
Abstract
The recent emergence of pandemic swine-origin influenza virus (H1N1) and the severe outbreaks of highly pathogenic avian influenza virus of the H5N1 subtype leading to death in humans is a reminder that influenza remains a frightening foe throughout the world. Besides vaccination, there is an urgent need for new antiviral strategies to protect against influenza. The innate immune response to influenza viruses involves production of interferon alpha and beta (IFN-α/β), which plays a crucial role in virus clearance during the initial stage of infection. We examined the effect of IFN-α on the replication of H5N1 and H1N1 in vitro and in vivo. A single pretreatment with low-dose IFN-α reduced lung virus titers up to 1.4 log10 pfu. The antiviral effect increased after multiple pretreatments. Low-dose IFN-α protected mice against lethal H5N1 viral infection. Further, IFN-α was also effective against H1N1 in vitro and in the mouse model. These results indicate that low-dose IFN-α treatment leads to the induction of antiviral cytokines that are involved in the reduction of influenza virus titers in the lung. Moreover, it might be possible that a medical application during pandemic outbreak could help contain fulminant infections.
Introduction
S
In the first stage of influenza infection, it is essential to protect the airway epithelium against infection. There are 2 major pathways that activate the host innate immune response against viral infection, namely, the RNA helicase RIG-I as a receptor for intracellular viral double-stranded RNA and the toll-like receptor family. Toll-like receptors induce interferons (IFNs) to activate the transcription factors NF-κB, IRF3, and IRF7 (Ludwig and others 2006; Seth and others 2006). These IFNs are one of the most potent known antiviral mechanisms of the host response that can limit virus replication and spread (Goodbourn and others 2000; Randall and Goodbourn 2008). The importance of this innate immune response against pathogens is highlighted by the fact that several viruses such as influenza virus, measles virus, Newcastle disease virus, and respiratory syncytial virus inhibit the synthesis of IFNs by interacting with the host IFN response (Huang and others 2003; Palosaari and others 2003; Lo and others 2005; Kochs and others 2007a). Type I IFNs, namely, IFN-α and IFN-β, are synthesized and secreted by cells in response to viral infection. They act in an autocrine as well as a paracrine manner to upregulate the expression of hundreds of IFN-stimulated antiviral genes (ISG). These ISGs are involved in the upregulation of the double-stranded RNA-activated protein kinase R (PKR), 2′,5′-oligoadenylate synthetase (OAS), and Mx proteins, which are all known to mediate resistance to viral infections (Silverman 2007). The MxA protein has been shown to directly interfere with influenza A virus replication (Haller and others 2009). In addition to the direct antiviral properties, IFNs-α/β modify the host immune response in different ways. IFNs-α/β upregulate MCP-1, MCP-3, and IP-10 gene expression, which results in additive recruitment of monocytes/macrophages to the site of infection. IFNs-α/β also enhance the antigen presentation of macrophages and dendritic cells by upregulating MHC gene expression (Stark and others 1998). Moreover, IFNs-α/β are important cofactors in the development of Th1-type immune response. IFNs-α/β are involved in T lymphocyte survival, enhancement of IL-12 receptor, and IFN-γ gene expression in human natural killer cells and T lymphocytes (Rogge and others 1997; Matikainen and others 1999). These data underline an essential role of IFNs-α/β in both innate and adaptive immunity against influenza A and other viral infections. During influenza A virus infection the nonstructural protein (NS1) of the virus is one of the most important antagonist for this antiviral state (Haye and others 2009). Influenza viruses that are unable to express NS1 (delNS1) induce large amounts of IFN in infected cells (Kochs and others 2007b). These findings demonstrate that type I IFNs play an important role in inducing the antiviral state in the first line of defense and to help protect the tissue against a fatal infection.
Treatments of high-dose IFN (3 × 106–9 × 106 U) are actually applied in a wide spectrum of diseases like hepatitis B and C, multiple sclerosis, and autoimmune disorders (Cooksley 2004; Pereira and Jacobson 2009; Sottini and others 2009). These long-term high-dose treatments are often accompanied by various severe adverse events such as depression, fever, chills, muscle aches, headache, and a general feeling of discomfort. The short-term intranasal administration of IFN-α treatment for human viral respiratory diseases has been shown in Eastern Europe and Russia to have promising antiviral effects (Imanishi and others 1980; Isomura and others 1982; Saito and others 1985). The question arises whether low-dose IFN-α is effective in vitro and in vivo to protect against highly pathogenic and seasonal influenza A virus infections.
In the present study, we examined the antiviral effect of low-dose IFN-α treatment on influenza A virus infection of cell cultures and in the mouse model. IFN treatment of human and mouse cell lines resulted in a reduction of progeny viral titers. Decreased lung viral titers of H5N1 and H1N1 were observed after intranasal IFN-α treatment of infected mice. The decrease of virus in the lung apparently resulted in prolonged survival of mice that were infected with an otherwise lethal dose of influenza H5N1 virus.
Materials and Methods
Virus strains
The highly pathogenic avian influenza A H5N1 virus strains A/mute swan/Germany/R1349/07 (SN1) and A/goosander/Bavaria/20/2006 (GSB1) were grown in embryonated chicken eggs. This isolate was originally obtained from the Bavarian Health and Food Safety Authority, Oberschleissheim, Germany. The H1N1 swine-origin influenza A virus A/Hamburg/4/2009 (SH4) isolated from a patient in Hamburg was originally obtained from Robert-Koch-Institute Berlin, Germany, and further propagated on MDCK cells at the Friedrich-Loeffler-Institut, Federal Research Institute for Animal Health, Tuebingen, Germany (FLI).
Virus inoculation of mice
Six- to 8-week-old BALB/c mice were obtained from the animal breeding facilities at the FLI. Before intranasal inoculation with H5N1 or H1N1, mice were anesthetized by intraperitoneal injection of 150 μL of a ketamine (Sanofi)-rompun (Bayer) solution [equal amounts of a 2% rompun solution and a 10% ketamin solution were mixed at the rate of 1:10 with phosphate-buffered saline (PBS)]. All animal studies were approved by the Institutional Animal Care and Use Committee of Tuebingen.
