
Abstract
The glycoprotein 130 (gp130) is a shared signal-transducing-membrane-associated receptor for several hematopoietic cytokines. Its activation is implicated in pain and in a variety of diseases via signaling of proinflammatory cytokines. These include interleukin-6 (IL-6) subfamily cytokines, many of which play important roles in the pathogenesis of diseases such as rheumatoid arthritis, Castleman's disease, and Kaposi's sarcoma. Several strategies have been developed to block gp130-receptor-mediated signaling. These include the application of monoclonal antibodies, the creation of mutant form(s) of the gp130 with increased binding affinity for such ligands as IL-6/sIL-6R complex, and the generation of antagonists by selective mutagenesis of the specific cytokine/gp130 receptor binding site(s). Other strategies include targeting gp130-mediated signaling pathways such as that involving signal transducer and activator of transcription-3. This review provides a summary of the latest research pertaining to the role of gp130 in the pathogenesis of inflammatory and other diseases in which the gp130 receptor is implicated. An overview of antagonists targeting the gp130 receptor is included with particular emphasis on their mechanism of action and their limitations and potential for therapeutic application.
Introduction
G
Gp130 activation is implicated in a variety of diseases. These include rheumatoid arthritis (RA), Castleman's disease, Crohn's disease, multiple myeloma, psoriasis, amyotrophic lateral sclerosis, several cancers, diabetes, asthma, and obesity (Table 1). Further, gp130 has also been shown to amplify pain, not only indirectly via the induction of proinflammatory cytokines, but also through a direct action on neurons in response to painful stimuli (nociceptors) (Andratsch and others 2009). Andratsch and others (2009) found that gp130 expression on the membrane of nociceptive primary afferent nerves is required for the development of pathological pain and hyperalgesia in inflammatory conditions and cancers. They showed that cytokines contribute to pain hypersensitivity via the gp130-Gab1/Gab2-PI3K-PKC-δ-TRPV1 pathway in peripheral sensory neurons. Importantly, IL-6 was shown to play a major part in the activation of this pathway.
IL, interleukin; LIF, leukemia inhibitory factor; OSM, oncostatin M; CNTF, ciliary neurotrophic factor; gp130, glycoprotein 130.
Accordingly, strategies to counteract the action of gp130, by targeting gp130 itself, its ligands, or the genes or proteins associated with its signal transduction pathway, have become an important focus of research, both from an immunopharmacological standpoint and also in the quest for development of pharmaceuticals to control pain and inflammation.
In particular, the recognition of shared molecular subunits (gp130) in the receptors for IL-6 cytokines has made it possible to use components of these receptors, either alone or in combination, as antagonists or as research tools. Most of the antagonists targeting IL-6 family cytokines, which have been developed so far, are based on the shared use of the gp130 receptor system. One approach has been to block the formation of IL-6/IL-6R/gp130. This strategy has been used to improve the symptoms in patients with RA, multiple myeloma, and Castleman's disease (Rebouissou and others 1998; Nishimoto and others 2000).
Interestingly, capsaicin, the pungent ingredient of hot peppers, has been found to inhibit the IL-6/STAT-3 pathway by depleting intracellular gp130 pools through endoplasmic reticulum stress (Lee and others 2009) Other antagonists that include those targeting the IL-6/gp130 complex have now been approved by the FDA for patients who have failed anti-tumor necrosis factor (TNF) therapy (Emery and others 2008; Genovese and others 2008). Examples include Tocilizumab, which is a humanized monoclonal antibody (mAb) with specificity for IL-6R. Its therapeutic efficacy in RA has been extensively tested and proven in clinical trials (Nishimoto and others 2004). In addition, ALD518, which is a humanized mAb directed against IL-6, has progressed to a phase II clinical trial for advanced cancer and to a phase IIa study for RA (Clarke and others 2009).
