
Abstract
Recent advances in our understanding of foreign nucleic acid sensing indicate an important role for the human Toll-like receptor (TLR) 8 in the initiation of immune responses to certain pathogens. However, TLR8, far too often grouped together with TLR7 for its common ability to detect RNA, has a function on its own in the initiation of specific proinflammatory responses to viruses and bacteria. Here, we present an overview of what is currently known of human TLR8 biology, from genetic regulation to its function in innate immunity, and discuss how TLR8 could present novel therapeutic opportunities in viral and cancer diseases.
Introduction
Human TLR8 Tissue Expression
Cell-specific expression and signal transduction
Original studies investigating the expression of TLRs in several human tissues showed that TLR7 and 8, although sharing a similar structure, genomic localization, and involvement in RNA sensing, are predominantly found in distinct peripheral blood lymphocyte subsets, with a preference for plasmacytoid dendritic cells (pDCs) and B cells for TLR7, and monocytes for TLR8 (Hornung and others 2002; Zarember and Godowski 2002). Genome-wide studies across 79 human tissues have since confirmed this and also demonstrated that pDCs (BDCA4+), although primarily expressing TLR7, also express TLR8, while monocytes (CD14+), although primarily expressing TLR8, also express TLR7 (Su and others 2004). This selective expression of human TLR7 and 8 in two distinct immune cell populations underlines their distinct biological function, given that pDCs will predominantly produce antiviral interferon-α (IFN-α), while monocytes/macrophages produce high levels of proinflammatory cytokines such as tumor necrosis factor α (TNF-α) or interleukin-12 (IL-12) (Gorden and others 2005). As such, TLR7 recruitment favors the generation of an antiviral state (through IFN signaling), while TLR8 activation favors a more Th1 cell-mediated antimicrobial immune response (Gorden and others 2005).
Human natural killer cells (NK cells) have also been shown to become responsive to TLR8 ligands, when cocultured with IL-12p70-producing monocytes and IL-18-producing macrophages (Hart and others 2005; Gorski and others 2006). RNA stimulation of human TLR8 in monocytes promotes production of IL-12p70 (Ablasser and others 2009), which presumably upregulates TLR8 expression in NK cells [where the basal expression of TLR8 is very low compared to monocytes (Su and others 2004; Gorski and others 2006)], and renders those cells responsive to TLR8 agonists. Critically, TLR8 recruitment induces production of IFN-γ and NK cell cytotoxicity to a greater extent than that of TLR7 activation (Gorski and others 2006).
In humans, neutrophils are the most abundant immune cells in blood and rapidly migrate to the site of infection. Neutrophils express low levels of TLR8 compared to monocytes (Hornung and others 2002; Hayashi and others 2003), but have been shown to respond to TLR8 agonists and induce IL-8 production when pretreated with the granulocyte/macrophage colony-stimulating factor (Hayashi and others 2003; Hattermann and others 2007; Wang and others 2008).
Human dendritic cells (DCs) also express and are responsive to TLR8 ligands, and this property is subverted by the human immunodeficiency virus type 1 (HIV-1) to activate p65 and viral transcription initiation (Gringhuis and others 2010).
Intracellular location and downstream signaling
TLR7 and 8 are located in intracellular endosomal/lysosomal compartments, as are TLR3 and 9 (Fig. 1). This localization is critical in limiting their function to the detection of phagocytosed ribonucleic acids, thereby preventing detection of endogenous cellular RNAs. Like TLR3, 7, and 9, TLR8 can interact with UNC93B1 to potentiate its function, presumably by regulation of shuttling to the endoplasmic reticulum and early endosome (Itoh and others 2011). TLR8 is comprised of an ectodomain of 25 leucine-rich repeats, a transmembrane domain, and a TIR domain, and low pH is required for its function (Gibbard and others 2006). This last property is critical to TLR8 function, which can be ablated using chemical inhibitors of endosomal acidification (such as chloroquine) (Sioud 2005; Zamanian-Daryoush and others 2008).
