
Abstract
Influenza pandemics are imminent and represent a major world health concern. Since vaccinations are expected to be less efficient in the coming years due to newly emerging influenza virus strains, novel antiviral therapies are urgently needed. Here, we show that influenza-infected mice, capable of clearing the virus in the early stages of infection, failed to control inflammation and death. Sequential administration of Interferon-γ (IFN-γ) at early stage of the infection protected infected mice from death in a NK cell-dependent manner. IFN-γ treatment stimulated NK cell proliferation and function and increased their number in the bone marrow, blood, spleen, and infected lungs, keeping viral clearance intact. In parallel, IFN-γ treatment significantly reduced the number of T cells and NKT cells in the lungs at the inflammatory phase following infection. Thus, rapidly clearing the virus and reducing inflammation by shaping the cellular and cytokine profiles in the early stages of infection may favorably change the fate of influenza pathogenesis.
Introduction
I
In this study, we show that in the course of influenza infection, treatment of mice with IFN-γ can stimulate NK cell proliferation and function and increase their number in the BM, blood, spleen, and infected lungs, keeping viral clearance intact. In parallel, IFN-γ treatment can attenuate inflammation by an as yet unclear mechanism(s).
Materials and Methods
Mice and viral infection protocols
C57BL/6 mice were maintained under SPF conditions at the Hebrew University Animal Facility. The study was approved by the Hebrew University Ethical Committee for animal experiments. The influenza A PR8 virus was generously provided by Prof. O. Mandelboim. Mice (female, 7–8 weeks of age) were anesthetized and inoculated intranasally with 50 μL of 1.5 × LD50 IV. IFN-γ (PeproTech Asia, Rehovot, Israel) was reconstituted in PBS and injected at the indicated concentrations; control mice were injected with PBS. NK cell mobilization protocol using IFN-γ included 2 injections of 10,000 units/mouse of IFN-γ i.p., 17 h apart. IFN-γ-treated mice were treated twice with this protocol, on the first and third days after influenza infection, as shown in Figure 1. In survival experiments, mice were monitored on a daily basis.
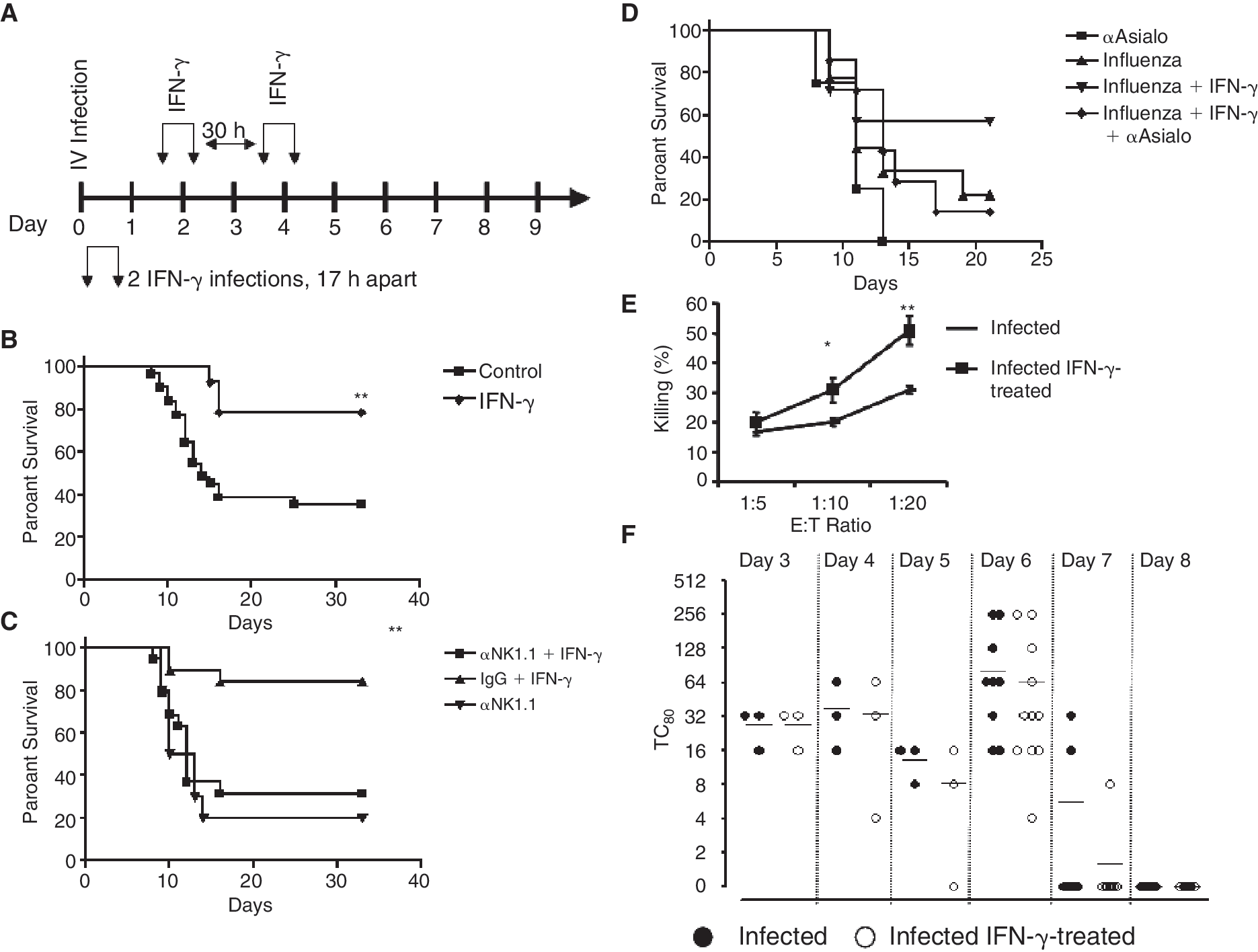
Interferon-γ (IFN-γ) injections rescue mice from lethal influenza infection in an NK cell-dependent manner, but do not alter viral titers in the lungs. (
In vivo NK cell depletion
Depletion of NK cells was done in C57Bl/6 mice using functional grade purified anti-NK1.1 (eBioscience, San Diego, CA), or anti-asialo antibody (Cedarlane, Canada) (Maruyama and others 1991; Szomolanyi-Tsuda and others 2006). Mice were injected i.v. with 50 μg of anti-NK1.1 or 50 μL of anti-asialo 1 day before influenza infection, and with 50 μg of anti-NK1.1 or 50 μL of anti-asialo i.p. 3 days post-infection depletion was confirmed by FACS analysis.
