
Abstract
The ISG56/IFIT1 family of genes is clustered on human chromosome 10 and is comprised of 4 members, ISG56/IFIT1, ISG54/IFIT2, ISG60/IFIT3, and ISG58/IFIT5, whose homologs are evolutionarily conserved from mammals to amphibians. While these genes are normally silent in most cell types, their transcription is strongly induced by interferons, virus infection, and molecular patterns such as double-stranded RNA or lipopolysaccharides. The encoded P56 family proteins are characterized by multiple repeats of tetratricopeptide repeat helix-turn-helix motifs mediating a variety of protein–protein interactions, which result in a multitude of effects on cellular and viral functions, such as translation initiation, virus replication, double-stranded RNA signaling, cell migration, and proliferation.
The Members of the ISG56 Family of Genes
A
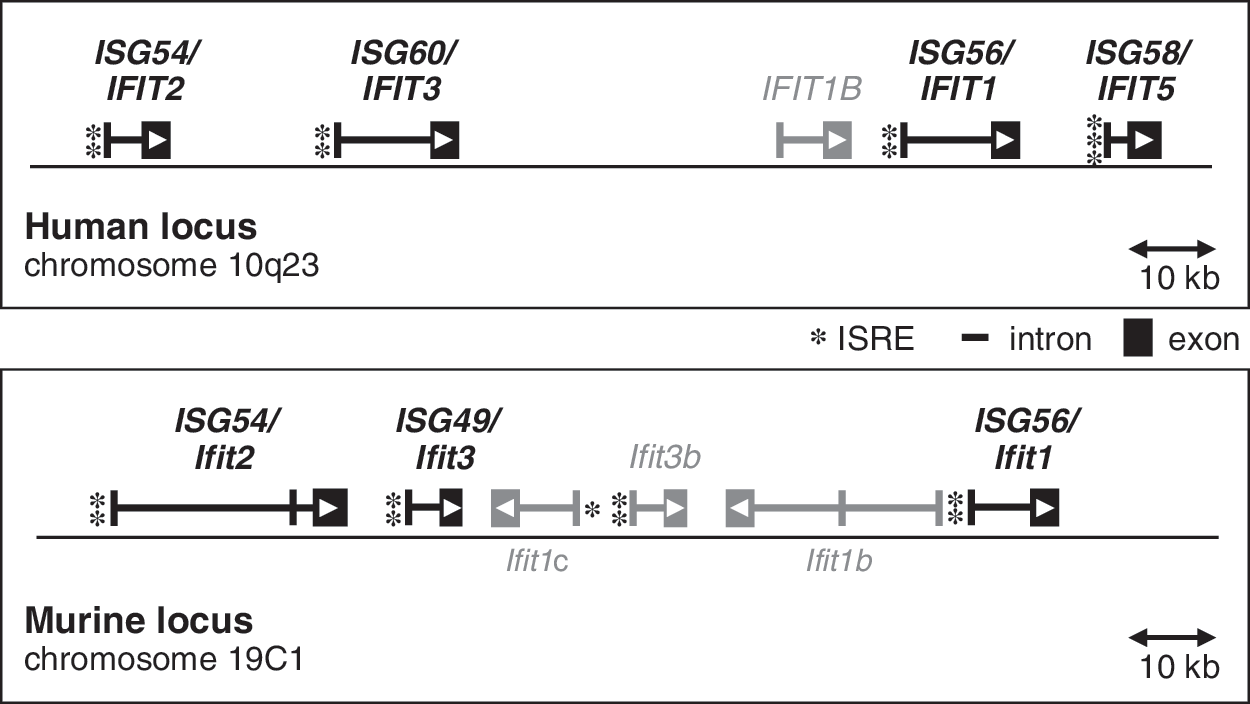
Chromosomal loci of the human and murine ISG56/IFIT1 family genes, depicting exon/intron structures, orientation of the open reading frames (arrowheads), and the positions and numbers of ISREs in the promoters (asterisks). Additional, uncharacterized IFIT genes are shown in gray. ISRE, interferon-stimulated response elements.
