
Abstract
Ten Toll-like receptor (TLR) family members have been reported in humans. Here, the endoplasmatic receptors TLR9, TLR8, TLR7, and TLR3 respond to nucleic acids and derivatives or to small molecules (TLR7 and 8). Another cytoplasmic RNA receptor, retinoic acid inducible gene I (RIG-I), is stimulated by 5′ triphosphate double-stranded RNA. We discovered that TLR7 small-molecule agonists inhibit nucleic acid-mediated TLR3, TLR7, TLR9, or RIG-I-dependent interferon-α (IFN-α) immune response. Other cytokines and chemokines stimulated by nucleic acid agonists remained unaffected. The observed blockage of TLR3, TLR7, TLR9, and RIG-I-mediated IFN-α response appears to be driven by a competitive mechanism at the type I IFN pathway. Besides type I IFN, IFN response genes such as IFIT-1, Mx1, OAS1, or IRF7 were affected, which indicates that the key element driving the inhibition is located in the type I IFN pathway. Indeed, the heterotrimeric complex formation of phosphor-signal transducer and activator of transcription factor 1 (STAT1), phosphor-STAT2, and IRF9 (called ISGF3, IFN-stimulated gene factor 3) is inhibited through the TLR7 small-molecule agonists by phosphor-STAT2 blockage. These findings provide novel insights into the use of synthetic TLR7 or TLR7/8 small molecules as ligands for immune activation and suppression.
Introduction
C
Another group of PRRs called RIG-I-like receptors (RLRs) consists of the cytoplasmic RNA helicases retinoic acid inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (Mda-5), and LGP2. RIG-I and Mda-5 contain 2 caspase-recruitment domains (CARD) and a DExD/H-box helicase domain, which recognizes viral RNAs while the CARDs are responsible for signaling (Yoneyama and others 2004; Gitlin and others 2006). RIG-I is selectively activated by dsRNA containing a 5′ triphosphate, such as in panhandle RNA viruses (Kato and others 2006; Hornung and others 2006; Schlee and others 2009). It was additionally revealed that RIG-I can also be activated by short (∼25 bp) dsRNAs with at least a single phosphate at either the 5′ or 3′ end (Takahasi and others 2008; Marques and others 2006; Zamanian-Daryoush and others 2008).
In this study, we discovered that TLR7 small-molecule agonists such as R-848 or 9-substituted-8-hydroxyadenine derivatives can act as inhibitors of TLR3, TLR7, TLR9, or RIG-I-induced type I interferon (type I IFN) production, but not of the proinflammatory immune responses. TLR3, TLR7, TLR9, or RIG-I were stimulated with the corresponding nucleic acid ligands. Only active TLR7 small-molecule agonists were capable of mediating type I IFN suppression. 9-substituted-8-hydroxyadenine derivatives that do not activate the immune system failed for TLR3, TLR7, TLR9, or RIG-I blockage. We identified that the blockage occurred at the type I IFN signaling pathway. Small-molecule TLR agonist blockage affects type I IFN and response genes such as IFIT-1, ISG15, Mx1, OAS, or IRF7 and also the formation of the IFN-stimulated gene factor 3 (ISGF3) complex on the basis of signal transducer and activator of transcription factor 2 (STAT2) phosphorylation.
Materials and Methods
Reagents
TLR7 and TLR8 agonist Resiquimod (R-848, S-28463) was commercially synthesized by GLSynthesis, Worchester MA. Small molecules 52455, 52457, 52459, 52763, 52542, 52587, TLR7/8/9 small molecule antagonist, ORN R-0006, and CpG ODN 2395 were provided by Coley Pharmaceutical GmbH. Small-molecule variants of 52455 (Sumitomo) were generated by altering the pyrimidine/imidazole ring system or side group exchanges. 3M-001 and 3M-003 were purchased from 3M Pharmaceuticals, and 3M-002 was purchased from Invivogen. All oligonucleotides and small molecules were controlled for identity and purity by Coley Pharmaceutical GmbH and had undetectable endotoxin levels (<0.1EU/mL) measured by the Limulus assay (BioWhittaker). Poly rI:rC was obtained from Sigma. Compounds were suspended in sterile endotoxin-free Tris-EDTA (ODN) (Sigma), in DNAse- and RNAse-free water (ORN) (Life Technologies), or in DMSO/DNAse- and RNAse-free water (small molecules) (VWR), and were stored and handled under aseptic conditions to prevent contamination. Structures and sequences are listed in Fig. 1.
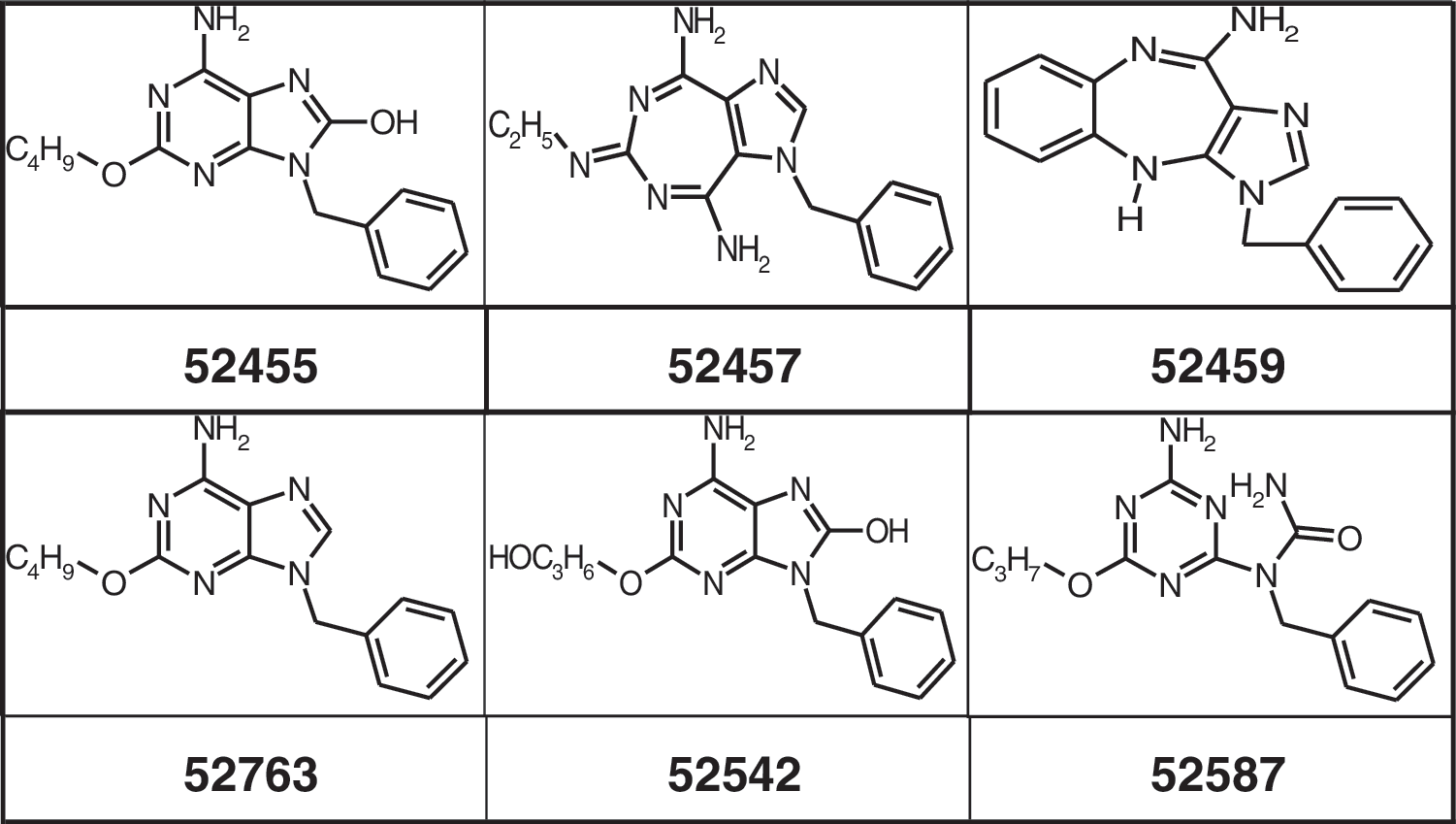
Structures of small molecules used.
Peripheral blood mononuclear cells
Peripheral blood mononuclear cell (PBMC) preparations from healthy male and female human donors were obtained from the Institute for Hemostaseology and Transfusion Medicine at the University of Düsseldorf (Germany). The PBMC were purified by centrifugation over Ficoll-Hypaque (Sigma). Purified PBMC were washed twice with 1× phosphate-buffered saline and resuspended in RPMI 1640 culture medium supplemented with 5% (v/v) heat inactivated human AB serum (BioWhittaker) or 10% (v/v) heat inactivated fetal calf serum, 1.5 mM
Cytokine secretion and detection of ISGF3 components
Freshly isolated PBMC were resuspended at a concentration of 3×106/mL to 5×106/mL and added to 96-well round-bottomed plates (200 μL/well), which had previously received nothing or small molecules and ORN complexed to DOTAP, or ODN, or DOTAP alone. The cells were cultured in a humidified incubator at 37°C for the indicated time points. Culture supernatants (SNs) were collected and, if not used immediately, were frozen at −20°C until required. The amounts of cytokines in the SN were assessed using commercially available ELISA Kits [IL-6, IFN-γ or TNF-α, Diaclone; IL-12p40, BD Pharmingen; or an in-house ELISA (IFN-α) developed using commercially available antibodies (from PBL)]. STAT1 and phosphorylated STAT1 or STAT2 and phosphorylated STAT2 were measured using cell extracts from human PBMC (Invitrogen or R&D Systems). IRF9 was detected using SN from human PBMC (Antibodies Online).