IFN-α treatment of cell cultures and experimental design of in vivo studies
Mouse fibroblast cells (MC57) or human lung adenocarcinoma epithelial cells (A549) were grown in minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal calf serum and antibiotics (penicillin and streptomycin). Before infection, cells were grown overnight in 24-well plates. Immediately before IFN treatment, the cells were washed with PBS and subsequently incubated for 8 h with 100, 1,000, or 5,000 U of IFN-α (mu-IFN-α or hu-IFN-α; PBL) in MEM containing 0.2% bovine albumin (BA) and antibiotics. After incubation, IFN-α was aspirated and cells were washed with PBS. Thereafter, cells were inoculated with the H5N1 virus strain or with the H1N1 strain at a multiplicity of infection of 0.001 diluted in PBS/BA (0.2% BA), 1 mM MgCl2, 0.9 mM CaCl2, penicillin, and streptomycin for 30 min at 37°C. After 30 min of incubation, the inoculum was aspirated and cells were incubated with 1 mL MEM containing 0.2% BA and antibiotics. Supernatants from MC57 and A549 cells were collected at 24, 48, or 72 h postinoculation (p.i.) and virus was titrated on MDCK cells by AVICEL® plaque assay.
The in vivo studies were performed in 6- to 8-week-old BALB/c mice. At 8, 32, and 56 h before viral inoculation, mice were anesthetized by intraperitoneal injection of ketamine/rompun and treated intranasally with 1,000 U murine IFN-α (PBL) in 50 μL PBS and 0.1% BSA. Five BALB/c mice were treated with murine IFN-α and inoculated with H5N1 virus. A single intranasal treatment of mice was performed with 1,000 U mouse IFN-α at 8, 24, 48, or 72 h before H5N1 inoculation. The lungs of IFN-α-treated and solvent-treated control mice were collected 48 h p.i. and viral titers were determined by plaque assay.
To examine the effect of an additive IFN-α treatment on the virus replication, experiments were conducted using a multiple pretreatment protocol. Eight mice per group were treated with 1,000 U IFN-α or solvent 56, 32, and 8 h before H5N1 inoculation, and one group of mice was treated only once with 1,000 U of IFN-α 8 h before inoculation with H5N1 virus. After IFN-α administration, animals from both groups were inoculated with a 10-fold MLD50 of the H5N1 influenza virus (2 × 102 pfu). Lungs were collected 48 h p.i. and the virus titers were determined by plaque assay. After mice were inoculated with 1 × 105 pfu of H1N1, the lung viral titers were analyzed 48 h p.i. by plaque assay.
To determine survival after IFN treatment, 8 BALB/c mice per group were given 1,000 U IFN-α or solvent. IFN-α was administrated 56, 32, and 8 h before inoculation of 10-fold mouse lethal dose 50% (MLD50) of H5N1. Treatment was extended to 24 and 48 h postinfection. In addition, to determine the blood level of IFN-α in treated mice, sera of individual animals were collected between treatments and the amounts of IFN-α were analyzed by enzyme-linked immunosorbent assay (ELISA). Four mice out of each group were killed 84 h p.i. The weights of livers and spleens after multiple IFN-α treatment were compared to solvent-treated controls. The bodyweight and visual clinical symptoms of the remaining 4 mice per group were monitored for a 14-day observation period. Clinical symptoms were scored as described earlier (Droebner and others 2008). To determine whether IFN-α given intranasally caused adverse events, 5 mice were treated once with 1,000 U of IFN-α. After 48 h, the spleens and livers of each mouse were weighed and compared to weights of control mice.
Influenza virus titration (AVICEL plaque assay)
To assess the number of infectious particles (plaque titers) in the samples, a plaque assay using AVICEL was performed in 96-well plates as described by Droebner and others (2008). Virus-infected cells were immunostained by incubating for 1 h with a monoclonal antibody specific for the influenza A virus nucleoprotein (Serotec) followed by 30-min incubation with peroxidase-labeled anti-mouse antibody (DIANOVA) and 10-min incubation with True Blue™ peroxidase substrate (KPL). Stained plates were scanned on a flat bed scanner and the data were acquired by Corel DRAW 9.0 software. To define the titer of progeny virus, the foci of infected cells for every sample in each well of the 96-well plates were counted and multiplied with the dilution factor. From the final number of foci in each well, the mean values were taken. The viral titers are shown as the logarithm to the base 10 of the mean values.
IC50 determination of IFN-α and oseltamivir
Oseltamivir carboxylate was obtained from Toronto Research Chemicals, Inc., and dissolved in sterile PBS and IFN-α as described above. For the determination of the IC50, cells were infected and treated with different concentrations of oseltamivir (0.01 nM–1 mM) or IFN-α (0–5,000 U). The viral titers of cell culture supernatants were calculated in percent. The number of pfu of untreated virus-infected control was set as 100% and the titers of IFN-α-treated sample were calculated as follows:
Percent inhibition = 100/(pfu virus − infected sample × IFN-α-treated sample). The IC50 value (ie, the concentration of IFN-α required to reduce the virus titer to 50%) was determined with the GraphPad Prism 5 Software by plotting the percent virus titer as a function of IFN-α concentration.
Enzyme kinetics and neuraminidase inhibition assay
For determination of Michaelis–Menten constant (Km) and V max for every virus, a standardized enzyme activity (0.5 OD units at λ = 540 nM) was incubated with 5 different fetuin substrate concentration for different time points. Both values were determined with the GraphPad Prism 5 Software by plotting the enzyme velocity against substrate concentration. Kinetic assays were performed in triplicates. Neuraminidase (NA) activity was measured by a colorimetric assay (Aymard-Henry and others 1973) with fetuin (Sigma-Aldrich) as a substrate. The inhibitory effect of oseltamivir on NA activity was determined by assaying for enzyme activity in the presence of a range of concentrations.