Transcriptional Induction of the ISG56 Family of Genes
A variety of stimuli can induce human and murine ISG56 family transcription (Fig. 2). The most prominent inducers are IFN-α/β, whereas IFN-γ is a weak inducer (Der and others 1998). Infection with a multitude of RNA- or DNA-viruses, such as Sendai virus, Respiratory syncytial virus, Vesicular stomatitis virus (VSV), West Nile virus, Lymphocytic choriomeningitis virus (LCMV), Influenza viruses, Reovirus, Herpes simplex virus, Cytomegalovirus, and Adenovirus, efficiently induces these genes (Zhu and others 1997; Nicholl and others 2000; Dorn and others 2005; Elco and others 2005; Terenzi and others 2005; Smith and others 2006; Daffis and others 2007; Janssen and others 2007; Wacher and others 2007). The use of cell lines with appropriate genetic deficiencies of JAK-STAT pathway components was instrumental for demonstrating that viral and bacterial molecular patterns (PAMP) can directly (ie, independently of IFN action) induce transcription of a subset of ISGs, including ISG56 family genes (Bandyopadhyay and others 1995; Elco and others 2005). These genes are hence termed viral stress-inducible genes (VSIG) (Sarkar and Sen 2004). The induction of these VSIGs by different stimuli is based on the activation of IRFs, which recognize the ISREs in the VSIG promoters and initiate transcription: ISGF3 composed of IRF-9 along with STAT1/2, activated by the IFN-α/β receptor, mediates the IFN response; IRF-3, activated by double-stranded RNA (dsRNA) via Toll-like receptor 3 (TLR3) or via the cytoplasmic receptors retinoic acid-inducible gene-I/Melanoma differentiation-associated gene-5 (RIG-I/MDA-5), and by lipopolysaccharides via TLR4, potently induces ISG56 family genes, especially ISG54/56 (Grandvaux and others 2002; Sarkar and others 2004; Ogawa and others 2005; Terenzi and others 2006). Therefore, ISG56 expression is frequently used as the readout for IRF-3 transcriptional activity (Chattopadhyay and others 2010); IRF-5 and IRF-7, activated by virus infection or by TLR7/8- or TLR9 signaling, also drive expression of these genes (Barnes and others 2004; Zhu and others 2010).
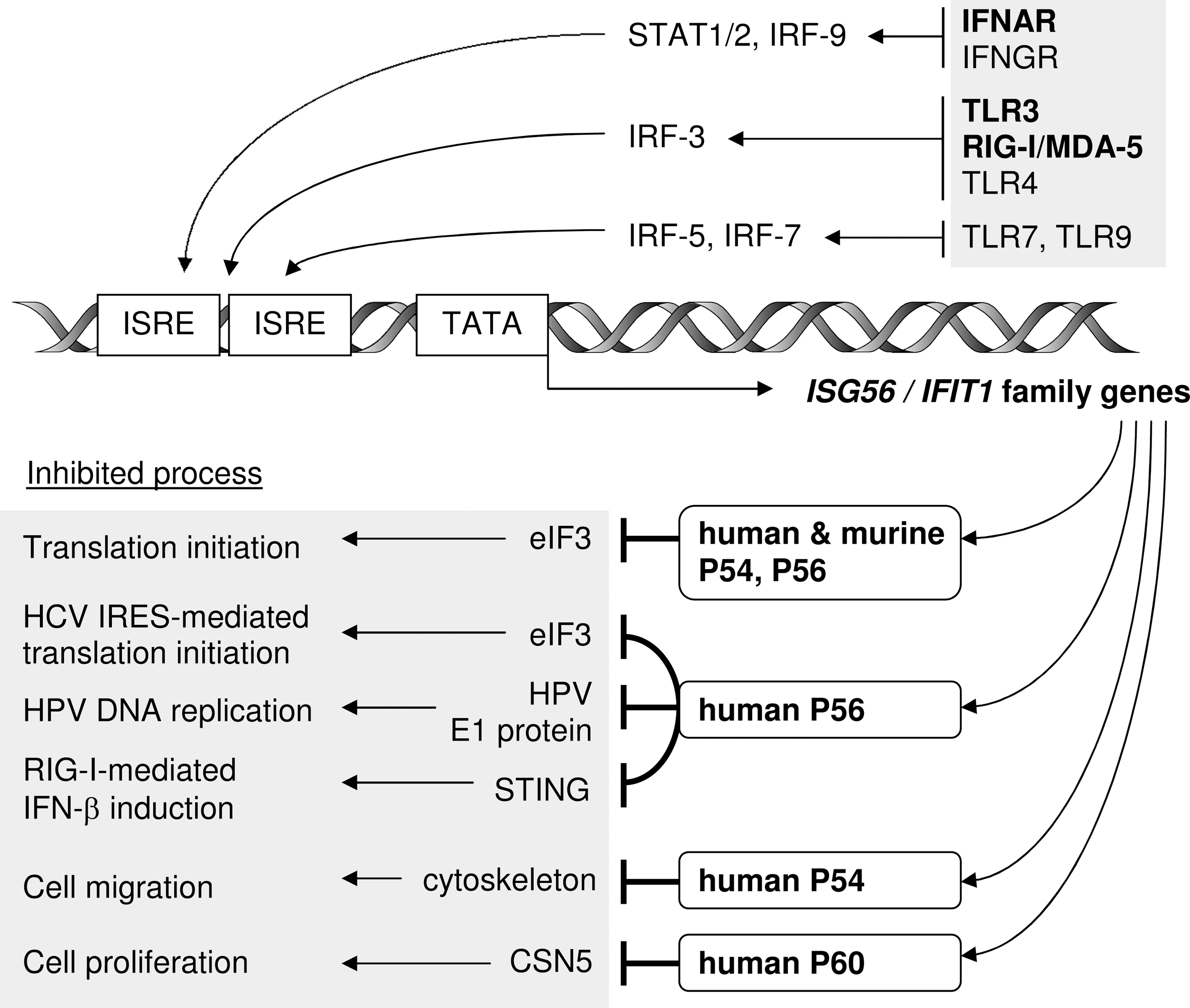