Gp130 Receptor
Gp130 receptor is a transmembrane protein, so called because of its molecular weight of 130 kDa. It is expressed widely in different organs, including brain, lung, liver, spleen, heart, kidney, and placenta (Hibi and others 1990). Signals from IL-6 subfamily members (IL-6, IL-11, LIF, and OSM) depend on gp130 (Ohtani and others 2000). While IL-6 and IL-11 binding leads to homodimerization of gp130, LIF-R and OSM-R heterodimerizes with gp130, forming gp130/LIF-R or gp130/OSM-R, which in turn leads to signal transduction (Fig. 1).
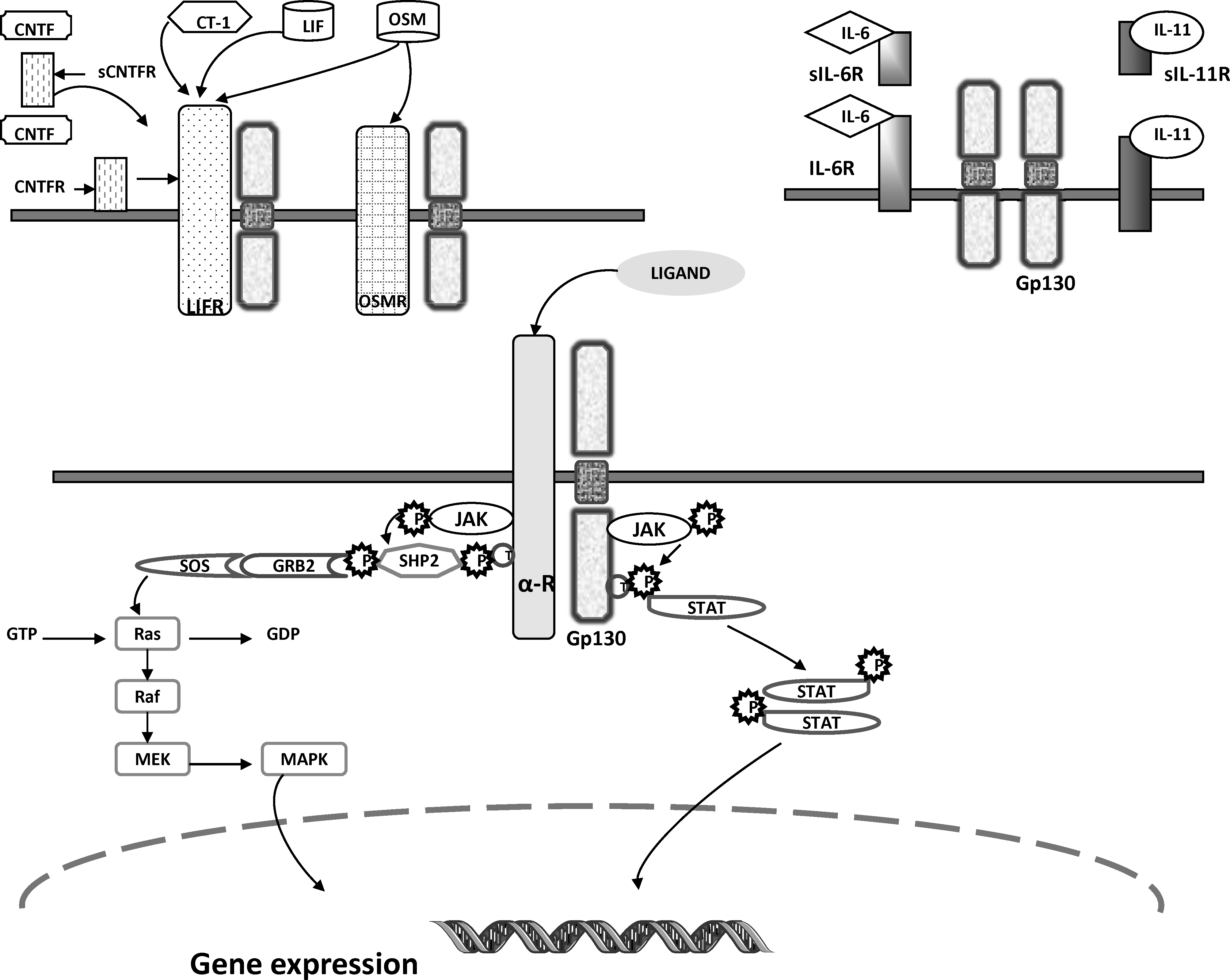
Gp130 signaling pathways. Gp130 receptor essentially signals through JAK/STAT and/or MAPK transduction pathways. The cytoplasmic component of gp130 is noncovalently associated with JAK molecules, which become activated upon ligand binding. The JAKs then phosphorylate the specific receptors and gp130 on certain highly conserved tyrosine residues in the membrane-distal cytoplasmic part, which in turn binds to adaptor protein molecules containing an Src-homology-2 (SH2). Depending on the tyrosine residue activated, the adaptor molecules can either be of the signal transducers of transcription (STAT) family triggering the JAK/STAT signaling pathway or SHP-2 (SH2 domain containing tyrosine phosphatase, binding to Tyr759 of gp130 and Tyr974 of LIF-R) and Shc (SH2 and collagen homology domain containing protein, binding to the Tyr861 of OSM-R), which are found to be crucial for the activation of the Ras/Raf/MAPK pathway. In the JAK/STAT signaling pathway, the phosphotyrosine–SH2 interaction paves the way for subsequent tyrosine-phosphorylation and homo/hetero-dimerization of the STAT molecules. Dimerized STATs are then translocated to the nucleus where they initiate relevant gene transcription and subsequent translation. Gp130, glycoprotein 130; JAK, Janus kinase; STAT, signal transducer and activator of transcription; MAPK, mitogen-activated protein kinase.