Measurement of type I IFN with ELISA
Sera of IFN-α-treated mice were collected at 57, 44, and 20 h before inoculation as well as 16, 36, 60, and 84 h p.i. The sera were stored at −70°C until the analysis of IFN-α. The IFN-α level in sera was measured using sandwich ELISA (IFN-α ELISA test kits were purchased from PBL). IFN-α was quantified on the basis of a standard curve obtained with recombinant IFN-α. The protein concentrations were given in x-fold secretion, compared to sera of untreated mice. ELISA was performed according to the manufacturer's instructions.
Quantitative real-time reverse transcriptase polymerase chain reaction
Total RNA was isolated from the lungs of IFN-α- or solvent-treated mice using TRIzol reagent (Invitrogen). Mice were treated 56, 32, and 8 h before RNA isolation either with 1,000 U IFN-α or solvent. The quantitative real-time reverse transcriptase-polymerase chain reaction (qRT-PCR) was performed as described previously (Vogel and others 2010). Briefly, a total of 50 ng RNA was used for qRT-PCR to determine the expression of IFN-α, OAS, RNaseL, and GAPDH using the QuantiFast SYBR Green RT-PCR Kit (Qiagen), and the SmartCycler System and software (Cepheid). The following specific primers for qRT-PCR were used: Mm_Ifna2_1_SG, Mm_Oas1a_1_SG, Mm_Rnasel_2_SG, and Mm_Gapdh_2_SG (Qiagen). The relative expression values (ratio) were normalized to the expression value of the housekeeping gene GAPDH.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software v5.02. For the investigation of significant differences between 2 groups, Mann–Whitney test as well as paired t test (P value < 0.05) were performed; for more groups, analysis of variance Newman–Keuls test (P value < 0.01) was performed. Error bars are given as the standard error of the mean.
Results
IFN-α treatment reduces progeny influenza virus titer in vitro
To investigate the antiviral potential of IFN-α against influenza A virus infection in vitro, MC57 cells were treated 8 h before infection with 100, 1,000, or 5,000 U mouse IFN-α. After removal of IFN-α, cells were infected with H5N1 influenza virus at a multiplicity of infection of 0.001. The progeny virus titers were determined by plaque assay 24 or 48 h postinfection. H5N1 virus replicated 24 h p.i. in mock-treated cells to 3.1 ± 0.24 log10 pfu/mL (Fig. 1A; white bar). Reduced virus titers were found in cell cultures treated with IFN-α. Treatment with 100 or 1,000 U of IFN lead to a virus titer reduction of 0.7 ± 0.1 log10 pfu; treatment with 5,000 U of IFN resulted in viral titer reduction up to 1.0 ± 0.1 log10 pfu (Fig. 1A). An enhanced reduction of viral titer after IFN-α treatment was observed 48 h postinfection depending on the dose of IFN-α used (Fig. 1B). The use of 5,000 U of IFN-α resulted in a 1.4 ± 0.11 log10 pfu reduction, whereas treatment of infected cells with 1,000 and 100 U IFN-α led to virus titer reductions of 1.2 ± 0.08 and 1.0 ± 0.05 log10 pfu, respectively, compared to control cells.

The titer of H5N1 and H1N1 virus was reduced by interferon (IFN)-α treatment. Cells were treated with 0, 100, 1,000, or 5,000 U of IFN-α for 8 h before virus inoculation. After aspiration of IFN-α, cells were inoculated with highly pathogenic avian influenza A H5N1 virus A/mute swan/Germany/R1349/07 (SN1). After 24 h
In addition to infection with the H5N1 influenza virus strain, experiments were performed with the pandemic H1N1 virus. Human lung adenocarcinoma epithelial cells A549 and human IFN-α were used for these experiments. Cells were treated 8 h before H1N1 infection with 100, 1,000, or 5,000 U human IFN-α. The progeny virus titer was determined 48 or 72 h postinfection, because of the slow replication rate of the virus. In control cells H1N1 replicated 48 h p.i. to a titer of 3.77 ± 0.44 log10 pfu/mL and 4.10 ± 0.22 log10 pfu/mL after 72 h. After IFN-α treatment, hardly any virus was detectable in the cell culture supernatants 48 and 72 h p.i. (Fig. 1C, D). These results demonstrated the potent antiviral effect of hu-IFN-α against the pandemic H1N1 influenza A virus strain. The IFN-α concentrations used in these experiments showed on both cell lines no toxic effects assayed by crystal violet staining assay (data not shown).
IFN-α is effective against oseltamivir-resistant H5N1 influenza A virus
To investigate the antiviral potential of IFN-α in comparison to oseltamivir, IC50 values based on reduction of either virus titer (IFN-α) or NA activity (oseltamivir) to 50% were determined for 3 different influenza A virus strains. The IC50 values for IFN-α ranged from 1.49 ± 1.37 to 250.3 ± 1.26 U (Table 1). IFN-α demonstrated the highest sensitivity against the H1N1 isolate with IC50 value of 1.49 ± 1.37 U. The IC50 values evaluated for oseltamivir ranged from 0.23 ± 0.15 to 346.20 ± 1.89 nM, indicating that the influenza A H5N1 virus isolate, GSB1 (346.20 ± 1.89 nM), can be considered resistant against oseltamivir (Gubareva and others 2002). In contrast, IFN-α is effective against this oseltamivir-resistant H5N1 isolate with IC50 value of 23.43 ± 1.20 U. Because of the differences in the responsiveness to oseltamivir, we also determined enzymatic parameters for the sialidase activities of the NAs. Km, which reflects the affinity for the substrate, ranged from 146.64 ± 11.87 to 603.58 ± 102.06 μM of fetuin. V max values, reflecting the activity of the enzyme for the H1N1 and H5N1 (SN1) isolates, similarly ranged from 2.77 ± 0.35 to 2.75 ± 0.11 × 10−3, except for the oseltamivir-resistant H5N1 isolate GSB1 with an V max of 40.01 × 103 (Table 1). The mean inhibition constant (Ki) values for oseltamivir ranged from 0.23 ± 0.17 to 346.12 ± 1.97 nM and were higher for less susceptible isolates.