Pathways of ISG56 family gene induction, and functions of the encoded P56 family of proteins. Stimulation of innate immune receptors, especially IFNAR (IFN-α/β receptor) and the double-stranded RNA receptors TLR3 and RIG-I/MDA-5, induces transcription of ISG56 family genes via ISREs in their promoters. The respective protein products mediate inhibition of various cellular and viral processes by specific binding to effector proteins. IFN, interferon; IRF, IFN regulatory factors; TLR3, toll-like receptor 3. RIG-I, retinoic acid-inducible gene-I; MDA-5, melanoma differentiation-associated gene-5.
Most cell types do not express detectable levels of ISG56 family transcripts in the absence of stimuli. Induction of the genes is generally rapid as well as transient, with human ISG56 mRNA levels peaking at 6 h after IFN treatment and decreasing quickly within 12–24 h of treatment (Kusari and Sen 1986). However, kinetics differ between genes, and depend on stimulus and cell type. For example, in HT1080 human fibrosarcoma cells, ISG56 mRNA levels remain high at 24 h of IFN-β treatment, whereas ISG54 mRNA levels are largely decreased. When dsRNA is used as TLR3 stimulator of the same cell line, both ISG56 and ISG54 mRNA levels drop between 12 and 24 h of treatment, and in the case of Sendai virus as inducer, which activates the RIG-I pathway, both mRNAs decrease before 12 h postinfection (Terenzi and others 2006). In other cell types, such as 293 cells, IFN-induced levels of both ISG56/54 mRNAs are maintained at high levels even after 24 h of treatment (Terenzi and others 2006). Hence, expression of the genes is regulated differentially depending on the inducer and its signaling pathway, with variations between cell types due to specific kinetics of postinduction regulation.
Recently, new insights into the transcriptional regulation at the chromatin level of murine ISG56 and other ISGs such as Oas1 and Stat1 were obtained; soon after IFN-β treatment, the histone 3 variant H3.3 is deposited to the internal parts of ISG56, resembling exon 2, but not to its promoter region. H3.3 enrichment continues for at least 48 h, long after transcription has ceased; importantly, knockdown of H3.3 impairs ISG56 induction by IFN-β. Since H3.3 deposition is indicative of transcriptionally active chromatin, the authors speculate that prolonged deposition of H3.3 may serve as an epigenetic mark, capable of modulating transcription after repeated IFN stimulation (Tamura and others 2009).
