
Abstract
Upon viral infection, the major defensive strategy employed by the host immune system is the activation of the interferon (IFN)-mediated antiviral pathway, which is overseen by IFN regulatory factors (IRFs). In order to complete their life cycles, viruses must find a way to modulate the host IFN-mediated immune response. Kaposi's sarcoma-associated herpesvirus (KSHV), a human tumor-inducing herpesvirus, has developed a unique mechanism for antagonizing cellular IFN-mediated antiviral activity by incorporating viral homolog of the cellular IRFs, called vIRFs, into its genome. Here, we summarize the novel evasion mechanisms by which KSHV, through its vIRFs, circumvents IFN-mediated innate immune responses and deregulates the cell growth control mechanism.
Introduction
K
KSHV is a large, double-stranded DNA virus that belongs to the gammaherpesvirinae subfamily of the genus rhadinovirus. It shares sequence similarities with a number of other rhadinoviruses, including the prototype virus herpesvirus saimiri (HVS), and is also one of the closest phylogenetic relatives of rhesus monkey rhadinovirus (RRV) and murine gamma herpesvirus 68 (MHV-68) (Neipel and others 1997; Jung and others 1999; Searles and others 1999; Alexander and others 2000). As with all herpesviruses, once the host is infected with KSHV, the virus persists throughout its lifetime in one of 2 replicative programs, known as latent and lytic replication. During latency, the virus exists as a multicopy circular episomal DNA in the nucleus, rigorously expressing a handful of viral genes to allow the virus to retain its lifelong persistent infection. In lytic replication, however, virtually the entire set of viral genes is expressed, resulting in the production of infectious viral progenies. Both lytically and latently infected cells coexist in KS tumors and PELs (Decker and others 1996; Zhong and others 1996; Staskus and others 1997). Hence, the viral proteins expressed during both phases contribute significantly to the efficient establishment of persistent infections, as well as KSHV-associated pathogenesis.
To maintain this balance, a large portion of the KSHV genome is dedicated to encoding immunomodulatory proteins that regulate different aspects of the innate and adaptive immune responses, with many of these immunomodulatory proteins sharing significant levels of homology with cellular proteins. Especially notable among these are the viral interferon regulatory factors (vIRFs), which are homologous to cellular interferon (IFN) regulatory factors (IRFs). Remarkably, the KSHV genome encodes 4 vIRFs (vIRF1–vIRF4) within a cluster of loci (Russo and others 1996; Neipel and others 1997; Cunningham and others 2003). Three of the 4 (vIRF1–vIRF3) have already been functionally characterized, but little is known about vIRF4 (Kanno and others 2006). Although vIRF4 has been poorly studied up to this point, the other vIRFs have been shown to inhibit both host IFN responses and growth control mechanisms, but differences in their activations result in distinctive evasion strategies against the host defense mechanisms. The goal of this article is to better understand how KSHV utilizes its vIRFs to escape the host immune system, specifically highlighting the roles of the vIRF-mediated inhibition of host IFN responses and growth control mechanisms to yield persistent viral infections.
Regulation of the IFN signaling pathway
Interferons, which have been described as cytokines since their discovery (Isaacs and Lindenmann 1957), are signaling molecules essential for regulating the activation of immune cells as an antiviral response. There are at least 3 distinct types of IFNs: type I (IFN-α/β), type II (IFN-γ), and type III (IFN-λ1, -λ2, and -λ3, also known as interleukin-29 [IL-29]), which are classified based on their amino acid sequences (Pestka and others 2004; Ank and others 2006). The IFN-α/β genes are induced directly as a response to viral infections. The roles of IFN-λ are less clear, but its antiviral activities, elicited by the induction of IFN-stimulating genes (ISGs), are similar to those of IFN-α/β (Vilcek 2003; Pestka and others 2004; Honda and others 2006; Honda and Taniguchi 2006). IRFs, through the induction of IFNs, have been shown to play central roles in host immune responses, and among the 9 members (IRF1 to IRF9) of the IRF family identified thus far, IRF3 and IFR7 are the key regulators of the expression of the IFN-α/β genes upon viral infection (Sato and others 2000; Honda and others 2005). Viral infection activates certain host pattern recognition receptors (PRRs), resulting in the phosphorylation of cytoplasmic IRF3 and its subsequent translocation to the nucleus, wherein it interacts with the transcriptional co-activator histone acetyltransferase (HAT) CBP/p300 to induce IFN-β gene expression (Lin and others 1998; Yoneyama and others 1998). IRF7 is highly homologous to IRF3, but unlike IRF3, IRF7 is constitutively expressed at low levels in most cells and is strongly induced by the type I IFN-mediated signaling stemming from an IRF9-dependent pathway (Marié and others 1998; Sato and others 1998; Honda and Taniguchi 2006). Like IRF3, IRF7 undergoes phosphorylation upon viral infection, which allows its dimerization and nuclear translocation (Marié and others 1998; Lin and others 2000). IRF7 either heterodimerizes with IRF3 or homodimerizes and these dimers induce the expression of chemokines and the IFN-α/β genes (Marié and others 1998; Lin and others 2000). The large set of genes then induced by IFN-α/β ultimately form the first line of the antiviral defenses in suppressing viral replication and propagation. In turn, viruses have evolutionally developed various strategies to subvert these pathways, to the benefit of their life cycles. As an example, KSHV expresses the vIRFs to act as dominant-negative inhibitors by targeting IRF3 and IRF7.