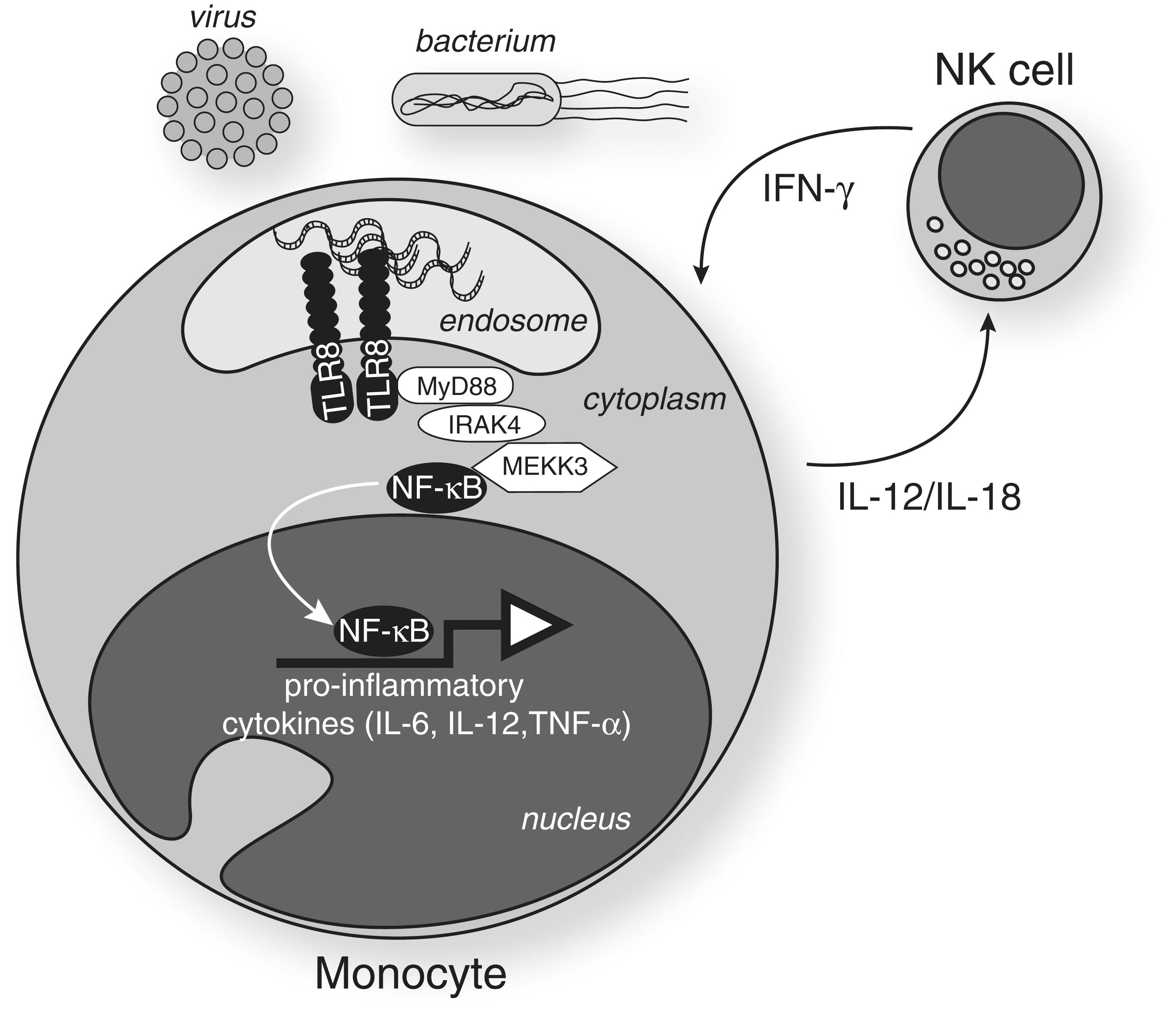
RNA sensing by the Toll-like receptor (TLR) 8. Following phagocytosis and endosomal degradation of bacteria and viruses, foreign RNA binds to TLR8—possibly in the form of a homodimer (Gibbard and others 2006). The conformational change results in the recruitment of MyD88 to the TIR domain of TLR8. Activation of components of the downstream signaling cascade (e.g., IRAK4 and MEKK3) promotes nuclear translocation of the nuclear factor κB (NF-κB) and the subsequent induction of proinflammatory cytokines. The figure also shows the positive regulatory loop that exists between natural killer (NK) cells and monocytes/macrophages, which potentiates TLR8 sensing. Interleukin (IL)-12 and IL-18 secreted by monocytes/macrophages induce the expression of TLR8 and the responsiveness of NK cells to TLR8 ligands. Engagement of NK cells by TLR8 ligands results in the secretion of interferon (IFN)-γ, which in turn activates monocytes and induces further TLR8 expression through STAT-1 binding to IFN-gamma activated sequence (GAS) elements of the TLR8 promoter (see Fig. 2).
Following TLR8 engagement, MyD88 is recruited to the TIR domain of TLR8. Downstream activation of a chain cascade involving components such as IRAK4 (Yang and others 2005), IRAK2 (Flannery and others 2011), and TRAF6 (Keating and others 2007) eventually promotes activation of nuclear factor κB (NF-κB), JNK, and interferon