Gp130 is a member of the cytokine-receptor superfamily. It is envisaged to consist of 6 parts in its extracellular domain, numbered from distal to membrane-proximal—D1–D6. The second and third parts comprise the cytokine-binding component highlighted by 4 conserved cysteines and a WSXWS motif (Pflanz and others 2000). Gp130 itself has no intrinsic tyrosine kinase activity, but it is phosphorylated on tyrosine residues after complex formation with any of the IL-6 subfamily cytokines. This leads to its association with JAK/Tyk tyrosine kinases and STAT protein transcription factors, specifically STAT-3, which when phosphorylated leads to the activation of a number of downstream genes (Heinrich and others 1998).
Mutational analysis has shed some light on the role of gp130. Homozygous mice deficient in gp130 activation have shown many defects such as impaired development of the ventricular myocardium. Ernst and colleagues (Ernst and others 2001) generated gp130 knock-in mice (gp130ΔSTAT/ΔSTAT), in which all STAT-binding sites were deleted from the gene encoding gp130, but the binding sites for both JAKs and protein tyrosine phosphatase-2 (SHP-2) were preserved. They found that this mutant mouse developed severe joint disease with features strongly resembling those found in RA, as well as a blastocyst implantation defect and gastrointestinal ulceration. Such studies highlight the potential importance of defects in gp130 signaling in the pathogenesis of arthritis.
Gp130R and IL-6 subfamily cytokines
The binding of cytokine(s) to gp130 receptor takes place at different sites (Table 1). In general, the IL-6 subfamily cytokines share and activate the gp130 receptor in different ways. In one scenario the cytokine binds to its own specific receptor with high affinity before heterodimerization with gp130. For example, LIF binds to LIF-R and LIF-R/gp130 heterodimerizes (Hudson and others 1996), and in the other, the cytokine binds to gp130 receptor with high affinity after it has first bound to another receptor; eg, IL-6 first binds to either membrane-bound IL-6R or soluble (circulating free) IL-6R (sIL-6R) and the complex then binds gp130 with high affinity causing it to homodimerize. Similarly IL-11 binds to sIL-11R. CNTF, in addition to binding to LIF-R, binds sCNTF (Taga and others 1989) (Fig. 1).
Methods Used to Block Gp130 Signaling
A number of strategies have been developed to block the gp130 activation, stimulated by the IL-6 subfamily cytokines. These are detailed below.