Cell isolation
Human pDCs and monocytes were isolated from PBMC by BDCA-4 or CD14 immunomagnetic bead positive selection according to the manufacturer's recommendations (Miltenyi Biotech). Purity was confirmed by staining with mAb to CD11c, CD14, HLA-DR, and CD123 (all from BD Pharmingen), and was typically >90%. Isolated cells were plated on 96-well plates at 5×105cells/mL and stimulated for 16 h with indicated amounts of ORN complexed to DOTAP, small molecules, or ORN complexed to DOTAP and small molecules. After 16 h, the SNs were harvested, and the cytokine or chemokine amounts were analyzed.
TLR reporter assay
Human embryonic kidney cells (HEK293) containing a nuclear factor kappa B (NFκB)-luciferase reporter construct and expressing human TLR7 or TLR8 or without TLR expression were used as described earlier (Jurk and others 2002; Jurk and others 2006b; Jurk and others 2006a). The cells were plated on 96-well plates at 1.5×104/well and allowed to attach overnight. The cells were subsequently incubated for 16 h with the indicated amount of ORN complexed to DOTAP (Roche), DOTAP alone, small molecules, poly rI:rC, or CpG ODN and then tested for luciferase expression. Each data point was done in duplicate.
Mouse in vivo studies
sv129 mice (6-8 weeks of age) were used for all in vivo experiments, purchased from Charles River Canada, and housed in micro isolators at the animal care facility of Coley Pharmaceutical Ltd. The ORNs formulated with DOTAP were administered IV to Sv129 mice (n=5 per group), 1 h post ORN dosed intraperitoneal with small-molecule R-848, 52455, or small-molecule TLR7/8/9 antagonist and after 4 h post ORN injection, the animals were bled, and the IFN-α levels in plasma were measured by ELISA (PharMingen).
Low-density TaqMan array
A 384-well micro fluidic card TaqMan array was pre-loaded with TaqMan expression assays by Applied BioSystems according to custom specifications. HPRT, PPIB, MyD88, TICAM, TRIF, STAT1, IL-6, IRF3, IRF5, IRF7, IFN-g, OAS1, OAS2, IFN-β, IFN-a2, IFN-a21, IFIT-1, ISG15, ISG20, Mx1, and Mx2 were selected due to their known influence on type I IFN pathways of TLRs and RLRs. Control genes such as housekeepers, adaptor proteins, type II IFN, or pro-inflammatory cytokines, were included (Honda and others 2005a; Choi and others 2009; Dai and others 2004; Elco and Sen 2007; Okumura and others 2006; Tamassia and others 2008; Jurk and others 2004, 2006a; Vollmer and others 2004b, 2004c; Forsbach and others 2007, 2008). mRNA was isolated from human PBMC samples with RNeasy Mini Kit (Qiagen) and used for cDNA synthesis with High Capacity RNA-to-cDNA Kit (Applied BioSystems) according to the manufacturer's protocol. 600 ng cDNA with a final volume of 100 μL was loaded from each sample to the low-density TaqMan array card and measured by a TaqMan instrument according to the manufacturer's protocol.
Computer modeling
Identification of the putative interaction sites was performed using the binding site tool in Accelrys's Discovery Studio (Discovery Studio 2.5; Accelrys Software, Inc.) on the x-ray structure of STAT1 (1BF5) and the homology model structure of STAT2. A binding site consists of a set of points on a grid that lie in the cavity of a receptor. The method (ref2) identifies the binding sites based on the shape of the receptors, that is, STAT1 and STAT2. An “eraser” algorithm was used to remove all grid points that were not in contact with the receptor. For the remaining grid points that were close to the receptor, a flood-filling algorithm was used to generate contiguous regions consisting of unoccupied, connected grid points.
Docking simulations were carried out using a Monte Carlo–based LigandFit method (ref2). The Monte-Carlo method was used in the conformational search of the ligand, allowing only rotatable torsion angles while bond lengths and bond angles remain constant. Therefore, the starting ligand conformation(s) should have a reasonably low-energy 3D geometry. We kept the protein structures fixed in this study. The internal nonbonded ligand energy was calculated for each new conformation. Bad conformations with high internal ligand energies (typically resulting from internal close contacts) were discarded. The internal ligand energy consists of a van der Waals (vdW) term and an electrostatic term. The nonbonded vdW energy was computed using a standard 9-6 potential. In-situ ligand minimization was performed using the smart minimizer that was optimized for ligand-receptor data. Scoring functions such as Ligscore1, Ligscore2, Piecewise Linear Potential (PLP1), PLP2, Jain, and Potential of Mean Force provided for the method were used for consensus scoring. The best 10 poses were retained per molecule for the analysis.
Results
Type I IFN immune activation by TLR7/8 ORN can be inhibited by TLR7 small-molecule agonists
Previous studies demonstrated sequence-dependent human TLR7 and TLR8 activation by short single-stranded GU-rich RNAs. Poly U or GU-containing phosphorothioate (PS) or phosphorodiester ORN induce high levels of IFN-α via TLR7 and TNF-α via TLR8 (Heil and others 2004). In addition, small molecules such as R-848 and derivatives (Jurk and others 2002), guanine nucleoside analogs such as loxoribine (Heil and others 2003), or 9-substituted-8-hydroxyadenine derivates (Isobe and others 2006; Kurimoto and others 2003) activate human and/or mouse TLR7 and induce IFN-α. To analyze the TLR7-specific immune response of 9-substituted-8-hydroxyadenine and our synthesized derivates (structures are shown in Fig. 1), we used stable transfected HEK-293 cells with TLR7, TLR8, and/or NFκB-luciferase read-out. All the TLR cell lines express constant levels of at least 10 copies of TLR7 or TLR8 normalized to 1,000 copies of PPIB (data not shown). As shown in Fig. 2 and Table 1, 9-substituted-8-hydroxyadenine and our synthesized derivates activate the human immune system via TLR7 but not TLR8. All tested substances demonstrated bell-shaped dose-response curves. Further increases in dose were associated with a decrease in the magnitude of response. The bell-shaped nature of these effects may be explained by negative feedback loops of the receptor in response to the stimuli. (Hartmann and others 2003; Marshall and others 2003; Vollmer and others 2004c; Abel and others 2005).
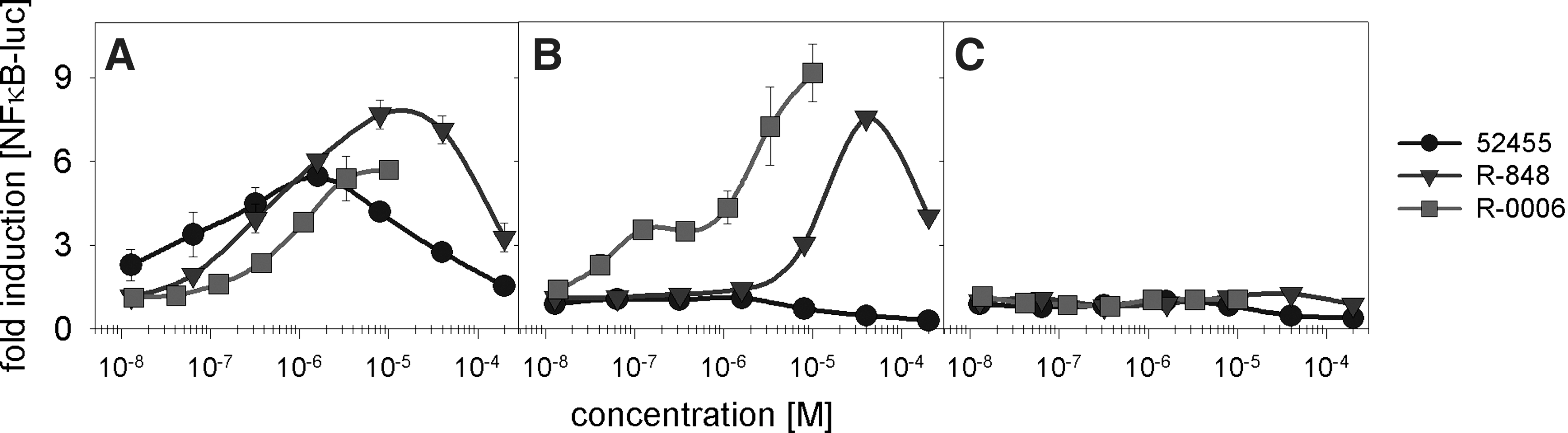
Toll-like receptor 7 (TLR7)-dependent signal transduction induced by small molecules activating the interferon-α (IFN-α) pathway. Human embryonic kidney cells (HEK293) stably transfected with human TLR7 and nuclear factor κ B (NFκB)-luc
HEK293 cells stably transfected with human TLR7 and NFκB-luc or TLR8 and NFκB-luc or NFκB-luc alone were incubated with the indicated small molecules (200 μM→1/5 dilution; full titration curve) or ORN R-0006 (10 μM complexed to 50 μg/mL DOTAP→1/3 dilution; full titration curve) for 16 h. NFκB activation was measured by assaying luciferase activity. EC50 values were calculated using a graphical evaluation by SigmaPlot 10.0. Mean and SD of 4 independent experiments (n=4).