Lymphocyte isolation and quantitation
Bones and spleen were splashed with PBS; blood was collected into heparin-containing tubes; cells were treated with RBC lyses solution (0.155 M NH4Cl, 0.01 M KHCO3, 0.01 mM EDTA, pH 7.4), washed, and resuspended with PBS containing 0.1% bovine serum albumin (BSA, Beit Hemek, Israel) (Biological Industries, Israel) and 0.01% sodium azide (FACS buffer). Cells were filtered through a cell strainer FACS tube (Becton Dickinson, Mountain View, CA) and stained for cytometry analysis.
Lungs were weighed, meshed through 70 μM cell strainers (BD FALCO N), washed with 1 mL of FACS buffer, and shortly centrifuged (500 rpm, 1 min) and supernatant was collected. Cells were washed again and resuspended in FACS buffer. Cells were filtered through a cell strainer FACS tube (Becton Dickinson, Mountain View, CA) and stained for cytometry analysis.
The total number and percentage of NK cells were calculated as follows: the number of NK1.1+CD3− NK cells in 100 μL of blood was counted by FACS and multiplied by 15 assuming a total blood volume of 1.5 mL. The number and percentage of NK cells in the tibia and femur were determined by FACS and the number of cells was multiplied by 5 assuming that these cells represent 20% of the total NK cells in the BM. The total number and percentage of isolated NK cells in the lung and spleen were also determined by FACS.
Phenotypic analysis of lymphocytes and flow cytometry analysis
To assess the number of total NK cells in the BM, spleen, blood, and lungs, lymphocytes derived from these organs were gated according to forward scatter and side scatter to exclude dead cells and include both activated and non-activated lymphocytes. Then, cells were stained with anti-CD45.2 FITC, NK1.1 PE, CD3 PE-Cy5 monoclonal antibodies; CD45.2+ cells were gated and subpopulations of NK1.1+CD3− (NK cells), NK1.1+CD3+ (NKT cells), and NK1.1−CD3+ (T cells) were defined. In CFSE experiments, and NK staining with NKG2D PE, APC-conjugated anti-NK1.1 (eBioscience, San Diego, CA) was used. Anti-IFN-γ FITC, IL-10 FITC, IL-4 FITC, IL-2R FITC, Mac-1 FITC, and NKG2D PE were purchased from eBioscience, San Diego, CA.
Immunostained cells were analyzed by flow cytometry using the FACSCalibur Flow Cytometer (Becton Dickinson, Mountain View, CA); the data were analyzed using the CellQuest 3.3 or the FlowJo 6.0 software.
Cell proliferation assay
CFSE experiments—Spleen cells were isolated on Ficoll, diluted to 107 cells/mL, and labeled with 5 μg/mL CFSE (BCECF/AM, Calbiochem, San Diego, CA). Each mouse received 5 × 107 labeled splenocytes, 1 day before influenza infection. Five days post-infection, cells were isolated, stained for NK1.1 (APC, eBioscience, San Diego, CA) and CD3e (PE-Cy5), CFSE (FITC fluorescence) were measured using the FACS to assess cell proliferation. In short experiments, 3 h after adoptive transfer, cells were loaded with 0.5 μg/mL CFSE. In BrdU experiments, mice were injected with 1 mg/mouse of BrdU (Sigma, Rehovot, Israel) in PBS 3 h prior to sacrifice. BrdU incorporation was evaluated using FITC BrdU Flow Kit (Pharmingen, San Diego, CA), as recommended by manufacturer.
Internal staining and ELISA assays
Intracellular staining for cytokines was done after staining with surface markers to NK cells by fixation of cells using 4% formaldehyde (Sigma, Rehovot, Israel) containing 0.1% saponin (Sigma, Rehovot, Israel) for 15 min. Afterward, cells were washed with FACS buffer supplemented with 0.1% saponin and stained with specific antibodies to IFN-γ, IL-10, and IL-4 in the same liquid for 30–60 min.
PolyI:C (Sigma, Rehovot, Israel) experiments were done as described by others (Wang and others 2005; Zhang and others 2007). In brief, polyI:C (150 μg/mouse in PBS) was injected intraperitoneally to the mice. Mice were sacrificed 5 h and 30 min post-injection and livers were removed.
Livers of polyI:C-injected mice were meshed through 70 μM cell strainers, and washed with PBS. Debris was removed by centrifugation (500 rpm, 1 min); supernatant was loaded onto Ficoll (Histopaque 1077; Sigma, Rehovot, Israel) and centrifuged for 20 min at 1,800 rpm. Then, lymphocytes were collected between the 2 phases, washed with FACS buffer, and stained for NK1.1 and intracellular CD3, and intracellular stainings were done for IFN-γ.
ELISA assays for murine CXCL9 and CXCL10 (Quantikine; R&D Systems Minneapolis, MN; ELISA sensitivity: 3.0 and 2.2 pg/mL, respectively), mouse IFN-γ, IL-2, IL-10, and IL-4 (Th1/Th2 10plex, Bender Medsystems, Vienna, Austria; ELISA sensitivity: mIFN-γ: 6.5 pg/mL, mIL-2: 8.8 pg/mL, mIL-4: 0.7 pg/mL, mIL-10: 5.4 pg/mL), murine IL-15 (eBioscience, San Diego, CA; ELISA sensitivity: 62 pg/mL), and IFN-α (PBL biomedical laboratories) were performed using the kit according to the manufacturer's instructions. In the in vivo experiments, spleen, lung, and BM were collected on specified days post-infection. BM and spleen were flushed with PBS containing a protease inhibitor cocktail (“Complete mini,” Roche Diagnostics, Mannheim, Germany). Lung extracts were obtained by mashing the tissue on 70 μM nylon mesh with PBS containing a protease inhibitor cocktail.
Viral titer determination using hemagglutination (HA) assay
HA assays were done as described by Hierholzer and others (1969). MDCK cells (generously provided by Prof. O. Mandelboim) were seeded in 96 wells at a concentration of 10,000 cells/well. The next day, 2-fold dilutions of each lung extract were layered on the cells for 3 h. Afterward, cells were washed twice with PBS and incubated in DMEM containing 2% FCS for 5 days. HA assay was done with 100 μL of supernatant and 100 μL of 1% chicken RBC in U-shaped wells.