Monoclonal antibodies
mAbs directed against gp130 have been shown to inhibit the biological activities of those cytokines that use gp130 receptor systems, eg, IL-6 subfamily cytokines. Gu and others (1996), for example, used human plasmacytoma cells, an IL-6 subfamily cytokine-dependent cell line, and generated anti-gp130 mAbs that specifically inhibited CNTF, but without affecting the biological activity of the other members of the IL-6 cytokine subfamily. Such antibodies, some of which are patented, have applications not only as research tools, to design anti-IL-6 cytokine inhibitors, but also in medicinal products and diagnostic reagents.
Soluble forms of gp130
As a means of blocking IL-6/gp130 activation, Jostock and others (1999) created recombinant soluble forms of gp130 (sgp130) proteins that were capable of binding to IL-6, in complex with sIL-6R, and blocked IL-6/sIL-6R-induced proliferation of gp130-expressing BAF/3 cells in vitro (at concentrations ranging from 10 to 100 ng/mL). Further, they showed that sgp130 partly inhibited LIF- and OSM-induced proliferation when used at higher concentrations (1,000 ng/mL). However, due to the bipartite nature of IL-6Rα, blocking IL-6 action using soluble receptors has, in general, been very challenging. This is because IL-6 alone does not bind to gp130, but it must first bind sIL-6Rα before it can be neutralized by sgp130. The gp130/sIL-6Rα complex thus formed can neutralize the agonistic complex of IL-6/sIL-6Rα. For this reason Jostock and others created a recombinant sgp130 that was capable of specifically blocking IL-6 responses dependent on sIL-6Rα, whereas membrane-bound IL-6R remained unaffected.
Muller-Newen and others (1998) showed that when cells lacking membrane-bound IL-6R were stimulated with IL-6/sIL-6R complexes, sgp130 was a more effective antagonist than on IL-6R-positive cells stimulated with IL-6 alone. This is not surprising given the low affinity of gp130 for IL-6 in the absence of IL-6R. They also argued that the sIL-6R, found in human plasma, could well be regarded as an antagonistic molecule that would possibly enhance the inhibitory activity of sgp130. To block IL-6 trans-signaling in vivo, a scenario in which the release of sIL-6R by one cell type renders other cells that only express gp130 responsive to IL-6, Rabe and others (2008) generated transgenic mice that overexpress sgp130. They showed that sgp130 specifically blocks all IL-6 responses mediated by the soluble IL-6R but that it does not affect IL-6 responses via the membrane-bound IL-6R. sgp130 has also been detected in human serum at high concentrations (in the order of ∼200 ng/mL) and has been correlated with blood pressure in stroke patients (Inta and others 2009).
Sgp130 mutants
To generate sgp130 mutant proteins (muteins), Tenhumberg and others (2008) analyzed the 3-dimensional structure of gp130 complexed with IL-6/sIL-6R and identified specific amino acid side chains in gp130 that interact with sIL-6R. These were then used as targets to produce mutants with increased binding affinity for the IL-6/sIL-6R complex. The mutants generated were tested for binding to the IL-6/sIL-6R complex as well as for inhibition of IL-6/sIL-6R-induced cell proliferation. Several mutants demonstrated marked increases in binding affinity of human sgp130 toward human IL-6/sIL-6R. In addition, some mutations showed an additive effect, improving the binding affinity of human sgp130, and its more stable Fc fusion counterpart sgp130:Fc, toward IL-6/sIL-6R. They also showed that one mutated form of sgp130:Fc, with 3 amino acid substitutions very effectively blocked IL-6/sIL-6R-mediated biological responses both in vitro and in vivo (using an animal model of arthritis driven by IL-6/IL-6R). The effect of sgp130 muteins on membrane-bound IL-6R was not examined in this study. Examples of glycoprotein 130 derivatives and their cytokine specificity and pharmacological attributes are shown in Table 2.
sgp130, soluble form of gp130; gp130, glycoprotein 130; IL, interleukin; RA, rheumatoid arthritis; PEG, polyethylene glycol; ELP, elastin-like peptide; STAT, signal transducer and activator of transcription.