νIRF-mediated negative regulation of the IFN pathway
The N termini of the vIRFs exhibit significant levels of homology to the N-terminal DNA-binding domain (DBD) of the cellular IRFs but lack several tryptophan residues that contribute to DNA binding (Takaoka and others 2008), which may make them unable to bind directly to DNA (Flowers and others 1998; Zimring and others 1998; Tamura and others 2008). Although they do not prevent cellular IRFs from binding to DNA, they are able to obstruct many of the functions derived from cellular IRFs (Flowers and others 1998; Zimring and others 1998; Tamura and others 2008). As an exception, a recent study has shown that vIRF1 binds directly to a specific DNA sequence in the promoter regions of K3, viral dihydrofolate reductase, and viral interleukin 6 of the KHSV genome, which may regulate viral gene expression (Park and others 2007)
vIRF1 (K9) is a nonstructural regulatory lytic gene that contains a DBD but does not compete with IRF1 for DNA binding, nor does it bind to or suppress IRF1 (Zimring and others 1998; Burýsek and others 1999a). However, vIRF1 directly targets p300 and regulates p300 activity. The association of vIRF1 with p300 interferes with the formation of the CBP/p300-IRF3 complex, as well as p300 HAT activity to prevent the IRF3-mediated transcriptional activation of IFN-α (Li and others 2000; Lin and others 2001). In contrast, vIRF1 does not block the transactivation activity of IRF7, although it interacts weakly with IRF7 (Lin and others 2001). Despite these efficient anti-IFN activities, the periods of time in which vIRF1 is able to block IFN responses in KSHV-infected cells are limited (Burýsek and others 1999a), suggesting that vIRF1 may have other functions during the multifarious stages of the virus life cycle or that other vIRFs and viral immunomodulators are required to completely inhibit IFN responses.
Previous studies have shown that the first exon of vIRF2 (K11.1) prevents the activation of PKR (Burýsek and Pitha 2001), reducing protein synthesis and blocking IFN-α/β signaling to decrease the ability of the cells to respond to viral infections (Burýsek and others 1999a, 1999b). Furthermore, full-length vIRF2, which is translated from exons K11.1 and K11, inhibits the expression of the IFN-inducible genes driven by IRF1, IRF3, and IFN-stimulating gene factor 3 (ISGF3), but not IRF7 (Fuld and others 2006). Although, vIRF1 and vIRF2 have similar activities, the specific mechanisms of vIRF2 activity have yet to be defined. Unlike vIRF1 and vIRF2, which mostly target IRF3-mediated interferon signaling, vIRF3, also known as latency-associated nuclear antigen 2 (LANA2), specifically interacts with either the DNA-binding domain or the central IRF association domain of IRF7, and this interaction leads to the inhibition of IRF7 DNA-binding activity and, therefore, suppression of IFN-α production and IFN-mediated immunity (Joo and others 2007). Remarkably, the central 40 amino acids of vIRF3, containing the double alpha helix motifs, are sufficient not only for binding to IRF7, but also for inhibiting IRF7 DNA-binding activity. Consequently, the expression of the double alpha helix motif-containing peptide effectively suppresses IRF7-mediated IFN-α production. This demonstrates a remarkably efficient means of viral avoidance of host antiviral activity (Joo and others 2007).