Determined by in vitro screening and represents the IFN-α units required to reduce the virus titer to 50%. IC50 values were determined for a 24-h infection period (multiplicity of infection 0.001) for each virus with the GraphPad Prism 5 software by plotting the percent virus titer as a function of compound concentration. The experiment was performed in triplicates.
The IC50 value (ie, the concentration of compound required to reduce the viral NA activity to 50%) was determined for each virus with the GraphPad Prism 5 Software by plotting the percent NA activity as a function of compound concentration. The inhibition assays were performed in triplicates. The IC50 values were determined for each virus with the GraphPad Prism 5 software by plotting the percent NA activity as a function of compound concentration.
For the determination of Km and V max for every virus, a standardized enzyme activity (0.5 OD units at λ = 540 nM) was incubated with 5 different fetuin substrate concentration for different time points. Both values were determined with the GraphPad Prism 5 software by plotting the enzyme velocity against substrate concentration.
Ki values were obtained using the following equation: Ki = IC50/[1 + (substrate concentration/Km)].
IFN, interferon; Ki, mean inhibition constant; Km, Michaelis–Menten constant; NA, neuraminidase.
Swine-origin H1N1 influenza A virus is susceptible to low-dose IFN-α treatment
Because of the strong antiviral effects of IFN-α even at low concentrations against influenza virus in vitro, we raised the question whether low-dose IFN-treatment was also effective in vivo against the pandemic H1N1 strain. Therefore, 1,000 U murine IFN-α were given 3 times (8, 32, and 56 h) before infection of 5 BALB/c mice. The animals were infected with 1 × 105 pfu of H1N1. Lung virus titers were analyzed 48 h p.i. by plaque assay. The administration of IFN-α was able to reduce the virus titers in lungs of infected mice up 1 log unit (mock 7.18 ± 0.28 versus IFN-α 6.19 ± 0.42 log10 pfu/mL) compared to solvent-treated animals (Fig. 2A). Thus, a 77% reduction of viral lung titer after low-dose IFN-treatment in pandemic H1N1 virus infected mice was found (Fig. 2B).
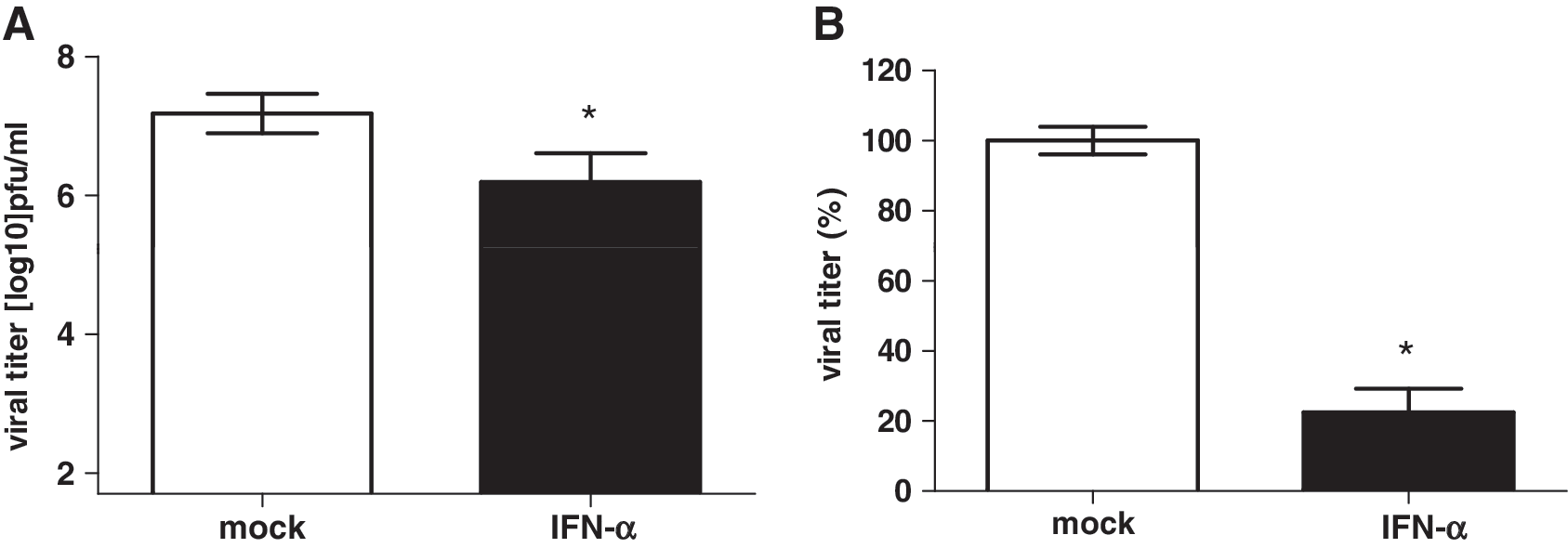
Intranasal IFN-α treatment reduced viral titers in the lungs of H1N1-inoculated mice. Five BALB/c mice were treated intranasally with 1,000 U of mu-IFN-α 56, 32, and 8 h before inoculation with 1 × 105 pfu H1N1. After an incubation period of 48 h, viral lung titers were determined by plaque assay. Viral titer in log10 pfu/mL
Single low-dose IFN-α administration reduces progeny virus titer in the lungs of H5N1-infected mice
Next, we were interested in the antiviral activity of IFN-α against the H5N1 influenza virus. Therefore, 5 BALB/c mice were treated with murine IFN-α and infected with the avian H5N1 influenza A virus. A single intranasal treatment of mice was performed with 1,000 U mouse IFN-α at different time points before H5N1 infection. The IFN-α application was carried out either 8 or 24 h or 48 or 72 h before infection. The lungs of IFN-α-treated and mock-treated control mice were collected 48 h p.i. and virus titers were determined by plaque assay. In the lungs of mock-treated mice a virus titer of 3.22 ± 0.17 log10 pfu/mL was noted (Fig. 3A, white bar). Mice treated with IFN-α once 72 h before infection showed a titer reduction of 0.8 ± 0.38 log10 pfu/mL in the lung to 2.41 ± 0.23 log10 pfu/mL. An even higher reduction in virus concentration resulted when IFN-α was applied 48 h (reduction of 1.1 ± 0.3 log10 pfu), 24 h (1.3 ± 0.36 log10 pfu), and 8 h (1.4 ± 0.04 log10 pfu) before inoculation (Fig. 3A). To investigate whether 1,000 U IFN-α shows adverse events, 5 mice were once treated with 1,000 U IFN-α. Spleens (Fig. 3B) and livers (Fig. 3C) of processed animals showed no significant differences in weight or appearance when compared to organs of mock-treated animals. Thus, the administration of a single dose of 1,000 U IFN-α did not appear to be toxic in mice.