Interestingly, human ISG58 and ISG60 were originally discovered as all-trans retinoic acid (ATRA)-inducible genes (Niikura and others 1997; Yu and others 1997). ATRA is used in treatment of acute promyelocytic leukemia, because it induces differentiation of immature leukemic cells and prevents their further proliferation. ISG60 induction by ATRA, however, is slow and starts between 24 and 48 h after treatment. The induction is not directly driven by ATRA, but in part by IFN-α, which is secreted after ATRA treatment and activates ISG60 transcription via the JAK-STAT pathway (Xiao and others 2006; Lou and others 2009).
The transcription of ISG56 family genes can be negatively regulated during anti-inflammatory therapy; the IRF-3-dependent induction of ISG56 family genes is attenuated by glucocorticoids such as dexamethasone, which activates glucocorticoid receptor (GR) (Smith and Herschman 1996). GR competes with IRF-3 for a mutual binding protein, GRIP1, thereby weakening the transcriptional activity of IRF-3 (Reily and others 2006).
Expression Patterns of Murine ISG56 Family Members
In spite of superficial similarity in the structure of ISG56 family genes, their expression patterns only partially overlap in different cell lines in vitro and in different tissues and cell types in vivo. A recent study utilized intracranial LCMV or intranasal West Nile virus infections of mice to examine expression of murine ISG49/54/56 in the brain (Wacher and others 2007). Overall mRNA levels of all 3 genes increase over the course of 6 days postinfection. Induction of ISG56 family mRNA expression by LCMV in neurons requires virus-induced secretion of IFN, which signals via STAT1/2 to induce the genes. In non-neurons the virus can directly induce these genes. The anatomic distribution of ISG56 family transcripts displays distinct patterns of preferential expression of different genes. For example, whereas deep cerebellar neurons express all 3 ISGs upon LCMV infection, astrocytes and hippocampal CA1/CA2 pyramidal cell layers mainly express ISG56; granule cells preferentially express ISG49, and in the olfactory bulb, ISG54 transcript levels are highest (Wacher and others 2007). Thus, expression of different ISG56 family genes in neurons and other cell types in the central nervous system is expected to have important nonredundant functions in the host response to viral infections of the brain.
The protein products of the ISG56 gene family are named P56, P54, and P49 (see below). Another study examined murine P56 and P54 expression in various tissues and cell types of mice treated with dsRNA or virus infection (Terenzi and others 2007). An example of a tissue-specific differential expression pattern is observed in case of infection with the RNA virus VSV, as compared to dsRNA injection: the virus induces P56 in both liver and spleen, whereas P54 is only induced in spleen and not in the liver, an effect confirmed on the mRNA level (Terenzi and others 2007).
Unexpected examples for cell type-specific differences are observed in B cells and plasmacytoid dendritic cells (pDC): After injection of IFN-α, dsRNA, or VSV, murine B cells express P54 but not P56, whereas CD4 or CD8 T cells express both proteins. Importantly, this effect is mirrored by the absence of ISG56 transcripts in ex vivo IFN-α-stimulated B cells. Murine pDCs also do not express P56 upon IFN-β stimulation, but express P54 and P49 (Terenzi and others 2007; Fensterl and others 2008). The selective unresponsiveness of ISG56 in murine B cells and pDCs highlights the surprising complexity of the regulation of this family of closely related genes.
Protein Products of the ISG56 Family: The P56 Family
The proteins encoded by ISG56/IFIT1 family of genes are named P56, P54, and so on (Table 1), with the number actually depicting the approximate molecular weight of the proteins, which is between 47 and 56 kDa (and was often miscalculated originally). All of them are cytoplasmic proteins that do not possess enzymatic activity. Instead, all share a single structural motif in multiple repeats, the tetratricopeptide repeat (TPR) (Fig. 3). The abundant TPR motifs invoked the official gene designation “IFIT” (which stands for IFN-induced protein with TPRs). The TPR is a 34 amino acid motif folding into a helix-turn-helix; it is most often present in cassettes of multiple repeats, and mediates protein–protein interactions and assembly of large protein complexes (D'Andrea and Regan 2003). However, the TPR is degenerate in sequence, requiring only 8 to 9 key position residues to be conserved, leading to large variability of TPRs even within the same protein.