HEK, human embryonic kidney; TLR, toll-like receptor
Since cells without TLR7 but with the NFκB-luciferase reporter read-out failed in response to 9-substituted-8-hydroxyadenine and our synthesized derivates, one can conclude a TLR7-specific reaction. As shown in Fig. 3A, by using a constant concentration of immune stimulatory PS ORN R-0006 (Forsbach and others 2008) and increasing the concentrations of 9-substituted-8-hydroxyadenine (Isobe and others 2006) (here referred to as 52455 or Sumitomo), 52455 is a potent inhibitor of R-0006-mediated IFN-α release on human PBMC activation. R-0006 w/o small molecules was used to measure the basic level of immune response on human PBMC activation and for comparison with the 52455 influence (dotted line). 52455 inhibits the R-0006-induced type I IFN at a level similar to or better than a TLR7/8/9 small-molecule antagonist. The TLR7/8/9 antagonist was previously identified in-house as inhibiting specifically TLR7, TLR8, and TLR9. It shows 100-fold higher potency compared with Chloroquine or Hydroxychloroquine (unpublished data). The IC50 for 52455 (1.1 nM) demonstrates 189-fold difference to the TLR7/8/9 small-molecule antagonist (208.9 nM). Beside 52455, 2 derivatives, 52542 and 52763 (structures see Fig. 1), also inhibit the ORN R-0006-induced IFN-α cytokine release on human PBMC activation with an IC50 of 29.7 nM and 1,463.7 nM, respectively. Interestingly, only 52455 and its derivatives that induce TLR7-mediated agonistic type I IFN (Fig. 3B and Table 1) are capable of functioning as antagonists against ORN R-0006. Although the agonistic effect is revealed to be less potent compared with the antagonistic effect (eg, 52455 IC50: 1.1 nM versus EC50: 538.8 nM; Fig. 3A, B), the order of potency is similar. Besides, 52455 and its derivatives also previously described that TLR7 or TLR7/8 small-molecule agonists, such as R-848, 3M-001, 3M-002, 3M-003 (Gorden and others 2005; Gorski and others 2006), or Loxoribine, can inhibit the ORN R-0006-mediated IFN-α response on human PBMC activation (Table 2). In summary, these data show the inhibition of TLR7 human PBMC-mediated type I IFN cytokine release for most, if not all, TLR7 and TLR7/8 small-molecule agonists.
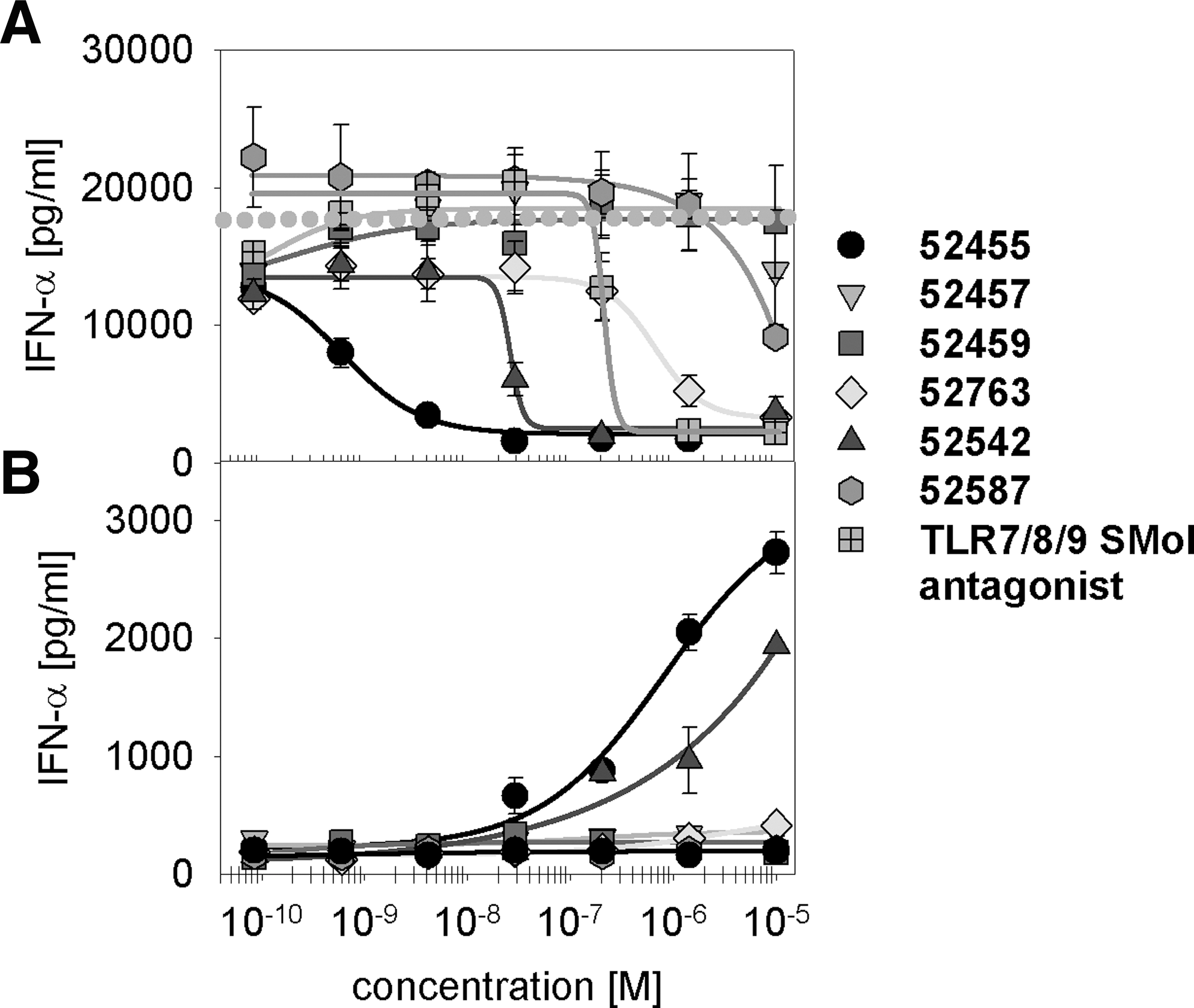
IFN-α production induced by a TLR7 ORN agonist is inhibited by TLR7 small-molecule agonists.
Human PBMC were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP and TLR7 and TLR7/8 small-molecule agonists (10 μM→1/7 dilution). After 16 h, the supernatants were harvested, and IFN-α was measured. IC50s were calculated using the SigmaPlot sigmoidal curve fitting parameters. Data shown are mean (±SEM) of 3 donors. One out of 2 independent experiments.
PBMC, peripheral blood mononuclear cell; ORN, oligoribonucleotide; SEM, standard error of the mean; IFN, interferon; IC50, half maximal inhibitory concentration.
TLR7/8 small-molecule agonists inhibit immune activation only at the TLR7 pDC level
Human TLR7 and TLR8 differ in their cell-type specific expression and, therefore, activate divergent cell types. This results in TLR7 and TLR8-specific cytokine patterns. TLR7 agonists directly activate pDCs and stimulate IFN-α and IFN-α regulated genes, while TLR8 agonists directly activate mDCs, NK cells, and monocytes, and induce proinflammatory cytokines and chemokines such as TNF-α, IL-12, IFN-γ, or IL-6 (Hornung and others 2005; Forsbach and others 2008). GU-rich ORNs, similar to R-0006, are TLR7 and 8 ligands and stimulate human TLR7 and TLR8 immune responses (Forsbach and others 2008).
To investigate whether both TLR7 and TLR8 are affected by the observed 52455 or R-848 antagonism, TNF-α, IL-6, IFN-γ, and IL-12p40 cytokine release on R-0006 human PBMC activation was analyzed (Fig. 4A–E). R-0006 w/o small molecules (dotted gray line) was used to measure the basic level of immune response on human PBMC activation and for comparison with the R-0006 blockage of 52455 and TLR7/8/9 antagonists. While the TLR7/8/9 antagonist suppressed all of these cytokines, 52455 inhibited only ORN R-0006-induced and TLR7-mediated type I IFN cytokine release on human PBMC activation. This indicates that TLR7 but not TLR8 stimulated by RNA agonists such as R-0006 can be suppressed by TLR7 small-molecule agonists. Similar effects were demonstrated in vivo (Fig. 4F, G). R-0006 mediated type I IFN but not IFN-γ cytokine release of SV129 mice was blocked by 52455 and R-848. TLR7/8/9 antagonist revealed the blockage of both type I IFN and IFN-γ cytokine release on SV129 mice activation.