regulatory factor (IRF) 7, which induce many cytokines including IFN-β, IL-1β, IL-6, IL-10, and TNF-α (Qin and others 2006; Cervantes and others 2011) (Fig. 1). Patients with deficiencies in IRAK4, a kinase downstream of MyD88, lacked the induction of IFN-α/β/λ in response to TLR7/8 ligands (Yang and others 2005), demonstrating a key role for IRAK4 in both TLR7 and 8 signaling. Importantly, TLR8-induced NF-κB and JNK activation appear to be MEKK3 dependent and TAK1 independent (Qin and others 2006), unlike the canonical IL-1 signaling pathway, which only moderately relies on MEKK3 (Fig. 1).
Genetic Regulation of TLR8 Expression
TLR8 chromosomal location
In humans and many other species, including mouse, TLR7 and 8 are located on chromosome X (in humans, specifically in region Xp22) (Chuang and Ulevitch 2000) (Fig. 2). The location of TLR8 on the X chromosome is 16 kb proximal from TLR7 (Edwards and others 2008). Notably, in some species of fish, such as the zebrafish (Danio rerio), TLR8 is expressed on chromosome 10. Genome-wide scanning studies have shown that genetic variations in chromosome Xp22 are linked to the risk of asthma development (Brasch-Andersen and others 2008). Further studies on asthmatic individuals revealed a significant association of single-nucleotide polymorphisms (SNPs) in TLR7 and 8 with asthma, rhinitis, atopic dermatitis, and increased levels of specific IgE (Moller-Larsen and others 2008).