TPR motifs in homologs of the P56 family proteins of human and mouse (left), and predicted 3-dimensional structure of human P54, amino acids 23–471 (right). The PHYRE server (Kelley and Sternberg 2009) was used to predict 3-dimensional structure, observation with MolMol 2K.2 software (Koradi and others 1996). TPR, tetratricopeptide repeat.
A suggested consensus defines a protein sequence as a TPR if the following residues are present in the given positions: W4G8Y11G15Y17A20Y24A27P32; however, even these key residues are not invariant and can be replaced by amino acids of similar class, ie, small/large/aromatic (D'Andrea and Regan 2003). Hence, software-based prediction of TPRs is difficult. Human and murine P56 family proteins consist of multiple TPRs and TPR-related motifs (Fig. 3). The comparison of predicted 2-dimensional and 3-dimensional structures of human P54 reveals that even if a putative TPR cannot be clearly identified, it may still fold into the TPR-like helix-turn-helix, yielding a spiral-shaped stack of antiparallel α-helical motifs (Fig. 3). The sequence identity between human and murine P56 is 52%, between P54 homologs it is 62% and between the human P60/murine P49 homologs it is 60%. Between different protein members in the same species, the identity is even lower: about 45% between the human P56/P54 and P56/P60. This suggests nonredundant functions of the different members of the family, which is indeed reflected in the different cellular functions of the different P56 family proteins (see below).
Functions of the P56 Family Proteins
Novel insights into the functions of P56 family proteins continuously emerge in the literature, and intriguingly, all of them characterize these proteins as inhibitors of cellular as well as viral processes, including translation, cell migration, proliferation, signaling, and viral replication (Fig. 2).
Inhibition of translation initiation by interaction with eIF3
The best-characterized cellular function of several, but not all, P56 family members is the inhibition of translation by binding to specific subunits of eIF3, presenting a mechanism of cell growth inhibition distinct from other ISGs such as PKR and OAS. For example, exogenous expression of human P56 can suppress overall cellular translation by 40% in HT1080 cells, being as effective as IFN-β treatment (Guo and others 2000). The underlying mechanism has been studied in great detail and is based on the inhibition of at least one of several functions of eIF3 during the initiation step of translation. eIF3 is a large protein complex of about 800 kDa and consists of 13 different subunits, termed eIF3a-m (Hinnebusch 2006; Damoc and others 2007). It fulfills several tasks during translation initiation, among them the dissociation of ribosomal 60S/40S subunits, promotion of the formation of the ternary complex eIF2/GTP/Met-tRNAi, binding of eIF4F and formation of the 48S preinitiation complex consisting of 40S ribosomal subunit, eIF3, eIF4F, ternary complex, and the mRNA (Hinnebusch 2006).
Both human P56 and P54 selectively inhibit the ternary complex formation step of translation initiation by binding to subunit eIF3e (=p48). Human P54 additionally binds to subunit eIF3c (=p110), enabling it to inhibit an additional function of eIF3, the 48S preinitiation complex formation (Guo and others 2000; Hui and others 2003; Terenzi and others 2006). The murine homologs P54 and P56 bind specifically to eIF3c, inhibiting translation initiation at the level of the 48S complex formation, without inhibiting other functions of eIF3, such as the eIF4F interaction (Hui and others 2005; Terenzi and others 2005). Murine P49, although capable of binding to eIF3c, does not inhibit translation in vitro (Fensterl and others 2008). Human P58 and P60 bind to neither eIF3c nor eIF3e (Terenzi and others 2006). Interestingly, the binding to eIF3c occurs via the N-termini of murine P54 and P56, and targets different domains of eIF3c, whereas the human P56 interaction with eIF3e utilizes the C-terminus of P56 (Guo and Sen 2000; Terenzi and others 2005). Thus, human and murine P54 and P56 share a similar function that is achieved through distinct molecular mechanisms, and is conserved in spite of considerable sequence variation between the homologs.