TLR7 small-molecule agonists are antagonistic only to type I IFN induction. Human PBMC were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP, and the indicated concentrations of TLR7 small-molecule agonist 52455, TLR7/8/9 small-molecule (SMol) antagonist, or as controls for the base level of immune activation 0.5 μM ORN R-0006 complexed to 6.25 μg/mL DOTAP alone (dotted grey line). After 16 h, supernatants were harvested, and IFN-α
Human TLR7 and TLR9 co-localize in the same subpopulations of immune cells, B-cells, and pDCs, whereas human TLR8 is strongly expressed in the myeloid compartment (Hornung and others 2002; Kokkinopoulos and others 2005; Berkeredjian-Ding and others 2005). To analyze TLR7 cell type-specific inhibition by 52455, pDCs and monocytes were purified from human PBMC. These cell subsets are the main source of TNF-α (monocytes) and IFN-α (pDCs) on ORN stimulation (Forsbach and others 2007; Forsbach and others 2008; Vollmer and others 2005). As shown in Fig. 5 and Table 3, R-0006 w/o small-molecule treatment (dotted line) induces the pDC-specific immune response of IL-1Rα, IL-6, TNF-α, IFN-α, MIP-1α, and MIP-1β (Vollmer and others 2005; Forsbach and others 2007, 2008) (Fig. 5). However, only R-0006-mediated type I IFN cytokine release was blocked by 52455 on human PBMC activation. Monocyte cytokine and chemokine secretion on TLR7/8 R-0006 ORN stimulation was not altered with 52455 treatments (Table 3). With human pDC being the main source for TLR7 R-0006-mediated IFN-α, the observed blockage effects can also be detected in human PBMC. The observed R-0006 immune response of IL-1Rα, IL-6, TNF-α, MIP-1α, and MIP-1β on human pDC activation is up to 100-fold lower than the main monocyte and B-cell–derived human PBMC immune response of this cytokine (Vollmer and others 2005; Forsbach and others 2007, 2008; Tluk and others 2009). This explains why the blockage of R-0006 by 52455 cannot be measured in human PBMC.
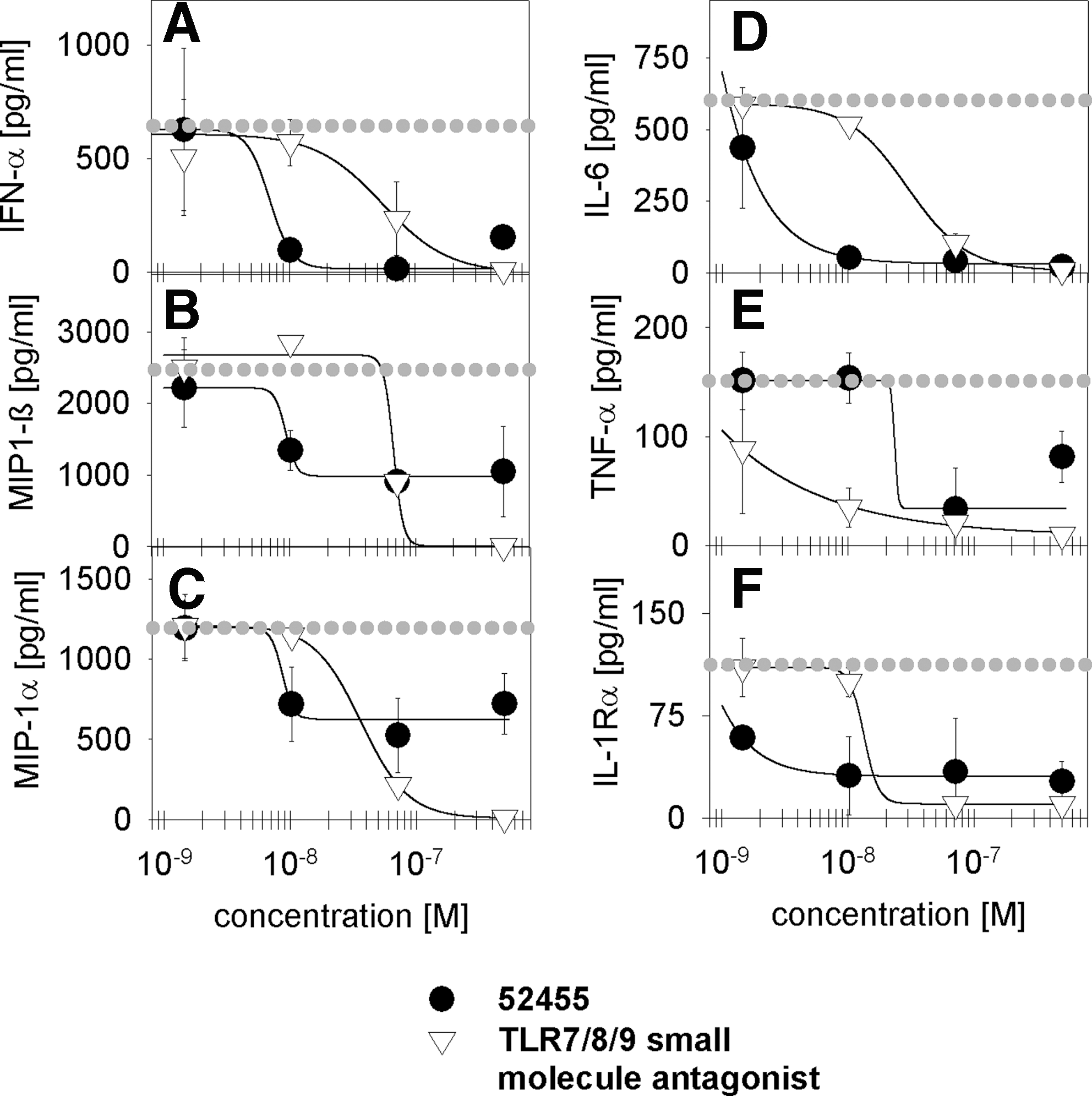
Effect of TLR7 small-molecule agonists on cytokine secretion from purified human CD123+ pDC-enriched immune cells stimulated by the TLR7/8 ORN agonist. CD123+ enriched cells were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP and indicated concentrations of the 52455 small molecule or the TLR7/8/9 small-molecule antagonist. As controls, CD123+ enriched cells were stimulated with 0.5 μM ORN R-0006 complexed to 6.25 μg/mL DOTAP (dotted gray line). After 16 h, supernatants were harvested, and IFN-α
CD123+ (pDC) or CD14+ (monocytes) enriched cells were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP and 4 concentrations of 52455 small-molecule (5 μM;>1/7 dilution) or TLR7/8/9 small-molecule antagonists. After 16 h, the supernatants were harvested, and the cytokines and chemokines were measured by Luminex technology. Data shown are mean (±SEM) of 9 blood donors. IC50s [μM] listed in the table just provided were calculated using the HillPlot 4 parameter (SigmaPlot10.0).
n.d., no detectable cytokine/chemokine response for R-0006 immune activation; 1, no inhibition of R-0006 immune response by small molecules; pDC, plasmacytoid dendritic cell.
Competitive inhibition and kinetic advantages for binding and blockage of the IFN-α pathway by TLR7 small-molecule agonists
Antagonists can actively change the physiological function of a receptor. This can be achieved either directly or indirectly by the modification of a physiological stimulus. Inhibitory molecules can bind orthosterically or allosterically to the receptor and either compete or not compete with the original agonist at the binding site. The determination of allosteric versus orthosteric receptor binding can be performed by direct binding assays. However, competitive or noncompetitive binding of the molecule to the receptor can also be observed by agonist/antagonist cross-titrations within a biological readout system.
Cross-titrations were performed by using full titration curves of ORN R-0006 (Fig. 6A,C) or CpG ODN 2395 (Fig. 6B, D) type I IFN cytokine release on human PBMC activation. Single different constant concentrations of R-848 (Fig. 6A, B) or 52455 (Fig. 6C, D) ranging from 4 nM up to 1 μM were added to a full titration curve of either R-0006 or CpG ODN 2395. By this method, one can analyze the influence of 1 single small-molecule concentration on the curve characteristics of R-0006 or CpG ODN 2395. Comparing these full titration curves with one another can help in the identification of effects such as decreased maximum cytokine release or potency shift. Above a certain concentration of 52455 or R-848, the full titration curve of R-0006 or CpG ODN 2395 demonstrated a depression of the maximal dose response without dextral displacement According to Kenakin (2002), this implies a competitive antagonism A competitive antagonism can occur at either the TLR7 or TLR9 receptor site or downstream at potential receptor sites of the type I IFN signaling pathway. The data demonstrate that even low concentrations of 52455 (4 nM) or R-848 (4 nM) lead to a decrease in the maximum read-out of the R-0006 IFN-α titration curve. A complete loss of R-0006-mediated type I IFN on human PBMC activation was reached at a 10 nM concentration of 52455 or R-848. Figure 6B and D show similar effects of TLR7/8 (R-848) or TLR7 (52455) small molecules inhibiting the TLR9 CpG C-Class ODN 2395-mediated IFN-α production. Here, the complete loss of the CpG ODN 2395 immune response was achieved at 0.3 μM. Comparable data were achieved for CpG A-Class or CpG B-Class ODN (data not shown).