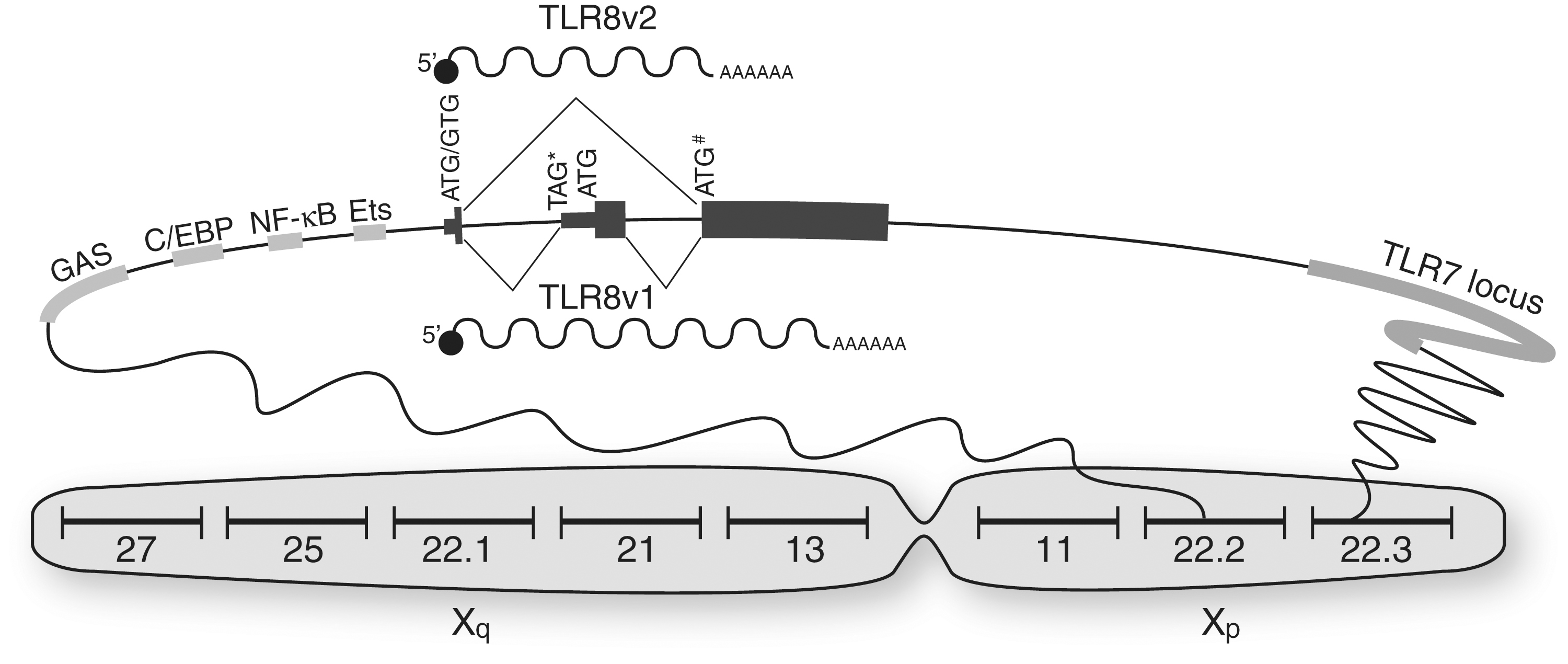
Genetic regulation of TLR8 function. TLR8 is encoded in the Xp22.2 locus of chromosome X (several other regions are indicated for visual reference). The figure focuses on the genomic region between Xp22.2 and Xp22.3, where TLR8 and TLR7 are located. The promoter of TLR8 bears several binding sites for specific transcription factors such as STAT-1 (GAS elements), C/EBPδ (C/EBP sites), p50/65 (NF-κB sites), and Ets-1 (Ets sites). Splicing of the two TLR8 variants occurs at the level of the translation start site of TLR8v2. TLR8v1, by virtue of its additional exon, bears an in-frame stop TAG* site, which postpones the initiation of translation of TLR8v1 to a downstream ATG site. Therefore, TLR8v1 has an identical amino acid sequence to TLR8v2, but with an additional 19 amino acids at the N-terminus. The rs3764880 SNP changes the ATG initiation codon of TLR8v2 to a GTG (indicated by ATG/GTG). However, an in-frame ATG site in exon 2 of TLR8v2 resumes TLR8v2 translation (symbolized by ATG#). Thus, the GTG allele of rs3764880 only truncates the N-terminus of TLR8v2 by three amino acids (Gantier and others 2010a).