Sgp130:Fc and sgp130his
To increase its potency, sgp130 has been fused with the Fc portion of human IgG1 creating sgp130:Fc. Fusion constructs of this type are commonly created to enhance the serum half-life of therapeutic molecules because the Fc protein is expressed as a disulphide-linked dimer (Jazayeri and Carroll 2008). In general, cytokines, in their monomeric form, have relatively small molecular size and short serum half-lives (5–50 min), and are not only rapidly cleared from the kidneys but also subject to degradation by proteolytic enzymes. This necessitates frequent administration and thus limits their clinical utility. In addition, since there is an inverse relationship between the size of these molecules and the rate at which they diffuse through mucus, Fc fusion constructs potentially have a lower rate of diffusion. Further, such constructs can also increase the avidity for multivalent ligands due to the resulting bivalency of dimeric fusion proteins.
The sgp130:Fc thus constructed was found to be 10 times more active compared to monomeric sgp130. In addition, the dimerized soluble form of sgp130 was shown to suppress the action of the soluble IL-6/IL-6R complex in vitro and in vivo (Jostock and others 2001a) without affecting the activity of the membrane-bound IL-6R. Jostock and others (2001b) constructed an sgp130 his, by replacing the Fc portion in the sgp130:Fc with 6 histidine residues; however, this construct was not as potent as sgp130:Fc. One possible reason for this lack of potency is its monomeric structure. Recently, mutated forms of sgp130:Fc have been generated and shown to have higher potencies for blocking biological responses mediated by IL-6/sIL-6R complex in vitro and in vivo (Tenhumberg and others 2008).
Fusion of Sgp130 sIL-6R
Recognition of the important role of sIL-6R together with sgp130 led to the idea of construction of an sIL-6R and sgp130 fusion complex. This was constructed mainly because, in cells expressing membrane-bound IL-6R, when treated with IL-6 alone, sgp130 was found to be a weak antagonist and sgp130 alone was inadequate to inhibit the IL-6 response. To overcome this, Ancey and others (2003) fused the 2 receptor subunit fragments creating an sIL-6R/sgp130 fusion complex. To achieve this, the ligand-binding domains of sgp130 (D1-D2-D3) and sIL-6R (D2-D3) were connected using 3 different linkers. Biological activity assays showed that the construct was capable of inhibiting IL-6 activity. It abrogated IL-6-induced phosphorylation of STAT-3, and inhibited the proliferation of transfected Ba/F3 cells expressing gp130 and IL-6R. These investigations also showed that the construct was much more effective than the separately expressed soluble receptor proteins.
Polyethylene glycolylated sgp130-dimers
To increase the serum half-life of sgp130 even further, the molecule was polyethylene glycol(PEG)ylated creating sgp130:PEG (patent application). The potency of this construct was found to be ∼2-fold higher than that of the sgp130:Fc molecule. Further, it was postulated that the same results, as those achieved with sgp130:Fc, would be possible with a lower dose of sgp130:PEG. This construct has several advantages, which include lower drug exposure for the patient, reduced application frequency, and lower therapeutic costs.
Gp130-RAPS
Gp130 of the RA antigenic peptide-bearing soluble form (gp130-RAPS) has been isolated as a disease-associated auto-antigen in RA. This soluble truncated form of gp130 has a unique sequence and has been identified as an auto-antigen in RA (Tanaka and others 2000). Antibodies against gp130-RAPS have been shown to neutralize gp130-RAPS activity. Gp130-RAPS have also been shown to inhibit IL-6 activity and to be neutralized by anti-gp130-RAPS antibodies derived from patients with RA. In addition, the concentrations of these antibodies in the serum of RA patients have been found to correlate positively with (i) serum C-reactive protein concentrations, (ii) erythrocyte sedimentation rate, (iii) platelet counts, and (iv) serum IL-6 concentrations. Thus, it is hypothesized that auto-antibodies to gp130-RAPS could play an important role in the pathogenesis of RA by stimulating IL-6 activity. Further, it is possible that auto-antibodies to gp130-RAPS may become clinically useful as a marker for disease activity in RA.