Killing assay
Splenocytes from IV-infected C57Bl/6 mice, untreated or treated with IFN-γ, were isolated on the fifth day post-infection and stained for NK1.1 PE and CD3 PE-Cy5. NK1.1+CD3− cells were sorted using the FACSaria (Becton Dickinson, Mountain View, CA) to >85% purity. Sorted NK cells were incubated with CFSE-labeled Yac-1 cells at the indicated ratio for 4 h. Thereafter, cells were supplemented with propidium iodide (PI; Sigma-Aldrich, Gillingham, UK) and evaluated by FACSCalibur. CFSE+PI+ Yac-1 cells were considered as dead cells.
Statistical analysis
Survival analysis was done using the GraphPad Prism software, using the Logrank test. Differences among groups of mice and different treatments on the number of NK cells were calculated using standard Student's t-test. Statistical tests were 2-sided. P values <0.05 were considered to be statistically significant.
Results
IFN-γ injections rescued mice from lethal IV infection in an NK-dependent manner
In this study, we tested the cross talk between IFN-γ cells and the immune response against the influenza virus. We hypothesized that sequential administration of IFN-γ would increase the number of NK cells in the periphery and the immune response against the influenza virus. To test this hypothesis, we treated influenza A PR8-infected mice with sequential administration of IFN-γ i.p. on days 1, 2, 3, and 4 post-infection, and monitored their survival (Fig. 1A). Sequential administration of IFN-γ significantly enhanced the survival of influenza-infected mice from 35% to 80% (Fig. 1B). The relative contribution of NK cells to the immune response against the influenza virus was further tested by depletion of NK cells with anti-NK1.1 or anti-asialo antibodies injected into mice on days −1 and +3 pre-/post-infection (Fig. 1C and 1D). Depletion of NK cells by NK1.1 or anti-asialo antibodies significantly increased the mortality of mice due to the IV infection (Fig. 1C and 1D). Furthermore, depletion of NK cells totally abolished the curable effect of IFN-γ on influenza-infected mice (Fig. 1C and 1D). Injection of control IgG antibodies alone or together with IFN-γ used as control did not affect the mortality of mice (Fig. 1C and 1D).
To characterize the cellular and molecular responses that regulate IFN-γ-dependent, NK cell-mediated response against the IV, we first characterized the effect of IFN-γ on the ability of sorted NK cells from viral and viral-treated mice to lyse target cells. NK cells from viral-infected mice treated with IFN-γ had increased killing capacity that may support faster clearance of the virus (Fig. 1E). To test this hypothesis, viral titers in the lung were tested. In untreated mice, we found that the titers of the influenza virus were increased until day 6 and then rapidly dropped to undetected levels on days 7 and 8 (Fig. 1F). Surprisingly, we found that IFN-γ treatment had no effect on viral titers. It is therefore apparent that in both treated and untreated mice, the IV was rapidly cleared; however, this rapid immune response that is dependent on NK cells (Fig. 1C and 1D) did not protect mice from the lethal effect of the IV.
Treatment with IFN-γ enhanced NK recruitment to the lung and abolished T and NKT cell infiltration to this organ after infection with IV
To further characterize the protective effect of IFN-γ and NK cells against the IV, we monitored the number of NK, T, and NKT cells in blood, lung, spleen, and BM of infected mice. Cells were stained with anti-CD45.2 FITC, NK1.1 PE, CD3e Cy3 monoclonal antibodies; CD45.2+ cells were gated and subpopulations of NK1.1+CD3− (NK cells), NK1.1−CD3+ (T cells), and NK1.1+CD3+ (NKT cells) were defined in each organ (Fig. 2A). NK, T, and NKT cell numbers were evaluated in influenza-infected mice, with or without IFN-γ treatment for 9 consecutive days post-infection. During the influenza viral infection, the number of NK and T cells in blood was dramatically reduced. Following IFN-γ treatment, the number of NK cells in blood on days 2–5 was significantly increased. In contrast, no effect was observed on the reduction in the number of T or NKT cells in blood, BM, or spleen (Fig. 2B). During infection, the number of NK and T cells in the spleen was also reduced. Following IFN-γ treatment, the number of NK cells in the spleen on days 3–5 was significantly increased, whereas the number of T cells did not change (Fig. 2C). The number of NK, T, and NKT cells in the BM was not significantly affected by the IV infection. However, an increased number of NK cells, but not T cells, was observed in the BM 3–7 days following IFN-γ treatment (Fig. 2D). In the lung, the number of NK cells gradually increased with time following infection. Between days 3.5 and 5.5, IFN-γ treatment significantly increased the number of NK cells in the lungs with a peak accumulation on day 4.5 (Fig. 2E). The increased number of NK cells in the lungs on days 2.5–5.5 was associated with an increased NK cell proliferation and numbers in the BM, spleen, and blood.