In summary, the down-regulation of the IFN regulatory pathway is a common characteristic of the 3 vIRFs, whose functions have been well studied, as IRF3 and IRF7 are the initial key factors of the host immune surveillance program against viral infections. vIRF4, the most recently identified member of the vIRF family, has yet to be studied mechanistically, but it may have the potential to affect IFN-mediated innate immunity. The apparent importance of the modulation of IRF3 and IRF7 activity by KSHV is further evident from the observation that the early protein K8 (K-bZIP) acts to obstruct IRF3, while 2 immediate–early proteins, ORF45 and ORF50 (RTA), negatively regulate IRF7. First, K-bZIP (also known as K8) directly interacts with the IFN-β promoter to impede its binding with IRF3, thus precluding efficient IFN-β gene expression (Lefort and others 2007). Second, ORF45 interacts with IRF7, preventing its phosphorylation and blocking its translocation to the nucleus, thus down-regulating type I IFNs (Zhu and others 2002). Furthermore, RTA promotes IRF7 ubiquitination, resulting in the induction of the proteasomal degradation of IRF7 (Yu and others 2005). Therefore, the manipulation of the modification, stability, and function of host IRF3 and IRF7 by the KSHV vIRFs provides novel regulatory strategies for circumventing the innate immune defense system (see Fig. 1).

An overview of the inhibition of the type I interferon (IFN) pathway of Kaposi's sarcoma-associated herpesvirus (KSHV). After endocytosis, Toll-like receptor (TLR) 3 responds to the uncoating of endocytosed viral particles in the endosome, resulting in the phosphorylation of TLR3. TLR3 then homodimerizes, binds to CD14, and activates TANK-binding kinase 1 (TBK1/IKKE), leading to the phosphorylation of IFN regulatory factor (IRF)3. IRF3 then homodimerizes and translocates to the nucleus, where it binds to p300/CBP, turning on IFN-β gene expression. TBK1 also phosphorylates IRF7, causing it to homodimerize and translocate to the nucleus, where it binds to the IFN-α promoter. Additionally, IRF7 can heterodimerize with IRF3, upon which the complex translocates to the nucleus to activate the IFN-β promoter. The newly expressed IFN-α and IFN-β genes then bind to the IFN-α receptor in both paracrine and autocrine manners, resulting in the phosphorylation and activation of STAT1 and STAT2, which recruit IRF9 to form the IFN-stimulating gene factor 3 (ISGF3) complex. ISGF3 binds to IFN-stimulated response elements (ISRE) and induces IFN-stimulating genes, such as IRF7. Among the KSHV-encoded vIRF proteins, vIRF3 specifically suppresses IRF7 transcriptional activity, whereas vIRF1 and vIRF2 inhibit the induction of the IFN-β promoter by IRF3.
Overview of the p53 pathway
p53, which is a transcriptional factor, responds to DNA damage and other cellular stresses, such as viral infections, by inducing cell cycle arrest or apoptosis as well as playing an important role in tumor suppression (Vogelstein and others 2000). In order to circumvent host scrutiny, viruses employ their products to disrupt and overcome p53-mediated irreversible cell cycle arrest and apoptosis that are parts of the overall host surveillance mechanisms to block viral replication and dissemination (Moore and Chang 2003; Collot-Teixeira and others 2004; Coscoy 2007; Liang and others 2008).
p53 is negatively regulated tightly by murine double minute 2 (MDM2) to maintain its low levels under normal conditions (Michael and Oren 2003; Bond and others 2005). It has been well established that MDM2, an oncogenic E3 ligase, is the major negative regulator of p53, which it modulates in two ways. First, MDM2 interaction masks the transactivation domain of p53, resulting in interfering with the transcriptional activity of p53 (Oliner and others 1993). Second, MDM2 promotes the ubiquitin-mediated degradation of p53 (Fang and others 2000; Honda and Yasuda 2000). Additionally, MDM2 and p53 are components of an autoregulatory negative feedback loop, wherein p53 induces MDM2 expression, while MDM2 represses p53 activity, with the feedback loop requiring the tight regulation for p53 to function properly (Moll and Petrenko 2003; Stommel and Wahl 2005). Furthermore, the activities of p53 can be differentially regulated through specific post-translational modifications. For example, upon DNA damage, p53 is phosphorylated by ataxia telangiectasia-mutated (ATM) and acetylated by p300/CBP, preventing MDM2–p53 interaction and ensuring p53 stabilization (Canman and others 1998; Tibbetts and others 1999; Maya and others 2001). This stabilization results in the engagement of p53-associated gene expression, leading to cell cycle arrest, DNA repair, and apoptosis (Bond and others 2005).