Antiviral effects
The general inhibition of cellular translation mediated by P56 family proteins can be considered as part of a nonspecific antiviral program elicited by exposure of cells to IFNs or viral molecular patterns, which induce the concerted expression of these and other ISGs and VSIGs. However, the repertoire of functions of P56 proteins extends to virus-specific inhibitory functions, both translation-related and unrelated.
Translation of the Hepatitis C virus (HCV) positive-sense RNA genome is initiated by IRES-dependent ribosome recruitment, which, similar to cap-dependent translation, requires eIF3 (Lukavsky 2009). This process is IFN-sensitive, because IFN induces human P56, which in turn inhibits HCV translation initiation both in vitro and within cells. Further, HCV-IRES translation is more strongly compromised than cap-dependent translation in the presence of human P56. The inhibition depends on binding of human P56 to eIF3, and P56 is found specifically in ribosomal initiation complexes containing eIF3 and HCV RNA (Wang and others 2003). Hence, IFN- or virus-induced human P56 contributes to the inhibition of HCV growth in infected cells.
An intriguing, newly identified function of human P56 is its antiviral activity against Human Papillomavirus (HPV) (Terenzi and others 2008). It is based on direct binding of P56, but not the other human family members, to a key viral protein, the E1 helicase, which is essential for viral DNA replication. The interaction with human P56 not only blocks E1 helicase activity and viral DNA replication, but it also sequesters the majority of HPV E1 in the cytoplasm, separating it from the viral genomes in the nucleus. The interaction with E1 is mediated by the N-terminus of human P56 and does not require its eIF3e-binding C-terminal portion to block E1 helicase activity. P56 binding to E1 is not virus strain specific; it binds E1 of both low- and high-risk strains of HPV, such as HPV11, 18, and 31. HPV DNA replication is highly sensitive to IFN treatment and this effect is largely diminished when P56 expression is knocked down by shRNA, establishing human P56 as the major IFN-induced antiviral protein against HPV (Terenzi and others 2008).
Evidence for antiviral activity of a murine P56 family member arose from studies with Sindbis virus, which is a positive-sense RNA genome Togavirus that can cause fatal encephalitis in newborn mice. When Sindbis virus genomes engineered to express murine P56, P54, or P49 are subcutaneously injected into 1 day-old mice, the total survival of all 3 groups of mice is increased from 25% to 40%. However, viral gene expression in cell culture is only inhibited by expression of one of the proteins, murine P56 (Zhang and others 2007). The underlying mechanism remains to be elucidated.
Inhibition of IFN-β induction
The transcriptional induction of IFN-β and VSIGs such as ISG56 and ISG15 by cytoplasmic viral RNA, especially dsRNA, is initiated by the cytoplasmic receptors RIG-I and MDA-5, which use TBK1/IKKɛ and IKKα/β kinases to activate transcription factors IRF-3 and nuclear factor κB (NF-κB), respectively, which then induce gene transcription (Sarkar and Sen 2004). The pathway originates from a signaling platform on the mitochondrial surface and requires the 2 proteins MAVS (=IPS-1, VISA, CARDIF) and STING (=MITA, ERIS, MPYS), which scaffold the formation of signaling complexes (Ishikawa and Barber 2008; Zhong and others 2008; Takeuchi and Akira 2010). Human P56 and P54 bind STING late after Sendai virus infection, which disrupts the STING/MAVS and STING/TBK1 complexes (Li and others 2009). Consequently, expression of human P56 inhibits IRF-3- and NF-κB-dependent gene induction and activation of the IFN-β gene by Sendai virus or dsRNA transfection. Further, the antiviral response to VSV infection is affected by P56: siRNA-mediated knockdown of P56 expression results in diminished virus replication, most likely due to increased virus-induced expression of IFN and antiviral VSIGs, both depending on IRF-3. Thus, human P56 is a negative feedback regulator helping in dampening virus-induced innate immune signaling (Li and others 2009).