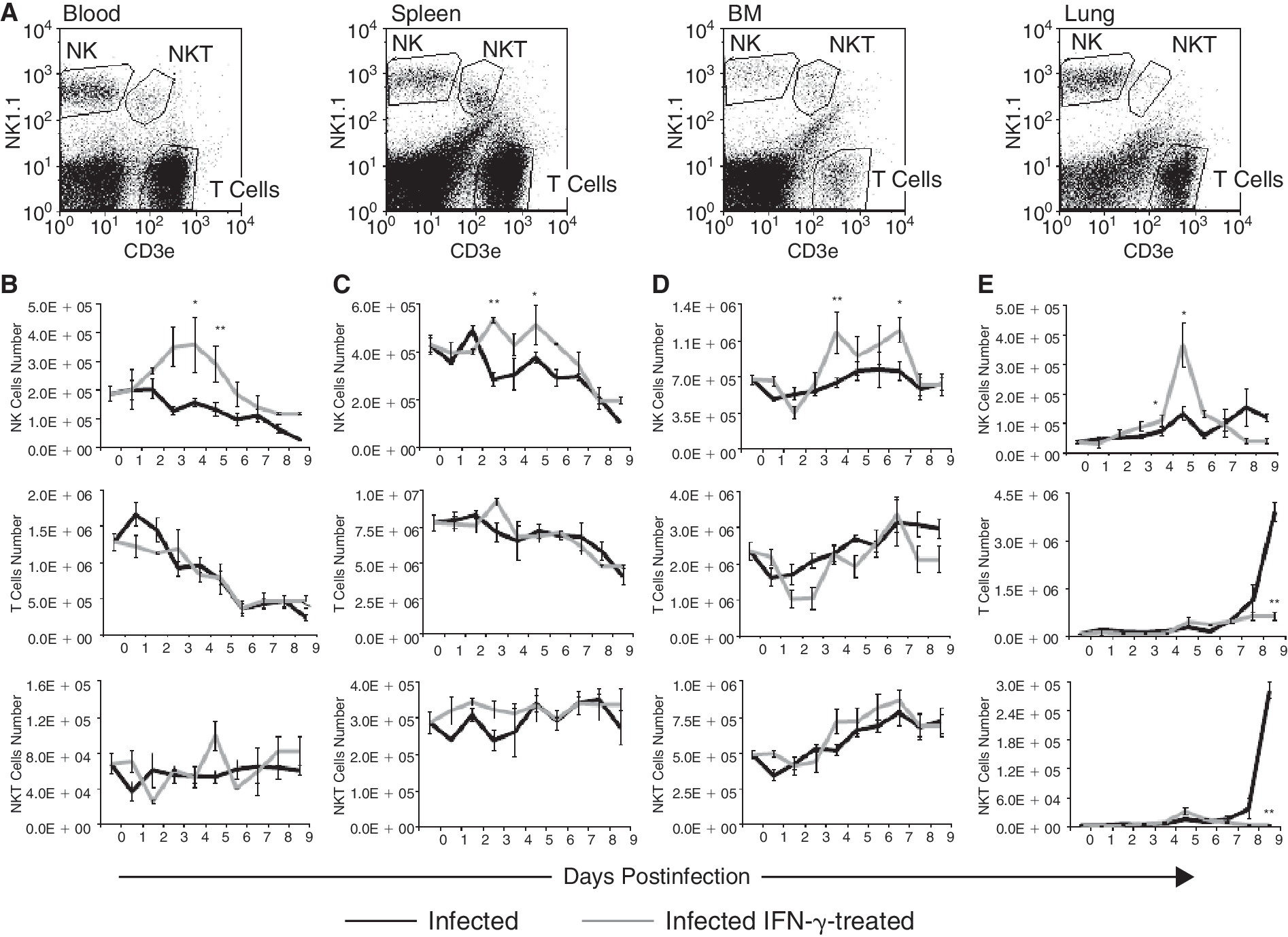
Treatment with IFN-γ enhanced NK cell recruitment to the lungs and abolished T and NKT cell infiltration to this organ after infection with influenza virus (IV). (
In both IV-infected and IV-infected IFN-γ-treated mice, T cells began to accumulate in the lung on day 5 and their number gradually increased toward day 9 (Fig. 2E). However, whereas in the untreated mice a 38-fold increase in the number of T cells was observed, in the IFN-γ-treated mice, only a 6.5-fold increase in the number of T cells was observed (Fig. 2E). Similar results were also obtained for NKT cells. The differences between treated and untreated infected mice were significant between days 7 and 9. T cells are the most abundant subpopulation in the lung of infected mice; however, using an in vivo BrdU assay, the rate of proliferation of these cells was found to be very low on day 5 following infection in both IFN-γ-treated and untreated mice, 4.2% versus 2.7%, respectively. Furthermore, 38- and 77-fold increases in the number of T and NKT cells, respectively, cannot be as a result of proliferation, but rather, as a result of a robust inflammatory reaction involving those 2 subsets of cells.
Hence, IFN-γ treatment, on the one hand, increased the number of NK cells in the periphery and lungs in the early stages of infection, and on the other, reduced T and NKT cell infiltration into the lung during the recovery phase of the infection. It is important to note that increased T- and NKT-cell-mediated inflammation was considered to play a major role in increased tissue damage and pathogenesis of the IV (de Jong and others 2006).
IFN-γ increased IV-dependent NK cell proliferation
Sequential injections of IFN-γ into naïve mice resulted in the mobilization of NK cells from the BM and spleen into blood, but failed to stimulate NK cell proliferation. To test whether an IFN-γ-dependent increase in the number of NK cells in the BM, spleen, and lung during IV was due to their proliferation, mice were pulsed with BrdU, 3 h before sacrifice and tested for the number of BrdU+NK1.1+CD3− proliferating NK cells. As expected, the percentage of proliferating NK cells in uninfected mice was much higher in the BM than in the spleen and lung (∼5%, 0.5%, 0.5%, respectively; Fig. 3A and 3B). The IV infection increased the percentage of cycling NK cells in the BM from 5% to 10%, in the spleen from 0.5% to 4%, and in the lung from 0.5% to 1%. IFN-γ treatment further increased the percentage of cycling NK cells in the BM from 10% to 20%, in the spleen from 4%, to 7%, and in the lung from 1% to 3%. It is therefore evident that the rate of proliferation of NK cells in the lung is low and that loading in vivo for 3 h with BrdU is sufficient to detect a highly proliferative population of NK cells. In addition, these results show that the BM is the main site for NK cell production and that IV infection alone or in combination with IFN-γ treatment enhances the proliferation of NK cells mainly in the BM, but also in the spleen and lung.