Reduction of viral titers in the lungs depended on the time point of IFN-α treatment.
Repeated low-dose IFN-α pretreatment of H5N1-infected mice increased the antiviral effect
As demonstrated in Fig. 3, a single low-dose treatment of mice with 1,000 U IFN-α resulted in a reduction of virus in lungs after H5N1 influenza A virus infection. Next, we asked whether a repeated IFN-α application would enhance the antiviral effect. Therefore, mice were treated with 1,000 U IFN-α or solvent 56, 32, and 8 h before H5N1 infection. To investigate whether multiple prophylactic treatment is beneficial compared to a single treatment, 1 group of mice was treated only once 8 h before infection with 1,000 U IFN-α. Additionally, the distribution of viral load in mice after multiple IFN-α or solvent treatment was examined by titrating lung, heart, spleen, kidney, liver, brain, and blood 2, 4, and 6 days p.i.
In the lungs of mock-treated mice, progeny virus replicated to a titer of 4.04 ± 0.05 log10 pfu/mL (Fig. 4, white bars) 48 h postinfection. The viral titer was reduced to 2.86 ± 0.35 log10 pfu/mL in the lungs of mice that were treated only once with low-dose IFN-α (1,000 U). In contrast, the viral titer in lungs of mice that were treated multiple times with 1,000 U IFN-α was reduced to 2.00 ± 0.12 log10 pfu/mL (Fig. 4). Thus, the single low-dose IFN-α treatment resulted in a viral titer reduction of 1.18 ± 0.61 log10 pfu/mL, whereas the multiple administrations IFN-α led to a titer reduction of 2.04 log10 ± 0.15 pfu/mL compared to mock-treated animals (Fig. 4). Table 2 shows the distribution of viral load 2, 4, and 6 days after multiple IFN-α or solvent treatment and H5N1 virus infection in mice. Progeny virus was only measurable in the lung, heart, and spleen. There was no virus detectable in kidney, liver, brain, or blood (data not shown). IFN-α-treated mice showed reduced virus titer in all tissues where progeny virus was detectable compared to control mice.
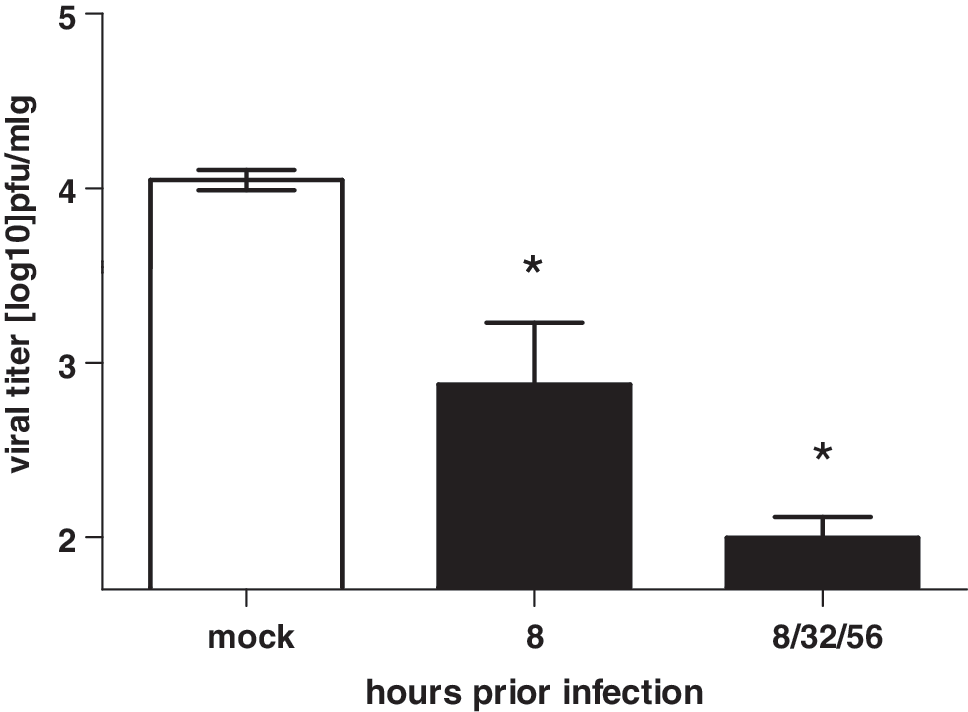
IFN-α treatment reduced viral titers in lungs. Mice were intranasally treated with 1,000 U mu-IFN-α once (8 h) or 3 times (8, 32, and 56 h) before inoculation with 10-fold MLD50 (2 × 102 pfu) of the H5N1 strain. Progeny virus titers in the lungs were determined 48 h postinfection. *P < 0.01 (Newman–Keuls test).