Thus, there exists a need for viruses to tightly regulate the expression and/or functions of p53, which KSHV accomplishes through its vIRFs, which hinder p53-mediated irreversible cell cycle arrest and apoptosis (Collot-Teixeira and others 2004; O'Shea 2005; Coscoy 2007; Liang and others 2008).
νIRF-mediated negative regulation of the p53 pathway
As noted earlier, in interfering with the activities of p53, the vIRFs mainly act as negative modulators. vIRF1 inhibits p53 through two coordinated mechanisms: first, vIRF1 interacts with the ATM kinase to block its activity, thereby reducing p53 phosphorylation at the serine 15 residue and increasing p53 ubiquitination (Shin and others 2006); second, vIRF1 interacts with p53 to suppress its acetylation, inhibiting the transcriptional activation of p53 and efficiently preventing p53-mediated apoptosis (Nakamura and others 2001; Seo and others 2001). In conjunction with vIRF1, the B-cell-specific, latently expressed vIRF3 (also known as LANA2) interacts with p53, thereby inhibiting p53-mediated transcriptional activity and apoptosis (Rivas and others 2001). Our recent studies have also demonstrated that vIRF4 interacts with MDM2, leading to a reduction of p53 via proteasomal degradation, thereby effectively suppressing p53-mediated apoptosis and establishing favorable circumstances for viral replication (Lee and others 2009). vIRF2, in turn, prevents the transcriptional activation of CD95L in activated mature T cells to suppress apoptosis (Kirchhoff and others 2002). Taken together, it is clear that the down-regulation of p53-mediated cell growth control is a common characteristic of the vIRFs.
In contrast to the anti-oncogenic properties of the vIRFs, as discussed earlier, they may actually possess oncogenic abilities as well, which accelerates tumorigenesis. Expression of vIRF1 in NIH3T3 cells fully transforms the cells and causes fibrosarcoma in nude mice (Gao and others 1997; Li and others 1998). Consistent with these observations, knocking down vIRF3 using RNAi in infected PEL cells reduces cell viability and increases apoptosis, indicating that the oncogenic activity of vIRF3 is one of the driving forces of the pathogenesis of PEL (Wies and others 2008). Given that the vIRFs are critical to two aspects of virus propagation—persistent infection and pathogenesis—a more detailed understanding of how the vIRFs disturb the host defense mechanisms may result in the identification of the vIRFs as therapeutic targets against KSHV infections (see Fig. 2).
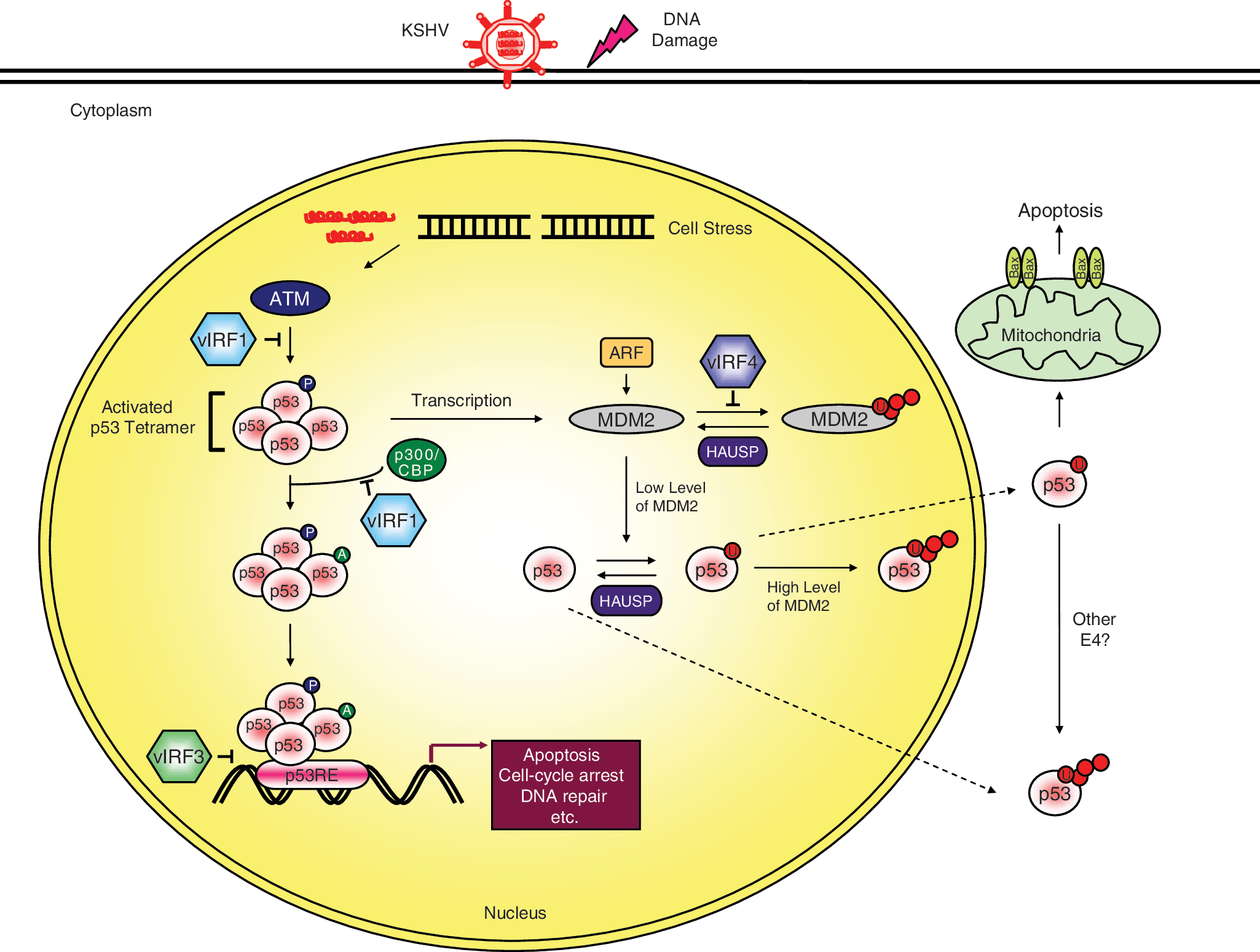