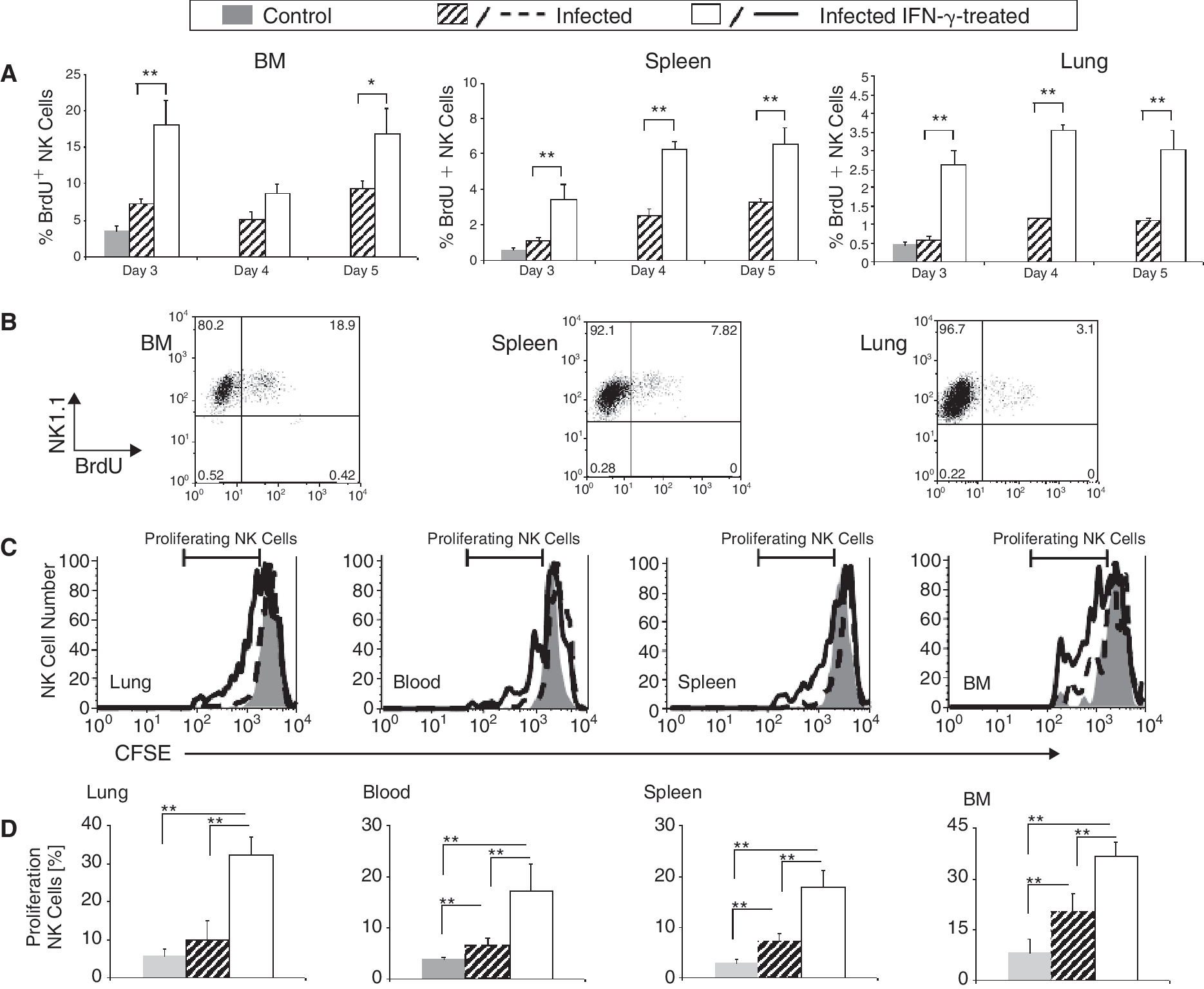
IFN-γ increased influenza virus (IV)-dependent NK cell proliferation and NK cells that proliferate in the bone marrow (BM) were mobilized and recruited to the lungs and the spleen. (
To further test the long-term proliferation potential of NK cells in the BM to the spleen, blood, and lung, we performed an adoptive transfer assay with NK cells loaded with CFSE. Total splenocytes were loaded with CFSE and injected onto mice 1 day prior to IV infection. Five days following infection, cells were collected from the lung, spleen, BM, and peripheral blood of IFN-γ-treated and untreated infected and uninfected mice and stained for NK1.1 and CD3, as described above. CFSE-labeled NK cells homing to the BM, spleen, and lung of uninfected mice proliferated at a low rate in the spleen and lung (<5%) and at a relatively higher rate in the BM (∼10%). Proliferating CFSE-labeled NK cells in the IV-infected mice increased their rate of proliferation to ∼10% in the spleen and lung and to ∼20% in the BM (Fig. 3D). Similar to the results shown in Figure 3A and 3B, the number of cycling CFSE-labeled NK cells increased significantly to 30% in the BM, spleen, and lung in the IV-infected IFN-γ-treated mice (Fig. 3C and 3D). By itself, IFN-γ treatment does not stimulate NK cell proliferation (Wald and others 2006). The current study provided evidence that the effect of IFN-γ on NK cell proliferation is dependent on additional stimuli provided by the IV infection.
IFN-γ treatment did not alter NK cell recruitment to the lungs of IV-infected mice
In order to prove that the trafficking of NK cells to the lung was regulated by IFN-γ, we chose a time point, that is, 3 h, wherein the contribution of proliferating NK cells to the overall number of CFSE-labeled NK cells in the lung is negligible.
Total splenocytes from uninfected mice, untreated and IFN-γ-treated infected mice, 5 days after infection, were isolated and loaded with CFSE. CFSE+ splenocytes were then injected into uninfected mice (control) and untreated and treated infected mice on day 5. Three hours after injection, cells were collected from the lungs of injected mice and the number of NK cells homing to the lung was measured by staining for NK1.1 and CD3. Three hours following injection, CFSE+ NK cells collected from the lungs did not proliferate and therefore considered as cells that home to the lung (Figs. 3A and 4A).
By comparing the homing of CFSE-labeled NK cells derived from uninfected mice, untreated, and IFN-γ-treated infected mice, 5 days after infection, to lung of uninfected mice (control) and untreated and treated infected mice on day 5, we were able to show that CFSE-labeled NK cells derived from all 3 groups had an equal, albeit superior homing potential to IV-infected or IV-infected IFN-γ-treated lungs compared to intact lungs. This clearly demonstrates that the homing potential of NK cells to the lung was mainly regulated by IV infection rather than by IFN-γ (Fig. 4B).