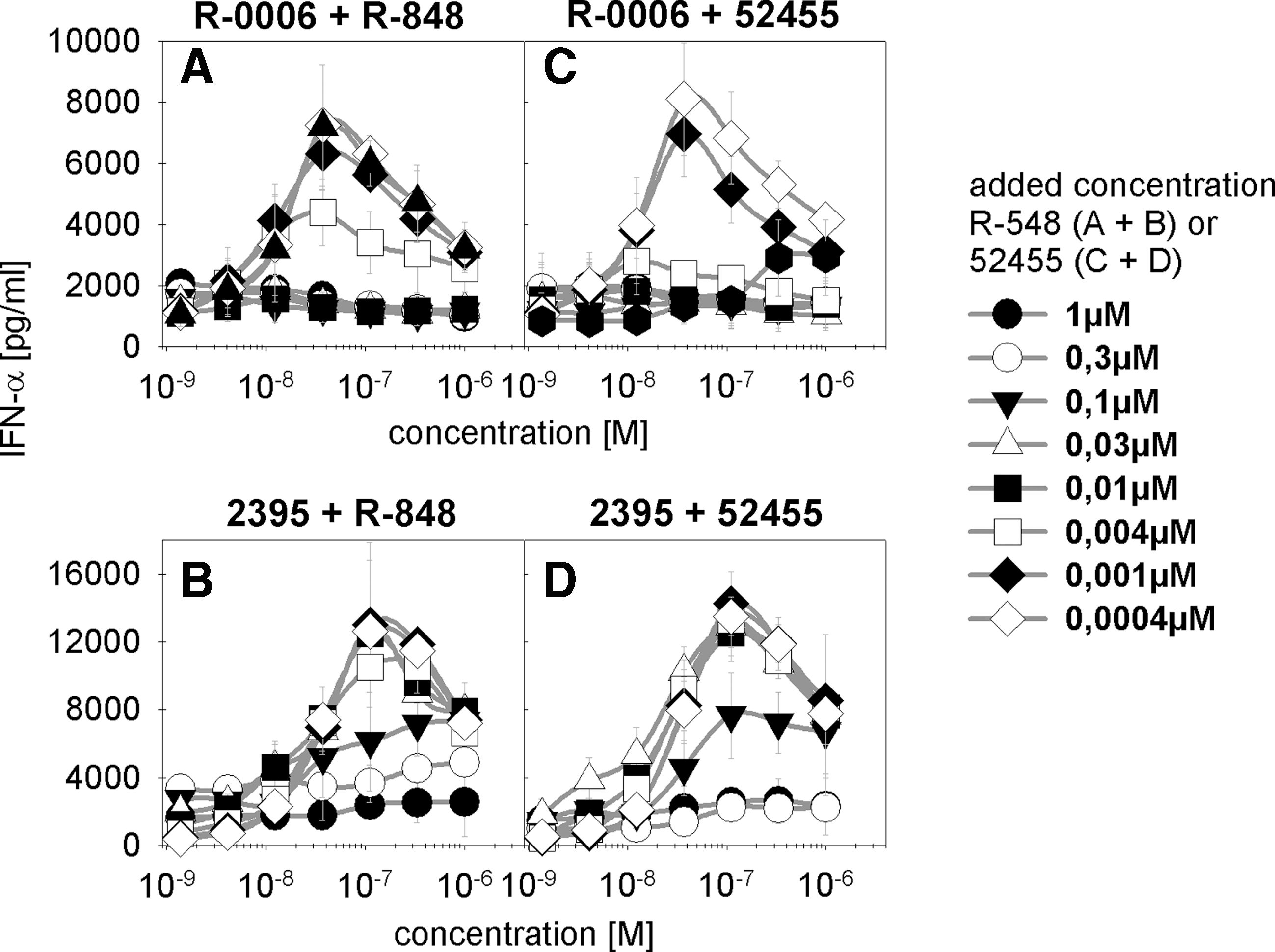
Cross-titration of TLR7 ORN and small-molecule agonists demonstrate competitive inhibition. Human PBMC were stimulated by full titration curves of ORN R-0006
TLR7 small-molecule agonists do have uptake and transport benefits due to their lower molecular weight compared with single-stranded ORN. Therefore, the possibility exists that small molecules may reach the receptor binding side and/or occupy the IFN-α signaling pathway earlier than nucleic acids. The uptake of nucleic acids and the interaction with TLR9 occur within 5–30 min (Latz and others 2004; Ahmad-Nejad and others 2002). Based on these kinetic data, the TLR7 small-molecule 52455 was added 30, 60, 120, 180, and 240 min later than the TLR7/8 ORN agonist R-0006 (Fig. 7) to allow appropriate binding of the nucleic acid to TLR7. The data demonstrate that after 4 h, the ORN R-0006-induced IFN-α production was no longer completely suppressed by 52455. This suggests that the suppression by TLR7 small-molecule agonists is dependent on the kinetic uptake advantage of the small molecules by faster occupation of the TLRs and/or the type I IFN pathway. Biacore binding studies (data not shown) showed no binding of 52455 to ORN R-0006 or CpG ODN 2395, excluding interactions between these molecules.
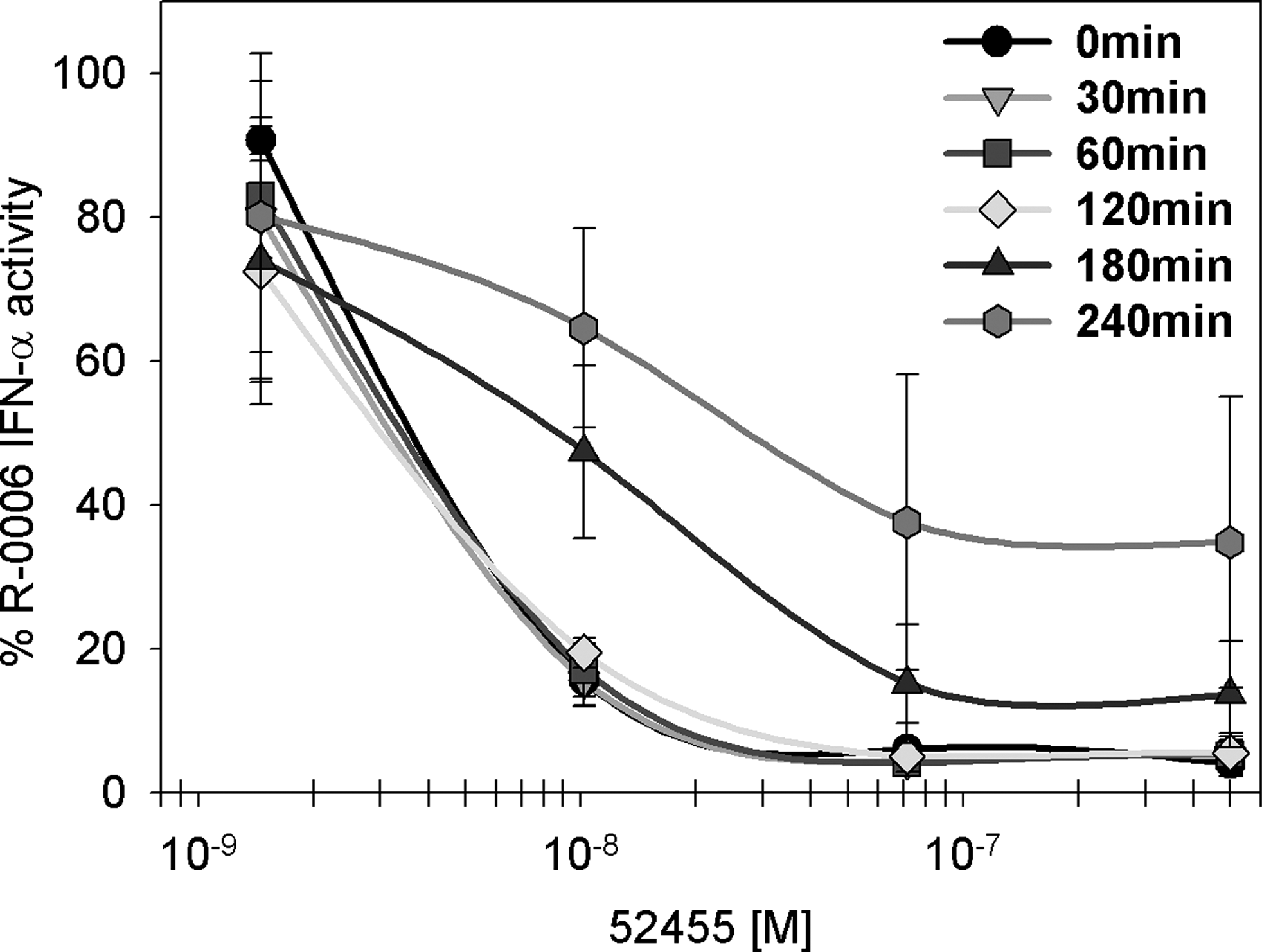
TLR7 ORN agonist and TLR7 small-molecule agonist compete for TLR7 binding. Human PBMC were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP, and the indicated TLR7 small-molecule agonist concentrations (52455) were added 0, 30, 60, 120, 180, or 240 min later. Cell culture media or 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP without TLR7 small molecules were used as assay controls. Data are graphed as % 0.5 μM R-0006 complexed to 6.25 μg/mL DOTAP w/o small molecules. After 24 h, supernatants were harvested, and IFN-α was measured. Data shown are the mean (±SEM) of 6 donors. Mean of 6 independent blood donors (n=6).
In summary, we showed that nucleic acids such as ORN R-0006 or CpG ODN 2395, and small-molecule TLR7 agonists compete for TLR7 or 9 receptor binding or the IFN-α signaling pathway. In addition, small-molecule TLR7 agonists have kinetic advantages compared with nucleic acids.