Regulation of p53 function by the viral interferon (IFN) regulatory factors (vIRFs). Activation of the network by stresses, including virus replication and DNA damage, stimulates the phosphorylation and acetylation of p53 to increase the levels of activated p53. The activated p53 then binds to its regulatory region, which activates the expression of several target genes. In turn, this transcriptional activation leads to diverse cellular responses, such as apoptosis, cell cycle arrest, or DNA repair. When p53 is no longer needed, it is ubiquitinated by murine double minute 2 (MDM2) and translocates from the nucleus to be degraded. Furthermore, MDM2 is capable of inducing both the monoubiquitination and polyubiquitination of p53 based on its protein levels in the nucleus. Alternatively, the Kaposi's sarcoma-associated herpesvirus (KSHV) vIRFs inhibit p53 protein levels and/or transcriptional activity in discrepant manners.
Conclusion and Perspectives
In this review, we have briefly described how the KSHV vIRFs impact various aspects of the host immune response to achieve persistent viral infection and pathogenesis. Most notably, studies on the vIRFs have revealed 2 remarkable functional redundancies in which they affect the transcription of type I IFNs and inhibit p53-mediated cell cycle arrest and apoptosis.
Stepping away from what we know about the functional consequences of the vIRFs in terms of immune evasion, the major challenge now is to integrate the in vitro observations of KSHV infection in elucidating the detailed molecular mechanisms of each viral immunomodulatory protein. However, difficulties still remain, as it is unclear what cell types permit viral replication even though multiple cell types can be infected transiently. In addition, understanding KSHV pathogenesis and developing immunotheraphies against KSHV-associated diseases remain secondary challenges, due to the restrictive host specificity of KSHV. Although, mice infected with MHV-68 and non-human primates infected with RRV provide animal model systems closely related to KSHV, these models are limited when trying to fully understand KSHV pathogenesis.
With the exception of KSHV, the only other herpesvirus known to encode vIRFs is RRV, a close relative to KSHV (Searles and others 1999; Alexander and others 2000). Future studies on the vIRFs will need to address why KSHV and RRV encode unique vIRF genes in a cluster of loci and the significance of their impact on KSHV viral replication and pathogenesis in a genetic context. Thus, understanding the biological foundations of the vIRFs may yield further information on previously undiscovered strategies used by KSHV to modulate host immunity and influence virus-induced pathology.
Footnotes
Acknowledgments
We thank Steven Lee for a critical reading of this manuscript. This work was partly supported by U.S. Public Health Service grants CA82057, CA31363, CA115284, AI073099, Korean KRIBB, and Hastings Foundation (J.J.), AI083841, the Lymphoma and Leukemia Society of USA, and the Baxter Foundation (C.L.).