Gp130-elastin-like peptide
Lin and others (2006) constructed a biologically active form of sgp130 variant that was produced in transgenic tobacco plants as an elastin-like peptide (ELP) fusion protein. The construct consisted of the first 3 domains of gp130 fused to 100 repeats of ELP. Expression of mini-gp130-ELP did not affect the growth rate or morphology of the transgenic plants, and purification was achieved using inverse transition cycling. With this approach Lin and others (2006) produced 141 μg of purified protein per gram of fresh leaf weight. The purified protein inhibited sIL-6R-mediated trans-signaling as demonstrated by 3 features: (i) bind to the IL-6–sIL-6R complex, (ii) its inhibition of proliferation in human hepatoma cells and murine pre-B-cells, and (iii) by its capacity to block sIL-6R-mediated activation of STAT-3 phosphorylation.
Cytokine-based antagonists targeting gp130
In addition to the application of mAb against sgp130, other approaches to blocking gp130 activation have been investigated and developed. These include Creation of mutations in the gp130-dependent cytokines that render them defective in respect to their capacity to bind specific sites within the gp130 receptor. For example, LIF mutants have been generated that can still bind to LIF-R with high affinity, but that lack the ability to efficiently bind to gp130 and in turn initiate its activation (Vernallis and others 1997). One potential disadvantage of this approach is the possibility that such mutants may be antigenic and provoke the elaboration of neutralizing antibodies that may in turn limit or completely abrogate efficacy or cause other toxicity. Creation of mutations in the gp130 cytokines that increase their affinities for the gp130 receptor. This is the opposite of the strategy outlined above. Examples of this approach include (i) the creation of a mutant form of human LIF that binds to human LIF-R with over 1,000-fold higher affinity than wild-type human LIF, but that does not bind gp130 as required for productive signaling, thereby blocking LIF-induced gp130 activation and (ii) the creation of IL-6 mutant antagonists with enhanced affinity for gp130 (Savino and others 1994; Fischer and others 1997). Exchanging biological specificities within the cytokine family. In yet another approach, the site III epitope of IL-6 was exchanged with its CNTF counterpart. This hybrid molecule acquired a requirement for LIF-R to activate signaling (Grotzinger and others 1999).
Antagonists targeting STAT-3 signaling
As outlined earlier, gp130 receptor essentially signals through JAK/STAT and/or mitogen-activated protein kinase transduction pathways (Fig. 1). Such pathways have also been targeted to inhibit gp130 signaling. For example, in a study conducted by Sobota and others (2000), it has been shown that parthenolide, a sesquiterpene lactone found in many medicinal plants, inhibits IL-6-type cytokine-induced gene expression by blocking STAT-3 phosphorylation on Tyr705. They showed that parthenolide prevents STAT-3 dimerization, which is required for its nuclear translocation and gene expression. In addition, based on the fact that prostate cancer cells have high levels of IL-6 (82% of prostate cancer patients), inhibition of STAT-3 may have therapeutic potential in patients with prostate cancer (Weerasinghe and others 2008). IL-6 also activates androgen-receptor-dependent gene expression in prostate cancer cells in the absence of androgen. Although the role of STAT-3 in prostate cancer is not clearly established, activated STAT-3 is considered to be an important signaling molecule that plays a critical role in progression of both androgen-dependent and androgen-independent prostate cancers (Nishimoto and Kishimoto 2008). Lee and others (2004) have used siRNA specific for STAT-3 and showed that inhibition of STAT-3 activation suppressed the growth of human prostate cancer cells and STAT-3-mediated gene expression, which also induced apoptotic cell death.