Inhibition of cell migration and antiproliferative effects
Several functions of P56 family proteins not directly related to the response to infections have been reported. Notably, human P54 was suggested to inhibit cell migration of oral cancer cells (Lai and others 2008). Several oral squamous cell carcinoma (OSCC) cell lines constitutively express human P54. P54 displays a cytoskeletal staining pattern in OSCC and other cell lines, and apparently interacts with cytokeratins. Importantly, in OSCC cells with low levels of constitutive P54, IFN treatment or exogenous P54 expression inhibits cell migration considerably, whereas knockdown of the endogenous P54 expression doubles OSCC cell migration rates. Interestingly, high expression levels of P54 in tumor tissues are associated with higher postsurgery survival of OSCC patients (Lai and others 2008).
IFNs have long been known for their antiproliferative effects in cell culture; P56 and P54 are likely contributors as inhibitors of translation, as described above. However, antiproliferative activity has been ascribed also to human P60, which uses a different mechanism (Xiao and others 2006). In monocytic U937 cells, P60 expression leads to upregulation of 2 cell cycle negative regulators, P27 and P21. Consequently, cells accumulate in the G1 phase. Normally, P27 turnover is regulated by CSN5 (=JAB1), which binds P27 in the nucleus and exports it to the cytoplasm for ubiquitin-dependent degradation. P60 sequesters CSN5 in the cytoplasm, thereby increasing nuclear P27 protein levels. On the other hand, upregulation of P21 by human P60 is suggested to depend on downregulation of c-Myc, a repressor of P21 expression (Xiao and others 2006).
Other IFIT-Like Genes in Human and Mouse
Besides the characterized members of the IFIT1 gene family described above, both human and mouse IFIT loci contain interspersed IFIT-like genes, whose mRNAs have fully or partially been sequenced. Nothing is known about their inducibility; however, the 3 additional murine genes present within the Ifit locus contain one or 2 ISREs in proximity of the transcriptional start site (Fig. 1) and are therefore expected to be IFN-responsive. We tentatively name these genes Ifit3b (official gene symbol = I830012O16Rik), Ifit1b (=2010002M12Rik), and Ifit1c (=Gm14446) (Fig. 1). The Ifit3b genomic sequence is 96% identical to ISG49/Ifit3, and the putative protein is 98% identical to P49. Ifit1b and Ifit1c are closely related to ISG56/Ifit1; the encoded proteins share 78% identical amino acids with each other and 60% with murine P56. Both of them are oriented reversely within the locus.
The human locus on chromosome 10 contains the additional family member IFIT1B (Fig. 1), which is located directly upstream of ISG56/IFIT1 and does not possess an ISRE; consequently, IFIT1B is unresponsive to IFN or dsRNA treatment in HT1080 cells (our unpublished observations). In addition, a nontranscribed IFIT1-related pseudogene, IFIT1P, is present on human chromosome 13 (Wathelet and others 1988).
The putative proteins encoded by human IFIT1B and murine Ifit1b/c and Ifit3 have full lengths and possess an identical distribution of TPR motifs compared to their respective relatives (Fig. 3).
Conclusions
The members of the ISG56 family of genes share a simple gene architecture and promoter structure, yet they exhibit complexity in cell type- and stimulus-specific gene induction patterns. Due to the amino acid sequence variability of TPR motifs, the encoded P56 family proteins fulfill a wide range of nonredundant functions. New binding partners and actions of P56 proteins are being identified; for example, murine P54 is reported to bind cytoskeletal β-tubulin, and murine P56 interacts with translation elongation factor eEF1α1 (Saha and others 2006; Li and others 2010). Interestingly, human P56 was noted to be posttranslationally modified by ISG15 conjugation (ISGylation) (Zhao and others 2005), as well as phosphorylation (Li and others 2009), hinting at the possibility of additional levels of regulation, by modulating the protein's function or changing its stability. The successful generation of ISG56 and ISG54 knockout mice will help to better understand the functions of the respective proteins at the organism level, and to identify new molecular mechanisms of action.
Footnotes
Acknowledgments
The relevant research in the authors' laboratory has been supported by the National Institute of Health grants CA068782, CA062220, and AI073303.
Author Disclosure Statement
No competing financial interests exist.