TLR7 small-molecule agonists inhibit the type I IFN pathway on blockage of STAT2 phosphorylation
We showed that TLR7 small-molecule agonists such as R-848 or 52455 block both the TLR9 (berghöfer, refman) and the TLR7 type I IFN cytokine release on human PBMC activation. To investigate whether besides TLR7 and 9 other nucleic acid PRRs can also be blocked in their immune response by R-848 or 52455, we analyzed the human PBMC IFN-α cytokine release of RIG-I, Mda-5, and TLR3 ligands with and w/o 52455. As shown in Fig. 8, type I IFN blockage by TLR7 small-molecule agonist 52455 is not limited to ORN R-0006-induced TLR7 activation. Type I IFN immune responses by RIG-I, Mda-5, or TLR3 nucleic acid ligands (5′ppp dsRNA or poly rI:rC) (Alexopoulou and others 2001; Schlee and others 2009) are blocked by 52455 in a concentration-dependent manner. Therefore, type I IFN blockage by synthetic TLR7 small-molecule agonists is not limited to TLR7 or TLR9. Blockage also includes the nucleic acid binding receptors TLR3, RIG-I, and Mda-5.
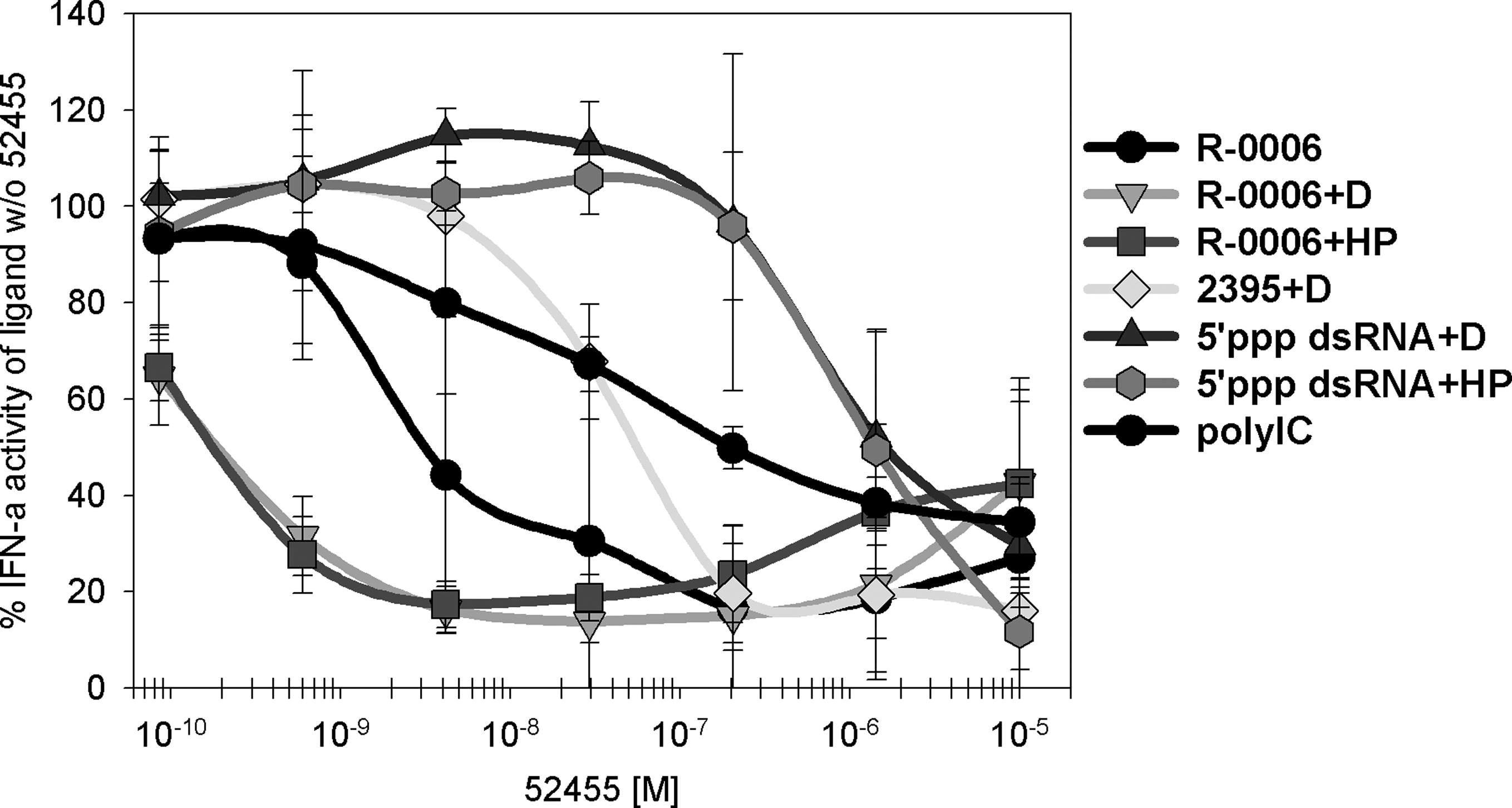
TLR7 small-molecule agonists suppress the IFN-α secretion induced by other nucleic acid binding receptors. Human PBMC were stimulated with 10 μM of ORN R-0006 alone, 0.5 μM ORN R-0006 complexed to 6.25 μg/mL DOTAP
As shown in Fig. 8, the blockage effect is independent of the usage of delivery systems: 52455 blocked type I IFN responses induced by unformulated R-0006, R-0006 on endosomal delivery (DOTAP), or on cytoplasmic delivery (HiPerfect). However, 52455 blockage of unformulated R-0006 was less effective than using delivery systems, probably indicating that small molecules may also use excessive delivery reagents as an uptake enhancer. Since we had depicted that the observed R-0006 type I IFN blockage is mediated via the direct interaction of TLR7 small-molecule agonists with pDCs (Table 3 and Fig. 5), we further raised the question whether the blockage occurs directly at the PRR or at the PRR-mediated type I IFN pathway. The binding of type I IFN to the IFN alpha receptor results in activation of the Jak-STAT pathway, including association with Janus protein tyrosine kinases Jak1 and Tyk2 and phosphorylation of downstream STATs, namely, STAT1 and STAT2 (Darnell and others 1994; Ihle and Kerr 1995). The tyrosine phosphorylation of these STATs leads mainly to the formation of ISGF3, a heterotrimeric complex of tyrosine-phosphorylated STAT1, STAT2, and IRF-9/p48/ISGF3γ (Bluyssen and others 1996; Bluyssen and Levy 1997). These complexes translocate into the nucleus and bind to specific DNA sequences containing IFN-stimulated regulatory elements, which leads to the activation of a large number of type I IFN response genes (Kessler and others 1990; Williams 1991).
Several type I IFN response genes have been described as being involved in TLR3, TLR7, TLR9, Mda-5, or RIG-I signaling to viral or bacterial pathogens (Honda and others 2005b; Takaoka and others 2005; Kong and others 2007; Tamassia and others 2008). ORN R-0006 up-regulates several type I IFN response genes such as OAS1, OAS2, IFIT-1, IRF-7, ISG15, ISG20, Mx1, and Mx2 but not IRF3 or IRF5. Pro-inflammatory cytokines such as IL-6 or type II IFN response genes such as IFN-γ are up-regulated as well, whereas TLR adaptor proteins (TICAM, TRIF, or MyD88) or housekeeper genes (HPRT or CyclophilinB) are not influenced by ORN R-0006 treatment (data not shown). Adding small-molecule agonist 52455 in a concentration-dependent manner to constant amounts of ORN R-0006 (Fig. 9; dotted line) results in mRNA down-regulation of several type I IFN response genes such as IFIT1, IRF7, ISG15, ISG20, Mx1, Mx2, OAS1, OAS2, IFN-α21, IFN-α2, and IFN-β, but IRF3 or IRF5. IFN-γ, HPRT, IL-6, or TLR adaptor protein mRNA levels remain unchanged. This indicates that 52455 does not cause type II IFN blockage, toxicity, or other unspecific effects but inhibits the mRNA expression of type I IFN and IFN-α response genes. The data imply that the effect of 52455 type I IFN blockage on activation of the immune system with nucleic acid ligands appears to take place upstream of the type I IFN response genes and downstream of the type I IFN receptor. The formation of the hetero-trimeric ISGF3 complex, phosphorylation of its members STAT1 or STAT2, down-regulation of the expression level of IRF-9/p48/ISGF3γ, and the association of the type I IFN receptor with Janus protein tyrosine kinases Jak1 and Tyk2 and their phosphorylation may be potential targets for an inhibitory effect. Human PBMC were treated with R-0006 complexed to DOTAP and the addition of 52455 or R-848. The response of R-0006 complexed to DOTAP w/o 52455 or R-848 was used as a reference for the calculation of % activity. Human STAT1 and STAT2 protein phosphorylation (Fig. 10) and Jak1 and Tyk2 protein phosphorylation (data not shown) were investigated by a cell-based enzyme-linked immunosorbent assay (ELISA) and normalized to total corresponding protein amounts. (Fig. 10). According to manufacturer's protocols, these STAT1/phosphoSTAT1, STAT2/phosphoSTAT2, Jak1/phosphoJak1, and Tyk2/phosphoTyk2 kit systems are comparable to Western blot analysis and use isolated fixed cells to detect the reactions based on HRP-anti-phosphotyrosin, 3,3′,5,5′-tetramethylbenzidine and stop-solution. The IRF9 protein was detected in the human PBMC SN. Figure 10 shows the data of phosphoSTAT1, phosphoSTAT2, and IRF9 graphed as % R-0006 activity complexed to DOTAP activity (dotted line). Interestingly, STAT2 but not STAT1 phosphorylation was blocked by small-molecule TLR7 agonists 52455 or R-848, whereas IRF9 remained unaffected. Upstream of the ISGF3 complex formation type I IFN receptor is associated with Jak1 and Tyk2 and leads to transphosphorylation. This event remained unchanged on 52455 or R-848 treatment (data not shown).