TLR8 splice variants
The TLR8 gene locus encodes for two splice variants referred to as TLR8 variant 1 (TLR8v1) and TLR8 variant 2 (TLR8v2) (Fig. 2). The longest transcript, TLR8v1, contains an additional exon encoding for an alternative transcriptional start site region to that of TLR8v2 (Fig. 2). This results in the addition of 19 amino acids at N-terminus of TLR8v1 compared to TLR8v2. Unlike TLR8v2, which appears to be the predominant form of TLR8 expressed in human monocytes (Zannetti and others 2010; Gantier and others 2010a), TLR8v1 is not evolutionarily conserved and is only found in higher primates, including rhesus, orangutan, chimp, and human (Gantier and others 2010a). TLR8v1 has been proposed to contribute to TLR8 function by positively regulating TLR8 signaling in specific cell populations, such as in mixed CD16+/CD14+ and CD16+/CD14dim cell populations (Gantier and others 2010a). Interestingly, CD16+/CD14dim monocytes have recently been shown to be a critical subset of monocytes that patrol the endothelium of blood vessels, and are the prime producers of TNF-α and IL-1β following TLR7/8 activation by viral nucleic acids (Cros and others 2010).
TLR8 polymorphisms
In addition to being the convergence point of alternative splicing of TLR8v1 and TLR8v2, the initiation codon of TLR8v2 is subject to important genetic variation (Fig. 2). A SNP (reference rs3764880) changes the first TLR8v2 ATG into a GTG codon. In humans, the GTG allele of rs3764880 SNP varies widely according to ethnicity and was observed to be as high as 85% among the Japanese population, and as low as 18% among individuals of Italian descent (Pickrell and others 2009). While the GTG allele of rs3764880 SNP results in a shorter TLR8v2 protein, this variant is only marginally shorter (it lacks three amino acids) and our experimental studies in stable cells overexpressing these two variants of TLR8v2 did not show any conclusive functional differences between them (Gantier and others 2010a). Nevertheless, the striking variation among populations suggests a possible role for rs3764880 in the modulation of TLR8 signaling and function. To date, the outcomes of three different infectious diseases have been correlated to the rs3764880 SNP (Davila and others 2008; Oh and others 2008; Engin and others 2010). For example, Davila and colleagues (2008) demonstrated that the G allele of rs3764880 correlated with protection against the progression of tuberculosis following Mycobacterium tuberculosis infection (Davila and others 2008). Although the mechanisms by which rs3764880 contributes to disease development are likely to be disease specific, our work implicates rs3764880 in the fine-tuning of TLR8v1 and TLR8v2 protein translation, through the regulation of 3′ upstream open reading frames and Kozak context variations (Gantier and others 2010a). In addition to rs3764880, there are 263 SNPs currently known for TLR8 that could also regulate its transcription, splicing, or function.
TLR8 transcriptional regulation
There is limited knowledge about TLR8 transcriptional regulation. This is, however, a critical component of TLR8 function, as it restricts TLR8 expression to very few cell types (such as monocytes, pDCs, NK cells, and neutrophils), as previously mentioned. In accordance with this, the core of the TLR8 promoter, in common with myeloid-type promoters, lacks a TATA box that contributes to efficient unwinding of DNA (Zannetti and others 2010). The original studies investigating TLR8 expression in monocytic-like THP-1 cells demonstrated that TLR8 mRNA levels were induced by phorbol 12-myristate 13-acetate (PMA) treatment and that was potentiated further following IFN-γ pretreatment of these cells (Zarember and Godowski 2002). It is noteworthy that while the effect of PMA treatment on TLR8 expression was most likely related to NF-κB regulation (as seen with other TLRs), IFN-γ pretreatment of PMA-activated THP-1 resulted in the specific upregulation of TLR8 and, to a lesser extent, TLR7 (Zarember and Godowski 2002). IFN-γ upregulation of TLR8 has also been shown in primary monocytes (Gantier and others 2008), and underlines a critical feedback loop between NK cells and monocytes, in TLR8 signaling. As such, following their prior activation by monocyte-secreted IL-12, NK cells feedback into TLR8 signaling through IFN-γ production, which upregulates TLR8 levels in circulating monocytes (Fig. 1). Mechanistically, IFN-γ exerts its effect on TLR8 expression through STAT-1 binding to four IFN-gamma activated sequence elements in the TLR8 promoter (Zannetti and others 2010) (Fig. 2). Both TLR8 variants have been shown to be similarly induced by IFN-γ and therefore appear to be under the same transcriptional control (Gantier and others 2010a; Zannetti and others 2010).