Further, it has been shown that G-quartet oligonucleotide, T40214, is a potent inhibitor of STAT-3 binding to DNA. At present, variations of this compound are being tested to determine if human tumors in nude mice are suppressed. The goal is to use such a compound to treat patients with metastatic prostate cancer. In addition, in a study conducted by Selander and others (2004), using an orthotopic nude mouse model, it was shown that inhibition of gp130 signaling in breast cancer blocks constitutive activation of STAT-3 and inhibits the development of malignancy.
Targeting STAT-3 blockade with gene therapy
Several gene-therapy-based approaches have also been taken toward activating/de-activating STAT-3-mediated signaling and thereby regulating gp130-associated signal transduction. Yu and others in 2009 patented many gene-delivery-based therapeutic strategies that have potential for prevention or treatment in ischemic disorders, proliferative angiopathies with neovascularization, such as diabetic microangiopathy, and for regulating immune responses before the advent of any disease.
Applications and Conclusions
The potential for targeting gp130 to favorably influence the activity and course of diseases in which the gp130 signaling system is implicated is summarized in Table 3. Targeting either gp130 itself or cytokines that depend on it for signaling, such as the IL-6 subfamily cytokines, constitutes a novel class of anti-inflammatory and anti-neoplastic drugs. Further, recent studies demonstrating the role of gp130 in sensory neuron function has opened up new possibilities for novel therapeutics that may be useful in pain management. IL-6 blockade, for which some antagonists have been developed, could also be applicable in this respect. The logic to inhibiting gp130 is that it will inhibit signaling dependent on all cytokines using the pathway. This may be beneficial in systems in which multiple ligands are present, eg, in RA in which IL-6, LIF, and OSM play a main role in the disease pathogenesis.
IL, interleukin; RA, rheumatoid arthritis; gp130, glycoprotein 130; TNF, tumor necrosis factor; JAK, Janus kinase; STAT, signal transducer and activator of transcription; LIF, leukemia inhibitory factor; OSM, oncostatin M; CNTF, ciliary neurotrophic factor; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase.
Treatment strategies for targeting gp130 include the use of mAbs, sgp130, and cytokine-based receptor antagonists against cytokines sharing gp130 and compounds targeting IL-6. Some of these are in advanced stages of development, but no gp130 inhibitor is ready yet for clinical application. One reason for this is that there are still concerns about safety. Blocking transmembrane receptors such as gp130, without affecting innate or acquired immune responses, remains a complex and challenging task. These and other safety issues will need to be carefully considered before gp130 antagonists can be used in clinical practice. If, however, the aim of anti-gp130 therapy is only to attenuate signaling, there may be a therapeutic window. Here it will be important to monitor residual gp130 signaling in a variety of tissues.
There are numerous studies of tissue-specific gp130−/− mice that demonstrate that a complete lack of gp130 compromises the animal's response to injury (gut, cardiac, neuronal, and immune) (Ernst and others 2001). Given the implicit role of the IL-6 cytokine subfamily in the innate immune system, the possibility of facilitating infections, provoking autoimmune phenomena, and compromising immune surveillance all need to be considered. Whether granulomas may be destabilized by gp130 signal blockade is unclear, but this would need to be kept in mind in view of the experience with TNF antagonists, where treatment has resulted in the reactivation of latent tuberculosis as a consequence thereof. Autoimmune disorders, including lupus and psoriasis, may be promoted, and skin, lymphoid, other bone marrow lineage, or solid organ malignancies may be induced or accelerated. Further, there will be a need for vigilance in respect to possible liver and CNS toxicity and the possibility of unforeseen allergic and metabolic consequences. Concern in respect to the latter is heightened by the observation of mild liver dysfunction and elevated cholesterol concentrations in recipients of the IL-6 receptor antibody, Tocilizumab. It is noteworthy that in the IL-6 null mouse, death occurs when a partial hepatectomy is performed, implying possible reduced hepatic reserve when the normal IL-6 pathway is compromised. There will also be a need for special care when considering therapeutic gp130 blockade in children and adolescents, since gp130 knockout studies demonstrate the importance of the gp130 signaling system in embryonic development.
Footnotes
Author Disclosure Statement
No competing financial interests exist.