Virus titers are given as the logarithm in pfu per 1 mL organ-homogenate.
Low-dose IFN-α treatment protected mice against a lethal H5N1 influenza A virus infection
From the reduction of progeny virus in IFN-α-treated mammalian cell cultures and the decreased viral loads in mouse lungs after intranasal low-dose (1,000 U) IFN-α application, we concluded that the antiviral effect of IFN-α is sufficient to prolong the survival of mice infected with influenza A virus. Mice were either treated with 1,000 U IFN-α or solvent. IFN-α was administered 56, 32, and 8 h before infection with 10-fold MLD50 of H5N1 influenza A virus. Further, treatment was performed 24 and 48 h postinfection. In addition, to investigate the level of IFN-α in treated mice, sera of individual animals were collected between treatments and ELISA analyzed the amounts of IFN-α. We also measured RNA level of IFN-α, OAS, and RNaseL after triple IFN-α treatment (-56, -32, and -8 h) in lungs of uninfected mice. To answer the question whether multiple treatments would lead to adverse effects, 4 mice out of each group were sacrificed 84 h p.i. to define the weight of liver and spleen after multiple IFN-α treatment compared to mock-treated controls. The bodyweight and visual clinical symptoms of the other 4 mice per group were monitored for 14 days.
Six days after inoculation, the control mice started to lose weight (Fig. 5A) and developed first clinical symptoms like ruffled fur or the reduction of their normal activity rate. Within the next 3 days all control mice died (Fig. 5B, black squares). Interestingly, only 1 out of 4 mice of the IFN-α-treated group showed clinical symptoms comparable to the control group; this 1 mouse died at day 11 p.i. (Fig. 5C). The other 3 IFN-treated-mice did not lose weight (Fig. 5A, white squares), remained normal, and survived the otherwise lethal challenge infection (Fig. 5B, white squares).
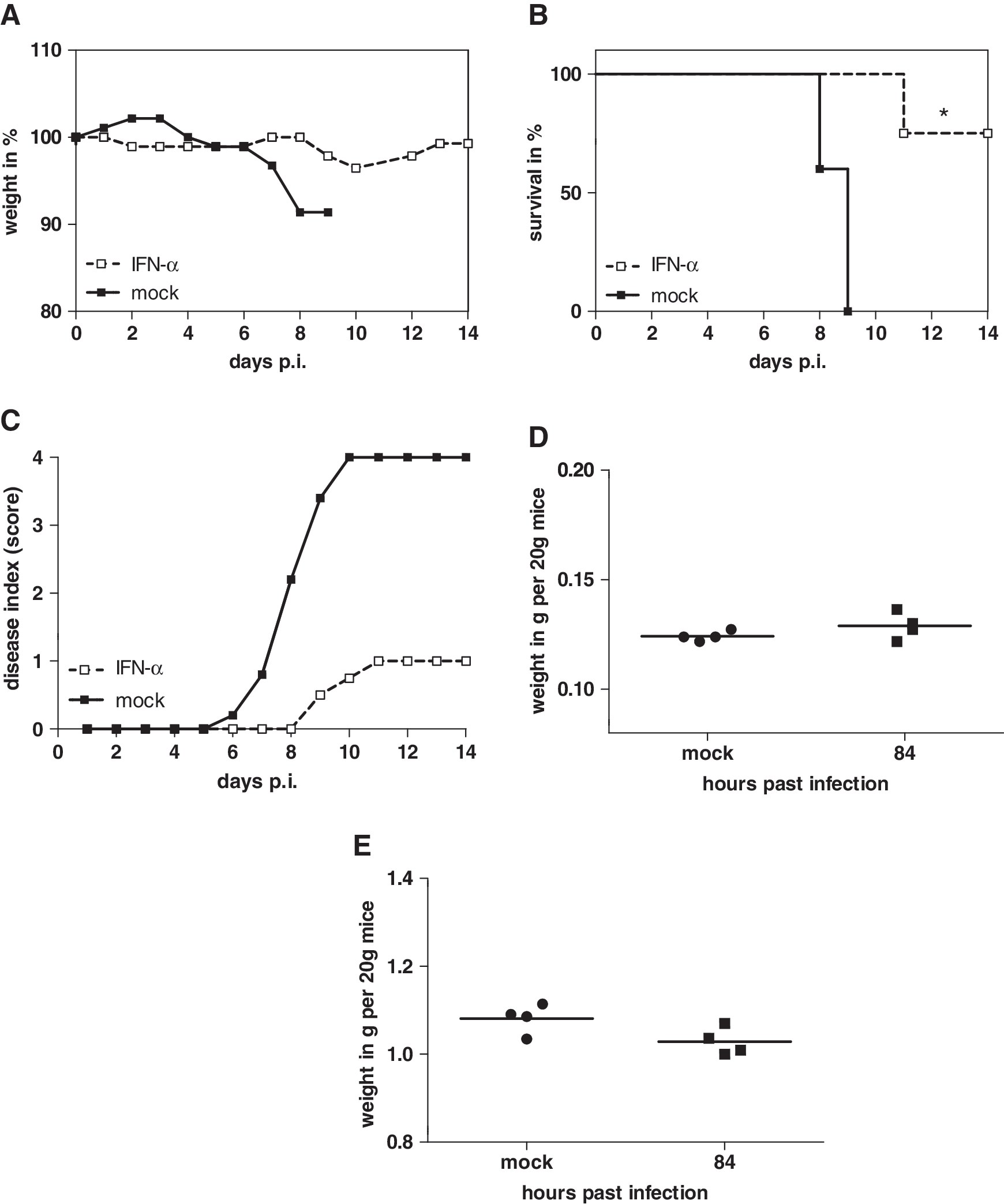
Multiple IFN-α treatment protected mice against lethal H5N1 influenza A virus infection. Eight mice from each group were treated with 1,000 U mu-IFN-α 3 times (56, 32, and 8 h) before and 2 times (24 and 48 h) after inoculation with 2 × 102 pfu H5N1.