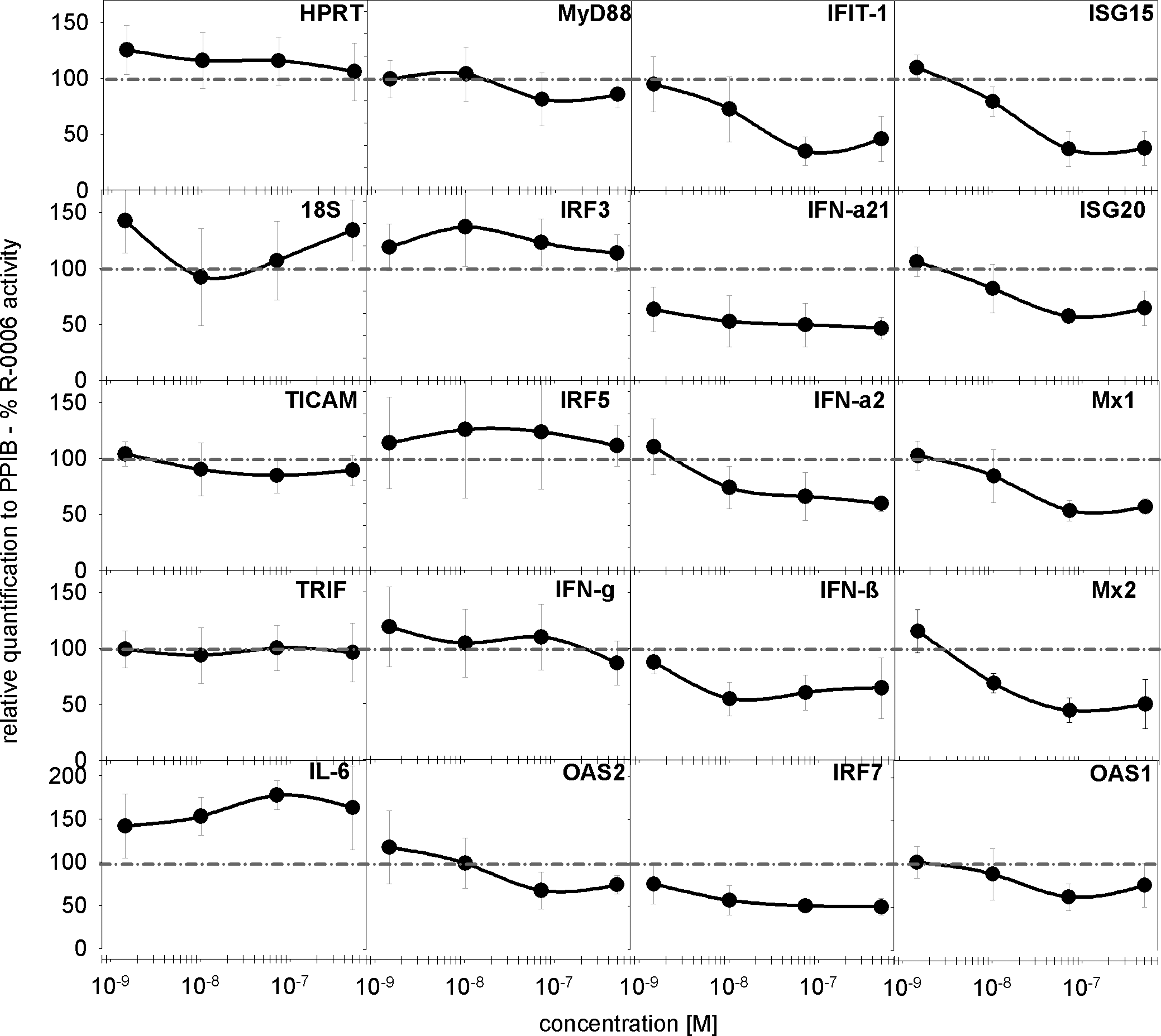
The TLR7 small-molecule agonist interferes with the activation of type I IFN response genes. Human PBMC (3 blood donors per experiment) were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP and the indicated TLR7 small-molecule agonist concentrations (52455) or ORN R-0006 complexed to 6.25 μg/mL DOTAP alone. After 16 h, the cells were harvested; RNA were extracted and analyzed via low-density TaqMan array. Expression levels were normalized to peptidylprolyl isomerase B/cyclophilin B (PPIB) and graphed as % R-0006 activity w/o 52455 (dotted line). Data are presented as the relative expression and mean of 6 independent blood donors (n=6).

The TLR7 small-molecule agonist blocks signal transducer and activator of transcription factor 2 (STAT2) but not STAT1 phosphorylation. Human PBMC were stimulated with 0.5 μM of ORN R-0006 complexed to 6.25 μg/mL DOTAP and the indicated TLR7 small-molecule agonist concentrations (52455). After 24 h, the cells were harvested and STAT2, phosphoSTAT2, STAT1, and phosphoSTAT1 protein amounts were measured. PhosphoSTAT2 and phosphoSTAT1 were normalized to total STAT2 or total STAT1 before being graphed. The IRF9 protein was measured after 24 h using the PBMC supernatant. Data are graphed as % R-0006 complexed to 6.25 μg/mL DOTAP w/o 52455 (dotted line). Data represent the mean (±SEM) of 15 independent blood donors (n=15).
In summary, our data suggest that small-molecule TLR7 agonists block type I IFN pathways of nucleic acid ligands by interfering with STAT2 phosphorylation. The observed competitive inhibition of TLR7 small-molecule agonists and TLR7/8 or TLR9 nucleic acid agonists (Fig. 6) seems to occur not at the TLR or RLR receptor site but at the receptor site of STAT2 phosphorylation. The interaction analysis of STAT2 and small-molecule TLR7 agonists is not possible with the existing measurement methods (eg, Biacore) due to the low molecular weight of, for example, R-848 or 52455 (around 314 g/mol) compared with the high molecular weight of STAT2 or STAT1 (around 113,000 g/mol). This signal-to-noise ratio is below the detection limit of Biacore instruments.
Modeling analysis confirms preferred binding of TLR7/8 small-molecule agonists to the STAT2 phosphorylation site
The following molecular docking analysis was performed to further investigate the hypothesis that small molecules such as 52455 and R-848 bind preferentially to STAT2 versus STAT1 and compete in the type I IFN pathway with the STAT2 phosphorylation event. The workflow was the first that generated a homology structure for STAT2; second, that identified all probable binding sites for both structures of STAT1 and STAT2; third, to dock and analyze the binding modes of both small molecules; and last, to rank these molecules based on the favourable energy.
To date, there are 7 available mammalian STAT family members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Based on the type I IFN pathway and our data, the modeling analysis was contextually focused on STAT1 and STAT2 for this study, where the crystal structure of a tyrosine phosphorylated STAT1 (1BF5) was used as a template to build the STAT2 homolog structure (Gerhartz and others 1996; Hemmann and others 1996; Scarabelli and others 2009). Figure 11A illustrates the alignment of STAT1 and 2 sequences with a 43.1% sequence identity. The STAT2 homolog structure was superimposed with the STAT1 structure (Fig. 11B) to demonstrate the STAT family similarity and the gaps of STAT1 due to the disordered loops.

Modeling analysis demonstrates the preferred binding of TLR7 and TLR7/8 agonists to STAT2.
There is no information about the specific binding site for the STAT family with small molecules. In order to understand the interactions of 52455 and R-848 with STAT1 and/or STAT2, all probable binding sites within these structures were to be identified and followed up by docking. We detected the binding site cavities of STAT1 and STAT2, respectively, by using the Accelrys program suite. Figure 11C and D illustrate the probable binding sites where small molecules might dock. STAT1 (Fig. 11C) has 5 probable binding cavities with 2 located in the SH2 domain, 1 in the linker domain, 1 in the DNA binding domain, and 1 in the coiled-coil domain. STAT2 (Fig. 11D) has 4 probable binding site cavities–2 equal binding cavities are in the SH2 domain. The other 2 cavities are in the same locations as STAT1. The Scr Homology 2 (SH2) domain is a structurally conserved protein domain that plays a central role in cellular communication. Its length is ∼100 amino acids, and the domain is found within 115 human proteins. SH2 domains typically bind a phosphorylated tyrosine residue, which is from Jak1 in the case of STAT2 or from Tyk2 for STAT1.
Proteins are well known to be flexible and conformationally dynamic. To minimize the computational intensity for this study, the protein targets were held fixed to better understand the causal effects of TLR7 small molecules on the inhibition of the heterotrimeric complex formation via preferred binding of the small molecules to STAT2. Docking calculations were performed for all probable binding sites; however, small molecules such as 52455 and R848 were only bound to the SH2 domain on the same site of either STAT1 or STAT2. Figure 11E shows the predicted binding affinities (-log Ki) for small molecules: 52455 and R-848 bound preferentially to STAT2. Figure 11F and G illustrates the orientations of 52455 with regard to the binding site of STAT1 (Fig. 11F) and STAT2 (Fig. 11G).