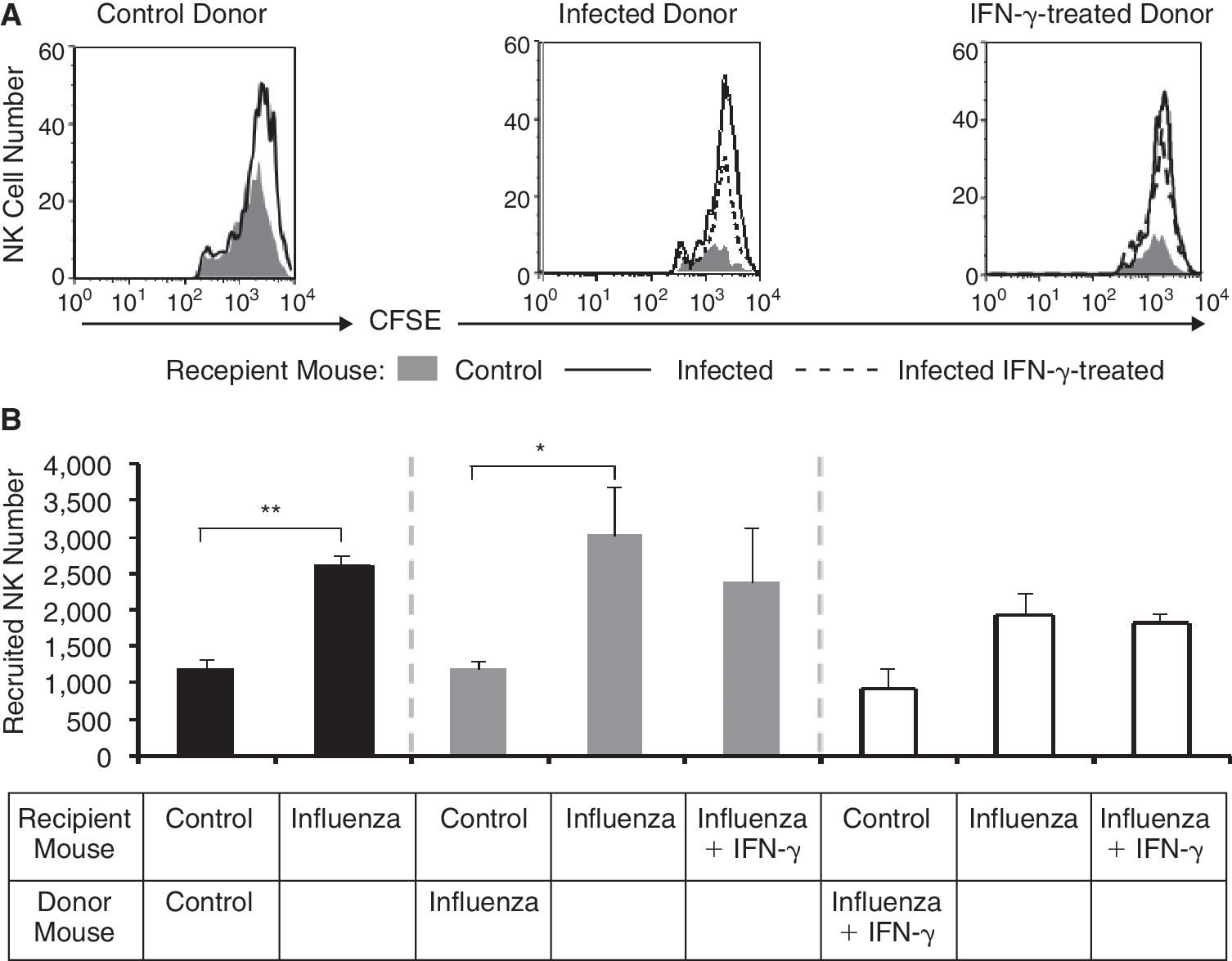
IFN-γ treatment did not alter NK cell recruitment to the lungs of influenza virus (IV)-infected mice. (
We therefore postulate that the increased number of NK cells in the lung was a consequence of a combined increased proliferation of these cells in the BM, spleen, and lung, an increased number of circulating NK cells due to their mobilization from the BM.
IFN-γ treatment combined with an unknown IV-dependent factor stimulated the proliferation of NK cells (Fig. 3A and 3D). Both IFN-α and IL-15 have been shown to regulate NK cell proliferation in vivo (Nguyen and others 2002). In order to test their potential involvement in NK cell proliferation, we monitored the levels of these cytokines in the lung, spleen, and BM of IV-infected and IFN-γ-treated IV-infected mice, on days 2–4 following infection, compared to control. The levels of IFN-α were significantly increased in all tested tissues; however, following IFN-γ treatment, we could not find any significant change in the levels of IFN-α treatment except for a reduction on day 3 in the treated BM (data not shown). The levels of IL-15 were slightly reduced in all tested tissues; however, following IFN-γ treatment, we could not find any significant changes in the levels of IL-15 treatment in all tested tissues (data not shown). Furthermore, we found that lung NK cells from untreated IV-infected mice or IFN-γ-treated IV-infected mice expressed similar levels of IL-2R, Mac-1, and NKG2D suggesting that NK cells from treated and untreated lungs have a similar phenotype and function (data not shown). We therefore hypothesize that sequential administration of IFN-γ stimulates an increased number of NK cells in the lung and in parallel induces a specific anti-inflammatory response.
IFN-γ treatment induced CXCL9 secretion in the lungs of IV-infected mice and IL-4 secretion from NK cells
The levels of IFN-α as well as IFN-γ were significantly increased during days 2–8 in the lungs of IFN-γ-treated and untreated, IV-infected mice (data not shown, Fig. 5A) creating a suitable environment for the clearance of IV-infected cells.
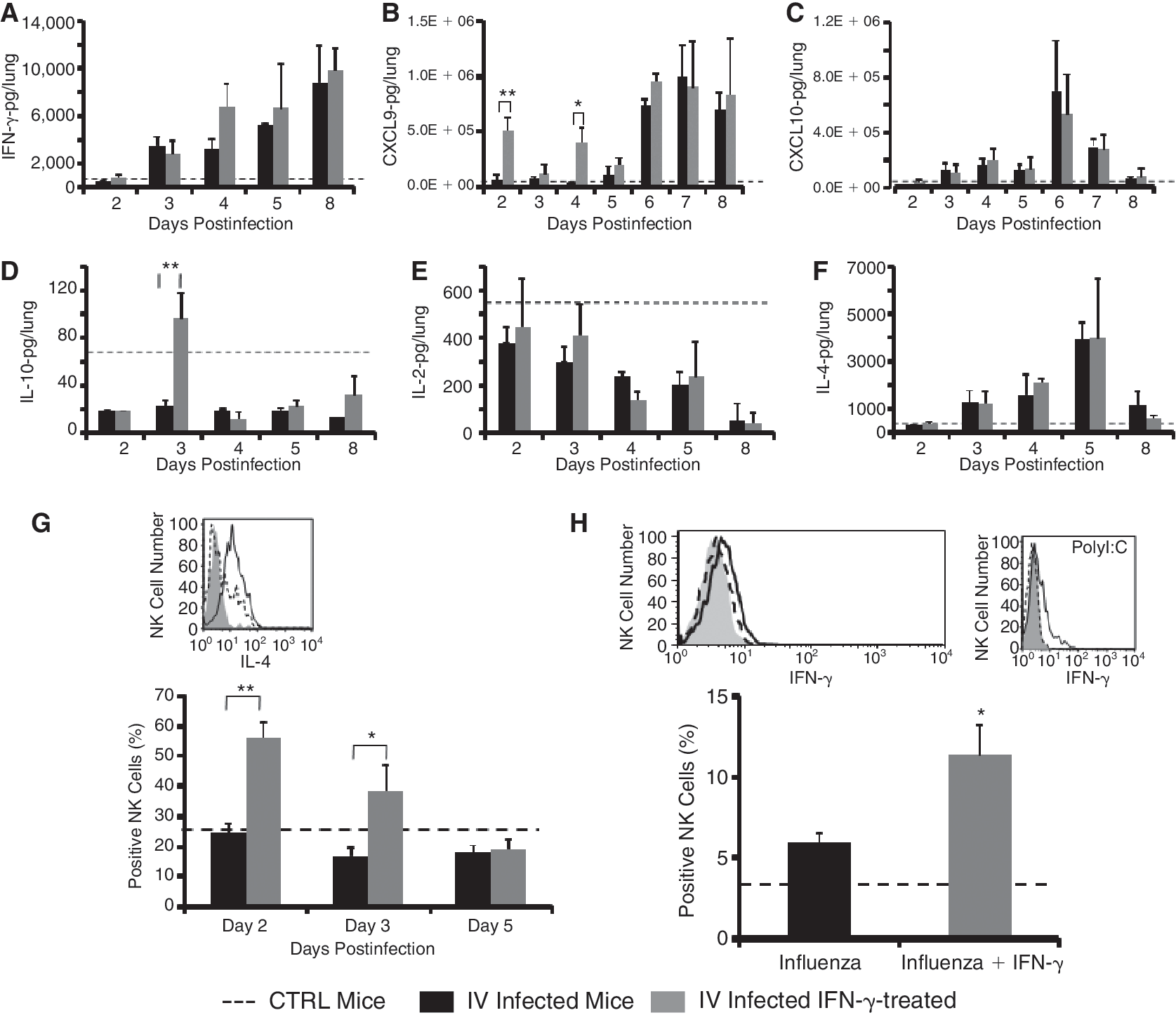
IFN-γ treatment induced CXCL9 and IL-10 secretion in the lungs of influenza virus (IV)-infected mice and IL-4 secretion from NK cells. (
In order to test whether NK cells in the lung of IV-infected mice are the source of the IFN-γ secretion, cells were harvested between days 2 and 4 from the lungs of infected mice and the intracellular levels of IFN-γ were determined. During this time frame, the level of IFN-γ in NK cells derived from uninfected and infected mice was low and the same in the 2 groups (Fig. 5H). Treatment of IV-infected mice with IFN- only slightly stimulated the production of IFN-γ by NK cells (Fig. 5H, black line). In order to test whether the inability of lung NK cells to secrete IFN-γ during IV infection was due to insufficient stimuli by IV, we injected mice with the TLR3 agonist, polyinosinic acid:cytidylic acid (poly(I:C)), and tested for the production of IFN-γ by liver NK cells (Wang and others 2005; Zhang and others 2007). Liver-derived NK cells stimulated with poly(I:C) produced higher levels of intracellular IFN-γ (Fig. 5H). It is therefore evident that IV infection in the lung did not stimulate significant amounts of IFN-γ secretion from NK cells and therefore exogenous administration may facilitate their regulatory role and killing capacity.