In silico analyses of a 3.5-kb promoter region upstream of the TLR8 mRNA start codon identified the potential binding of several important transcription factors involved in innate immunity, including NF-κB, Ets, STAT, IRF, and C/EBP responsive elements (Zannetti and others 2010) (Fig. 2). Nevertheless, most of the associated transcription factors did not have any impact on TLR8 expression when overexpressed with the 3.5-kb TLR8 promoter reporter in THP-1 cells, with the exception of C/EBPδ and C/EBPβ [with a >10-fold impact of C/EBPδ over C/EBPβ (Zannetti and others 2010)]. Although the exact transcriptional regulation of TLR8 by C/EBPδ is currently unknown, it is likely that C/EBPδ recruits the histone acetylase CREB-binding protein by binding its promoter, leading to the chromosomal opening and nucleosome remodeling (Weinmann and others 2001; Kovacs and others 2003; Zannetti and others 2010). In addition, NF-κB heterodimers p65/p50 and c-Rel/p50 also modulated TLR8 expression, confirming a role for NF-κB in TLR8 expression (Zannetti and others 2010). This is consistent with the previous observations that stimuli activating NF-κB (TNF-α, IL-1β, and lipopolysaccharide) resulted in TLR8 induction in PMA-activated THP-1 cells (Zarember and Godowski 2002; Gantier and others 2008). Finally, overexpression of Ets-1 was also shown to upregulate expression of a 1-kb TLR8 promoter reporter in HEK293 cells (Gantier and others 2010a). Although many transcription factors tested by Zannetti and others (2010) did not affect TLR8 expression in reporter experiments in THP-1 (Zannetti and others 2010), it is likely that other transcription factors in addition to C/EBPδ, Ets-1, and NF-κB heterodimers are at play in the cell-specific fine-tuning of TLR8 expression.
TLR8 Ligands
TLR8 chemical agonists
Before the discovery of their natural ligands, single-stranded (ss) RNA, TLR7, and 8 were found to be activated by nucleoside analogs such as imidazoquinolines, previously known for their antiviral activity (Hemmi and others 2002; Jurk and others 2002; Heil and others 2003). Here these will be referred to as chemical agonists. Although several chemical agonists with selective activity for TLR7 were rapidly identified (imiquimod [R-387], loxoribine, and S-27609), exclusive chemical ligands for TLR8 were originally not described and studies were limited to the use of R-848 (or resiquimod), which displayed both TLR7 and TLR8 activity (Hemmi and others 2002; Jurk and others 2002; Doxsee and others 2003; Heil and others 2003). Further development of chemical ligands with selective activity for TLR8 (3M002; also known as CL075) or TLR7 (3M001) confirmed that selective TLR7 and TLR8 activation was related to the induction of different cytokine profiles in human peripheral blood mononuclear cells (PBMCs) (Gorden and others 2005). This nonreciprocal activation of TLR7 and TLR8 by chemical ligands suggested that different natural ligands could also selectively activate one or both receptors, thereby modulating the immune response to the pathogen detected.
