain may have a higher affinity for TC than BC,(9) thereby making it more difficult to crystallize the BCC-BC complex. Alternatively, various forms of PC may be more suitable for crystallization than others. Kinetic studies using carboxybiotin-PC have shown that in solution this form of the enzyme preferentially exists with the BCC domain in complex with the BC subunit.(45) This was also observed in electron microscopy studies.(6) Binding of pyruvate in the second partial reaction site functions as the signal for carboxybiotin-BCC to move out of the BC domain and translocate into the TC domain.(6,46) However, if the active site is vacant, or in the presence of pyruvate analogues unable to accept the carboxyl anion, then spontaneous decarboxylation of biotin occurs by hydrolysis.(45) As yet carboxybiotin-PC has not been crystallized, possibly due to the labile nature of the intermediate. One possible application for the antibodies reported here is in co-crystallization studies that attempt to lock the enzyme into a more stable BC-BCC complex, thus mimicking carboxybiotin. Our current work complements the recent advances in our understanding of PC that have been made through structural biology, and assist in further understanding protein structure and function relationships for this important enzyme.
Footnotes
Acknowledgments
We gratefully acknowledge Dr. Dennis Rylatt, Dr. Peter Bundersen, and Ms. Jennifer Brazier for their assistance with the generation of the monoclonal antibodies.
Author Disclosure Statement
The authors have no financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.