In addition to their immediate effects, NK cell responses can have immunoregulatory roles that provide conditions shaping downstream adaptive responses (Lee and others 2007; Moretta and others 2008). NK cells were recently shown to secrete IL-4 and IL-10 (Lidström and others 2003; De Maria and others 2007). Treatment with IFN-γ stimulated IL-4 secretion from NK cells derived from IV-infected mice treated with IFN-γ on days 2 and 3 (Fig. 5G). In contrast, NK cells derived from either untreated IV-infected mice or IFN-γ-treated IV-infected mice on day 5, 24 h after the last treatment with IFN-γ did not secrete IL-4 (Fig. 5G). Although the cytokines IL-4 and IFN-γ have often opposite effects and mutually suppress each other's production by T cells, each of these cytokines can also stimulate the other's production. IFN-γ can have a role in Th2 priming in vitro and in vivo, and IL-4 can contribute in vitro and in vivo to the priming of an IFN-γ response. Interestingly, IL-4 was shown to stimulate the secretion of IFN-γ from NK cells and may therefore amplify the immune response against IV in the early stages of infection (Morris and others 2006). IL-10 production was not detected in NK cells (data not shown).
Next, we tested the effect of IV infection and IFN-γ treatment on the secretion of pro- and anti-inflammatory chemokines and cytokines in the lungs of treated mice compared to control (dotted line). We first tested the effect of IFN-γ on the secretion of IFN-γ-inducible chemokines CXCL9 and CXCL10 in the lungs on days 2–8 following infection (Fig. 5B and 5C). We have already shown that injection of IFN-γ stimulated a potent and higher production of CXCL9 than CXCL10 (40 ng versus 1 ng, respectively) in the liver of C57BL mice (Wald and others 2006). Similarly, 3 h after injection of IFN-γ, a robust increase in CXCL9 production, but not CXCL10, was observed in the lungs on days 2 and 4 (Fig. 5B and 5C). This CXCL9 increase was reversible and was reduced almost to baseline after 24 h, on days 3 and 5 (Fig. 5B and 5C). Furthermore, on days 5–8, there were increased levels of CXCL9 production in parallel to an increased IFN-γ production in a similar way in both IFN-γ-treated and untreated infected lungs. In contrast, the levels of CXCL10 increased on days 3–6 and then decreased dramatically to control levels (dotted line) on day 8. It is important to note that CXCL10 production paralleled IV titers, whereas the production of CXCL9 paralleled the production of IFN-γ (Figs. 5C and 1D). This correlation between CXCL10 and viral titers and the inability of IFN-γ to stimulate CXCL10 production may suggest that other molecular pathways such as activation of toll-like receptors may be involved in this phenomenon (Wuest and others 2006). To further elucidate the role of Th2 cytokines in regulating inflammation in the lung, we tested the production of IL-10 and IL-4 in the lung of IFN-γ-treated or untreated IV-infected mice compared to control (Fig. 5D and 5F, dotted line). Similarly to IL-2 (Fig. 5E), the levels of IL-10 were dramatically reduced starting as early as day 2. Interestingly, IFN-γ treatment increased the levels of IL-10 on days 3, 5, and 8 compared to IV-infected lungs (Fig. 5D).
In contrast to IL-10, the level of IL-4 production increased significantly between days 3 and 5 and then decreased to baseline levels on day 8 (Fig. 5D and 5F). No significant differences were found in IL-4 levels of IFN-γ-treated or untreated IV-infected lungs.
Treatment of PR8 virus-infected mice with IL-4 during primary infection greatly suppressed the generation of cytotoxic T-cell precursors. In addition, culture supernatants of secondarily stimulated spleen cells from IL-4-treated mice contained significantly less IFN-γ and more IL-4 than did spleen cells from controls. More importantly, the treatment of mice with IL-4 resulted in a significant delay in virus clearance (Moran and others 1996). These results correlate well with the fact that viral clearance was only achieved on day 8 when the levels of IL-4 reached the baseline levels (Fig. 5F).