No statistically significant differences in the weights of spleens and livers were found when comparing treated animals to untreated controls. The multiple IFN-α treatments did not provoke noticeable toxic side effects (Fig. 5D, E). When the IFN-α level in sera of treated mice was compared to untreated mice, an additive effect of IFN-α induction was observed (Fig. 6A). After the first 2 treatments 56 and 32 h before infection, an increased amount of IFN-α (4.86 ± 0.3-fold) was detected in treated mice compared to untreated controls. After inoculation of H5N1 virus, the level of IFN-α was reduced until 24 h p.i. Nevertheless, the next 2 IFN-α treatments (24 and 48 h p.i.) increased the level up to 1.6 ± 0.01-fold (36 h), 2.0 ± 0.01-fold (60 h), and 2.7 ± 0.14-fold (84 h) postinfection, compared to IFN-α levels in untreated mice. The relative quantification of IFN-α, OAS, and RNaseL RNA after triple IFN-α treatment in the lung showed for all genes an enhanced amount in IFN-α-treated mice compared to untreated controls. IFN-α treatment led to >1.5-fold higher IFN-α, 3-fold OAS, and 6.5-fold RNaseL RNA level in contrast to solvent-treated mice. From these results, one might speculate that the antiviral effect of intranasal administration of low-dose IFN-α enhances endogenous IFN production in vivo.
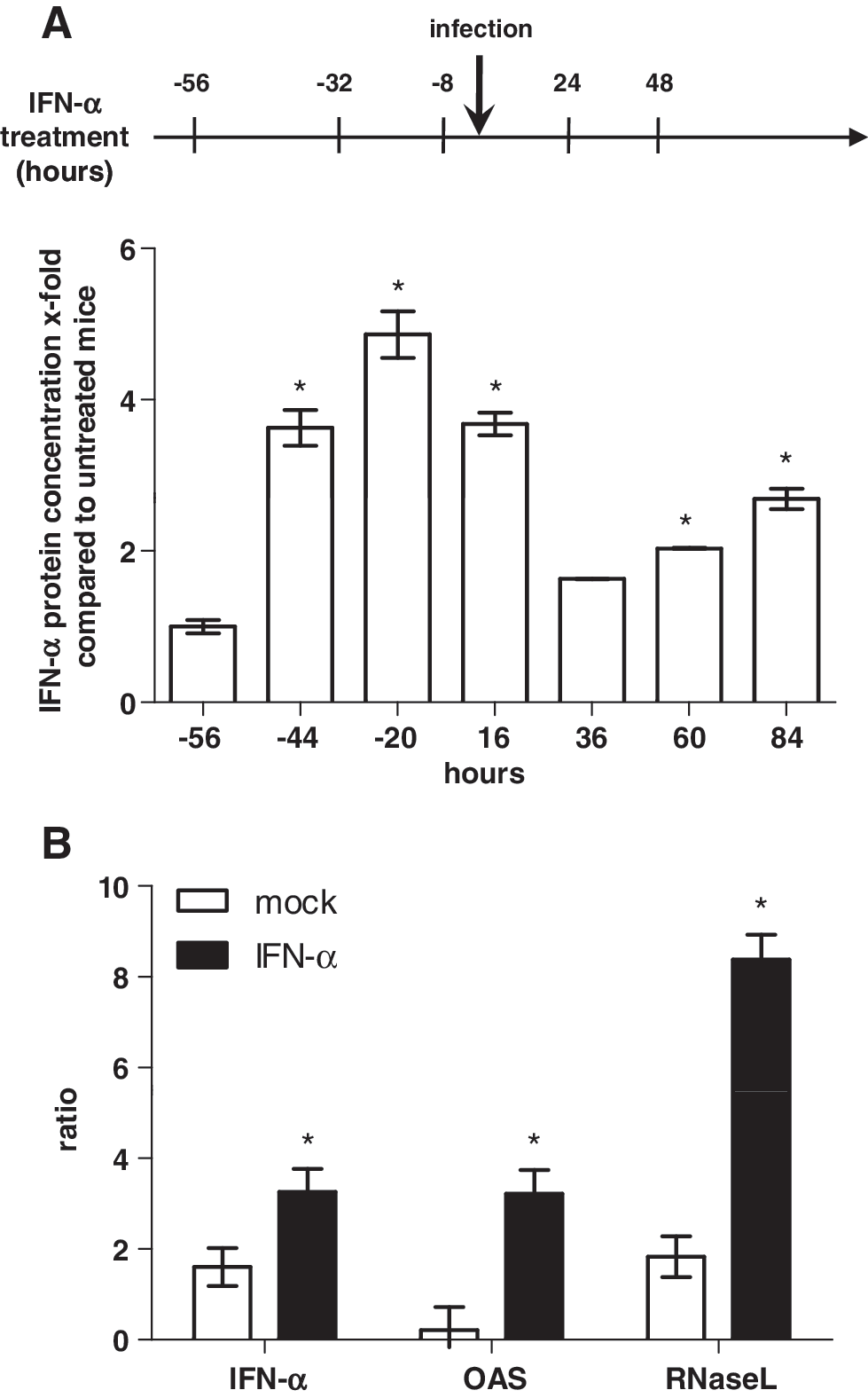
IFN-α level in sera of mice treated with IFN-α was measured by enzyme-linked immunosorbent assay and is given as x-fold expression relative to solvent-treated mice [*P < 0.01 (Newman–Keuls test)].