In summary, the modeling prediction of these small molecules demonstrates the preferred binding to STAT2. Further computational studies need to be performed to better understand the interactions of these molecules in the context of induced fitting and/or molecular dynamics. Since Biacore binding analyses are impossible due to high signal-to-noise ratios, crystal structure analysis as well as size exclusion chromatography as described for TLR3 (Bell and others 2003, 2005) would prove that this modeling hypothesis of SH2 is the TLR7 small-molecule binding domain.
Discussion
Immune activation by TLRs and RLRs with either nucleic acid ligands or small molecules results in type I IFN induction and is one of the most extensively studied events. However, the combinatorial effects of 2 different ligand types such as nucleic acids and small molecules have not yet been addressed in much detail. Co-stimulation with TLR7 small molecules and TLR9 CpG ODN was expected to result in higher IFN-α release. In contrast, the opposite effect was observed: type I IFN stimulated via TLR9 was suppressed on co-culture with a TLR7 small-molecule ligand (Berghofer and others 2007; Marshall and others 2007). One hypothesis to explain this observation was that small-molecule TLR7/8 ligands would suppress TLR9-induced IFN-α via the direct influence of TLR9 (Berghofer and others 2007; Marshall and others 2007). No other report investigated the combinatorial effects of 2 different ligands, a nucleic acid and small molecule ligand, for other nucleic acid binding receptors such as TLR3, TLR7, or RIG-I. The activation of TLR3, TLR7, TLR9, or RIG-I with either the natural ligand (Krieg and others 1995; Alexopoulou and others 2001; Bauer and others 2001; Krieg 2002; Heil and others 2004; Lund and others 2004; Tabeta and others 2004; Yoneyama and others 2004; Gowen and others 2007; Kalali and others 2008; Faul and others 2010) or its corresponding nucleic acid derivative (Krieg and others 1995; Klinman and others 1996; Alexopoulou and others 2001; Krieg 2002; Diebold and others 2004, 2006; Heil and others 2004; Jurk and others 2004, 2006a, 2006b; Vollmer and others 2004a, 2004c; Forsbach and others 2007, 2008) induces the production of proinflammatory cytokines and chemokines and signal transduction via the type I IFN pathway.
In this study, TLR7/TLR7, TLR3/TLR7, TLR9/TLR7, and RIG-I/TLR7 co-stimulation on human leukocytes and purified pDCs was systematically analyzed for small molecules and nucleic acid ligands. In contrast to earlier reports (Berghofer and others 2007; Marshall and others 2007), we demonstrate that blockage of type I IFN by small-molecule TLR7 agonists such as R-848 or 52455 is not limited to TLR9 nucleic acid ligands. Besides TLR9, also TLR3, TLR7, and RIG-I IFN-α immune responses are blocked by TLR7 small-molecule agonists such as 52455, R-848, Loxoribine, 3M-001, 3M-002, or 3M-003. Type I IFN but no other proinflammatory cytokines or chemokines are blocked by TLR7 small-molecule agonists on human PBMC activation with TLR7/8 ORN, TLR9 CpG ODN, RIG-I 5′ppp dsRNA, or TLR3 dsRNA ligand. Only small molecules that activate the immune system, but not other small molecules, are capable of blocking TLR7-mediated type I IFN responses. Increased apoptosis or potential toxic effects are unlikely to account for the inhibition of type I IFN after co-stimulation (Berghofer and others 2007). Cell viability and apoptosis measurements of human PBMC incubated with ORN R-0006 and TLR7 or TLR7/8 small-molecule agonists did not reveal any differences to the untreated cells (data not shown), and the mRNA levels of housekeeper genes are comparable among untreated cells and cells with ORN R-0006 and TLR7 or TLR7/8 small-molecule agonists.
Cross-titration and kinetic experiments imply competitive inhibition between small molecules and nucleic acid ligands, as well as a kinetic advantage of small molecules to reach the endolysosomal compartments. Competitive inhibition seems to occur not at the TLR or RLR receptor site but at the type I IFN pathway, and here at the STAT2 phosphorylation site. Adding a TLR7 small-molecule ligand to the cell culture 4 h after the stimulatory nucleic acid resulted in a loss of IFN-α suppression. Kinetic differences occurred only at 3 and 4 h. Small molecules and TLR3, TLR7, TLR9, or RIG-I synthetic nucleic acid ligands differ in their molecular weight, with the TLR7 small-molecule ligands (eg, with a molecular mass of 313.36 g/mol) being considerably smaller than the TLR7 nucleic acid ligands (eg, with a molecular mass of 6,600 g/mol). This implies that the TLR7 small-molecule agonists occupy the PRR or the type I IFN pathway better than the nucleic acids do even in unfavorable kinetic circumstances.
Besides IFN-α, other type I IFN response genes such as IFIT1, IRF7, ISG15, ISG20, Mx1, Mx2, OAS1, and OAS2 are blocked by TLR7 small-molecule ligands, whereas type II IFN remains unaffected. We hypothesized and demonstrated that blockage at the level of formation of the heterotrimeric ISGF3 complex in the form of inhibition of the STAT2 phosphorylation is the reason for TLR3, TLR7, TLR9, and RIG-I type I IFN blockage by TLR7 small molecules. Here, the stronger binding affinity of the small molecule agonist to the binding site at STAT2 would be a possible explanation. The direct binding of TLR7 and TLR7/8 small-molecule agonists to, for example, TLR9 nucleic acid agonists was excluded using Biacore experiments.
Modeling of STAT1 and STAT2 hypothesized that R-848 and 52455 preferentially bind to STAT2 and here to the SH2 domain, which is also known as the phosphorylation site. Crystallization as well as size exclusion chromatography of STAT2 would be needed for final proof of this hypothesis. So far, these techniques have been described for the identification of the TLR3 binding domain (Bell and others 2003, 2005). Biacore binding studies are excluded for this experimental setting due to the high signal-to-noise ratio for the high-molecular-weight STAT2 protein and the low-molecular-weight TLR7 small molecule.
Blockage of the STAT2 phosphorylation at the SH2 domain interferes with the formation of the heterotrimeric ISGF3 complex and downstream signaling of type I IFN. SH2 domains are protein modules found in many proteins involved in the binding of phosphorylated sequences and activation of downstream signaling. So far, only 3 SH2 domains have been directly implicated in human diseases: in SAP [SLAM (signaling lymphocyte activation molecule)-associated protein] causing X-linked lymphoproliferative disease (Coffey and others 1998; Nichols and others 1998), in Brutons tyrosine kinase causing X-linked agammaglobulineamia (Saffran and others 1994), and in Noonan's syndrome causing autosomal dominant disorder (Tartaglia and others 2001). Although examples of the direct implications of SH2 domains in diseases are a few, several drug targets, including cancer, allergy, AIDS, and osteoporosis, use the SH2 domain for the development of clinical small-molecule inhibitors. In these cases, the small-molecule inhibition of STAT and, especially, STAT3 phosphorylation is the key focus in human cancer (Borges and others 2008; Lin and others 2010a, 2010b; Fletcher and others 2011). Activation of the immune system has not been described for SH2 STAT3 inhibitors. To date, no study has been performed using STAT2 SH2 blockage in human diseases. Our data suggest the binding of R-848 or 52455 to the SH2 phosphorylation site of STAT2 and, therefore, blockage of the type I IFN pathway of the PRRs TLR3, TLR7, TLR9, RIG-I, and Mda-5. This implies the dual immune effects of TLR7 and TLR7/8 small-molecule agonists—depending on the concentration range—on the activation and inhibition of the immune system. These results will have important impacts in clinical research and on the development of small-molecule TLR7/8 agonists. The therapeutic potential of synthetic TLR7 ligands such as 9-substituted-8-hydroxyadenine derivates has been demonstrated in experimental animal models in infectious as well as autoimmune diseases or as adjuvants to vaccines against cancer (Thomas and others 2007; Dharmapuri and others 2009; Hayashi and others 2009). R-848 is in clinical use as a topical cream for the treatment of skin lesions such as those caused by the herpes simplex virus and as an adjuvant that increases the effectiveness of vaccines (Tomai and others 1995, 2000, 2007; Wu and others 2004, 2007; Fife and others 2008; Szeimies and others 2008). The dual in vivo and in vitro effects of synthetic TLR7 or TLR7/8 small molecules are as follows: Activation and inhibition of the type I IFN depending on the specific environment and conditions can cause unwanted side effects. Type I IFN immune response is one of the strongest antitumor factors for TLR ligands in cancer that negatively regulates tumor outgrowth and activates the tumor immunosurveillance process (Brassard and others 2002; Picaud and others 2002; Thyrell and others 2002; Takaoka and others 2003; Dunn and others 2005; Wenzel and others 2005). Blockage of type I IFN by very low concentrations of TLR7/8 small-molecule agonists may be sufficiently effective in cancer and may cause potentially progressive disease. On the other hand, the observation of type I IFN blockage by specific TLR7/8 small-molecule agonists may help in the development of drugs against autoimmune diseases such as lupus or rheumatic arthritis.
In summary, we show that small-molecule TLR7/8 agonists act, on the one hand, simialr to classical agonists, but, on the other hand, they may potentially act as antagonists that suppress type I IFN-mediated immune effects induced by bacteria or viruses via TLRs or RLRs. The clinical development of TLR7 and TLR7/8 small molecules, therefore, needs to be well controlled to avoid potential unwanted side effects such as an inhibitory effect on the immune system.
Footnotes
Author Disclosure Statement
No competing financial interests exist.