Discussion
Our previous study demonstrated that administration of IFN-γ stimulated the mobilization of NK cells from the BM and spleen into the circulation, but not their cell death or proliferation (Wald and others 2006). The results presented in this study demonstrate that in the early stages of IV infection, IFN-γ can stimulate the function and a robust proliferation of NK cells primarily in the BM and to lesser extent in the spleen and the lung (Fig. 3A). IV infection stimulates some NK cell proliferation in the BM; nevertheless, while the levels of NK cells in the blood and spleen are dramatically reduced, they remain the same in the BM and lung during IV infection. In contrast, sequential administration of IFN-γ increases the number of NK cells, but not T or NKT cells, in the BM, spleen, lung, and blood of IV-infected and treated mice. Upon careful review of the extensive literature on NK cell differentiation and effector functions, the factors that influence proliferation of the mature and immature NK cell pool during viral infection have received little attention. It has been shown that IL-15 is mandatory for the proliferation and survival of immature and mature NK cells, and that this IL-15 dependence is, at least in part, mediated through responses by other IL-15Rα-bearing cells (Koka and others 2003; Ranson and others 2003). Moreover, IL-15 also activates NK cell cytotoxicity and cytokine production and regulates NK cell/macrophage interaction (Fehniger and Caligiuri 2001). During the first 4 days following IV infection, IL-15 is similarly produced in the BM, spleen, and lung of uninfected mice and IFN-γ-treated and untreated IV-infected mice, and its secretion is slightly reduced following IV infection. We therefore conclude that both IV infection as well as IFN-treatment did not regulate NK cell proliferation through an increased production of IL-15.
Oral administration (Beilharz and others 2007) of low levels of IFN-α can protect mice from lethal influenza virus challenge. IFN-α induces NK cell trafficking and cytotoxic activity, and has also been shown to stimulate proliferation of these cells in vivo (Biron and others 1984; Biron and others 1999; Lee and others 2007). During the first 4 days following IV infection, the production of IFN-α in IFN-γ-treated and untreated IV-infected mice was dramatically increased in all tested tissues. Although this increase may have contributed to the maintenance and proliferation of NK cells in the BM and lung, it could not prevent the reduced number of NK cells in the spleen and blood of IV-infected mice. IL-2 has also been shown to induce NK cell proliferation and expansion in vivo (Biron and others 1990). We found that IL-2 was dramatically reduced in the lungs of IFN-γ-treated and untreated IV-infected mice. Interestingly, the reduction in IL-2 production paralleled the reduction of NK cell numbers in the spleen and blood. During the first 4 days after IV infection, the levels of IFN-γ were low whereas increased levels of IFN-α were already evident. While the exact mechanism by which IFN-γ stimulates NK cell proliferation is not known, it is possible that sequential administration of IFN-γ during this time frame coupled with the presence of IFN-α and IL-15 synergized to increase the proliferation and survival of NK cells.
Although it is evident that the intense inflammatory responses and hypercytokinemia resulting from IV are central to the lethal pathogenesis of the virus, most of the strategies explored to date to control IV infection focus on containing the spread of the IV infection (Rainsford 2006). There has been relatively little discussion about treating the lung and systemic inflammatory reactions occurring during IV infection. In both IFN-γ-treated and untreated mice, IV was cleared on day 8 following infection. However, this rapid IV clearance did not protect mice from the lethal effect of this virus (Fig. 1C). Interestingly, a huge influx of T and NKT cells to the lung started 1 day after the virus was cleared from the lungs and preceded death (Figs. 1 and 2). The earlier days following IV infection are important in shaping the type of cellular and cytokine inflammatory response that will be developed in the infected lungs. Sequential administration of IFN-γ during the first 4 days following infection changed the cellular response as well as the cytokine response in the lung without harming the ability of the immune system to clear the virus. During the first 4 days, IFN-γ stimulated NK cell proliferation in the BM, spleen, and lungs and allowed for greater numbers of NK cells to accumulate in the lung. These NK cells secrete low levels of IFN-γ and high levels of IL-4 and may therefore have a local anti-inflammatory effect. In addition, sequential administration of IFN-γ stimulated IL-10 production on day 3. IL-10 has been recognized as a major anti-inflammatory cytokine. Recently, IL-10 has been found to mediate virus persistence during chronic viral infections. Multiple cell types have been reported to produce IL-10 either constitutively or in response to inflammatory stimuli, most notably regulatory T cells and dendritic cells and, recently, CD4+ effector T cells during protozoa infections and T-effector cells during acute influenza infection (Sun and others 2009). Increased IL-10 levels together with the high IL-4 levels on that same day may induce an anti-inflammatory effect that will eventually prevent inflammation and damage to the lung at later stages. Similar to our observation, it has recently been shown that multiple injections of the IFN-γ-inducing factor, IL-18, promoted a peak production of both IL-4 and IL-10 after sub-lethal Escherichia coli infection (Kinoshita and others 2006). Furthermore, multiple injections of IL-12, which is also an IFN-γ-inducing factor in NK cells, also promoted sustained IL-10, but not IL-4 production (Portielje and others 2003).
Using anti-NK1.1 or anti-asialo antibodies, we depleted NK cells and have shown that NK cells independent of IFN-γ treatment are important for the immune response against IV and that the effect mediated by IFN-γ is also dependent on the presence of NK cells (Fig. 1C and 1D). Therefore, NK cells seem to play a critical role in our experimental system; however, it is possible that IFN-γ acts in a synergistic fashion through a variety of unknown mechanisms with NK cells or NK-derived cytokines.
Overall, the results presented here demonstrate the pivotal and dual role of IFN-γ in the treatment of IV infection by both promoting the function and proliferation of NK cells and stimulating antiviral response. Moreover, treatment with IFN-γ may contribute to a lesser proinflammatory milieu in the lung, thus enabling the clearance of the virus and preventing the deadly cost of inflammation. This study may therefore lead to the development of novel approaches that may generate balanced and sufficient immune responses against IV infection.
Footnotes
Acknowledgments
We thank Mery Clausen (Gene Therapy Institute, Hadassah Hospital) for technical assistance, Gazit R. for technical support, and Mr. Ranan Kulka for moral support and judicious advice. This work was supported by grants from the Blum, the Harold Grinspoon, the Horowitz, and the Wolfson Foundations.
Author Disclosure Statement
Authors declare no conflict of interest or financial interests.