Discussion
Infections with human and avian influenza A viruses are a major burden in human health care, and the options for control and treatment of the disease are limited. Viral resistance against the common influenza antivirals, amantadine, and oseltamivir underlines the urgent need for new antiviral drugs (McKimm-Breschkin and others 2007; Sheu and others 2008; Bai and others 2009). The continuous circulation and reassortment of influenza viruses represents a chronic public health threat. In this study, we demonstrated that treatment with low-dose IFN-α reduced progeny virus replication in cell culture. Reduction of highly pathogenic influenza H5N1 and the pandemic H1N1 influenza virus was also observed in mice treated with intranasal low-dose (1,000 U) IFN-α. Increased survival from IFN treatment was observed without any adverse effects. Moreover, we are able to induce ISG in the lung after low-dose IFN-α treatment.
After entry of a pathogen into the host, IFNs form the first line of defense and establish an antiviral state. This results in the induction of a large number of ISG. These genes are involved in expression of cytokines/chemokines and enzymes that interact with cellular and viral processes to avoid viral replication and spread (Stark and others 1998; Goodbourn and others 2000; Randall and Goodbourn 2008). IFN-α-mediated antiviral status in the innate immune response involves 3 mechanisms. All 3 mechanisms are important for the development of an efficient foreign pathogen clearance. First, activated double-stranded RNA (dsRNA)-dependent PKR that detects dsRNA inhibits protein synthesis by phosphorylating the eukaryotic initiation factor 2 (eIF2a) (Garcia and others 2006). Second, the OAS triggered by dsRNA activates the cellular endoribonuclease, RNaseL, which degrades cellular but also viral single-stranded RNA, resulting in inhibition of protein synthesis (Silverman 2007). The third mechanism by which IFN-α induces an antiviral state is the induction of the transcription of the Mx protein that interacts in a direct way with viral components to trap and sort them to cellular compartments where they become unavailable for the production of new virus particles (Haller and others 2007). In the present study wild-type mice were used that lack the Mx protein. Thus, our present results demonstrate that Mx protein is not a prerequisite to assure the antiviral effect mediated by IFN-α treatment. Further, the results give rise to the assumption that the IFN-α-mediated antiviral effect is even more pronounced when Mx protein is present.
The question then arises how intranasal delivery of IFN-α contributes to protection against highly pathogenic influenza virus infection. The molecular basis of IFN-α-mediated action after oral administration is not fully understood. It is well accepted that oral IFN-α treatment leads to the induction of OAS that functions as a molecular marker for IFN-induced cellular activation (Cummins and others 2005). In this concern, it is of interest whether influenza A virus developed several ways to evade the host immune response and virus clearance. One way is the interaction of viral NS1 protein with several cellular factors. NS1 can directly block the function of OAS and the dsRNA-dependent PKR (Min and Krug 2006; Min and others 2007; Wolff and Ludwig 2009). Besides OAS activation, orally administered IFN also leads to a local increase of MHC class I expression, which is a prerequisite for CD8+ T-cell effector function. Oropharyngeal delivery of IFN-α leads to activation of the IFN-activated natural killer cells, B-cells, and subpopulations of the cellular immune response (Cummins and others 2005). Investigations in animals and humans using radiolabeled IFN-α demonstrated a mucosal binding of IFN-α after oropharyngeal delivery in the salvia, oral cavity, nose, and in the paranasal sinuses (Diez and others 1987; Schellekens and others 2001). Taken together, these data suggest that the type I IFN detected in the blood of our mice treated via the intranasally route was of endogenous origin since the low-dose IFN would not be detectable systemically in the amounts shown in Fig. 6A. In addition, the enhanced RNA levels of ISG (IFN-α, OAS, and RNaseL) after IFN-α treatment underline the endogenous origin (Fig. 6B). IFN-α treatment did not provoke noticeable toxic side effects since there were no significant differences in the weights of spleens and livers (Van Loveren and others 1994; Meng and others 2008). Moreover, the data also demonstrate that orally administered IFN-α leads to an induction of an antiviral state in the region of the body where influenza virus replication takes place first.
Pretreatment with low-dose IFN-α reduced influenza virus in the lung of mice and protected these animals against a lethal infection. Beilharz and others (2007) already demonstrated the potential of low-dose IFN-α prophylactic treatment of mice infected with mouse-adapted human influenza virus A/Puerto Rico/8/34. In addition, oromucosal administration including low-dose IFN treatment was demonstrated to be protective against various virus infections, including vesicular stomatitis virus, encephalomyelitis virus, vaccinia virus, and cytomegalovirus (Bosio and others 1999; Lawson and Beilharz 1999; Tovey and Maury 1999; Sonnenfeld and others 2001).
Low-dose IFN-α treatment is a novel way to deal with potential pandemic outbreaks of new emerging influenza A viruses. The importance for new antiviral drugs especially after development of partial viral resistance to the available antiviral drugs against influenza virus is a prerequisite to assure human health. Related to development of antiviral drug resistance, we could show that the oseltamivir-resistant H5N1 virus strain A/goosander/Bavaria/20/2006 (GSB1) is susceptible to low-dose IFN-α treatment. Therefore, low-dose IFN-α may be an effective antiviral drug for viruses resistant to oseltamivir.
In contrast to hepatitis B and C long-term therapy with high concentrations of IFN-α, which leads to serious adverse effects, tissue-specific intranasal treatment with low-dose IFN does not cause adverse events. Thus, our findings demonstrate that low-dose IFN-α is a potential antiviral drug to induce the antiviral state as part of the first line of defense to protect against a fatal influenza virus infection.
Footnotes
Acknowledgments
The authors thank Carmen Mueller for her excellent technical assistance. The authors also thank Martin and Joseph Cummins for their editorial support and for providing some IFN for the studies. This work is part of the EUROFLU consortium activities and of the VIRGIL European Network of Excellence on Antiviral Drug Resistance supported by a grant (LSHMCT-2004-503359) from the Priority 1 “Life Sciences, Genomics and Biotechnology for Health” program in the 6th Framework Program of the EU. Further, this research was partially supported by the Federal Government of Germany under the Influenza Research Program “FSI” and by the BMBF Zoonose program “FluResearchNet.”
Author Disclosure Statement
No competing financial interests exist.