TLR8 sensing of RNA
Early observations suggested that unlike in mice, where TLR7 was found to be the sensor of uridine-containing ssRNA in knock-out studies, human TLR8 was the sensor of ssRNA (Diebold and others 2004; Heil and others 2004). This impression relied on the lack of responsiveness of human TLR7 to ssRNAs in overexpression studies in HEK293 cells (Heil and others 2004). Nevertheless, human TLR7 directly participates in ssRNA sensing in human THP-1 cells (Gantier and others 2008) and was also found to respond to various stabilized RNA molecules (with phosphorothioates, or deazaguanosine moieties) in TLR7-expressing HEK293 cells—suggesting that the stability of ssRNAs is an important parameter in this system (Lan and others 2007; Forsbach and others 2008). These findings thus established that RNA is the natural TLR7/8 ligand.
Although primarily sensing ssRNAs, TLR7/8 have also been implicated in sensing of short double-stranded (ds) RNAs such as ∼21-bp duplexes (small interfering RNAs; siRNAs) used in RNA interference (RNAi) (Hornung and others 2005; Judge and others 2005; Sioud 2005, 2006). Crucially, these studies demonstrated important variations of responses between sequences, identifying uridine-rich motifs coined “immunostimulatory motifs” (GU rich) for their ability to induce strong IFN-α or TNF-α production by immune cells. Although uridines are critical for immune sensing by TLR7 and 8 (Sioud 2006; Gantier and others 2008), both their location in the sequence as well as the number of these immunostimulatory motifs contribute to the level of immunostimulation (Diebold and others 2004; Heil and others 2004; Gantier and others 2008). In addition, studies with siRNAs and their single-stranded components suggested that different sequences could induce varying levels of IFN-α or TNF-α, independently of one another (Forsbach and others 2008; Gantier and others 2008; Zamanian-Daryoush and others 2008). This led to the understanding that TLR7 and TLR8 recognize different motifs, with TLR8-specific recognition of AU motifs (Forsbach and others 2008; Gantier and others 2008).
While specific uridine motifs can indeed modulate the selective recruitment of TLR7 or 8 (Forsbach and others 2008; Gantier and others 2008), it is hard to delineate whether immune stimulation directly relates to these motifs, or their impact on inter- and intra-molecular secondary structures. Studies have shown that annealing of anticomplementary oligoribonucleotides with an inherent immunostimulatory activity strongly reduced TNF-α/IL-12 production, but IFN-α less so (Zamanian-Daryoush and others 2008; Ablasser and others 2009), thereby suggesting a stronger impact of the secondary structure of RNAs on TLR8 sensing. We have shown that localization of uridines within various regions of such self-secondary structure has a strong impact on TLR8 sensing (Gantier and others 2008) and that variation of the self-secondary structure of ssRNAs plays a role in TNF-α, but not IFN-α production (Gantier and others 2010b). It can therefore be postulated that TLR8 more specifically senses ssRNAs through the ability of ssRNAs to form intramolecular structures and expose selective immunostimulatory motifs, which is prevented when two complementary oligoribonucleotides are used (Eberle and others 2008; Ablasser and others 2009; Gantier and others 2010b).
Endosomal delivery of TLR8 RNA agonists
Synthetic RNAs—be it ssRNAs or siRNAs—require some form of modification or duplexation with a transfection reagent to reach the cell endosomal compartment and activate TLR8. Naked ssRNAs are rapidly degraded after in vitro addition to cultured cells or in vivo injection (Lan and others 2007). Delivery vehicles provide the RNAs with ribonuclease protection and enhanced cellular uptake, which are crucial for efficient immunostimulation (Judge and others 2005). Many commercial delivery reagents are available with varying efficacy in ssRNA delivery to the endosomal compartment and associated TLR7/8 activation. Most delivery systems consist of lipids, polymers, or dendrimer-based reagents. DOTAP (Roche), ESCORT (Sigma), and Lipofectamine (Life Technologies) are lipid-based transfection reagents that preferentially deliver ssRNA to the endosomal compartment (Sioud 2005; Zamanian-Daryoush and others 2008; Forsbach and others 2011). Conversely, HiPerfect (Qiagen) and Dharmafect 1 (Thermo Scientific) are other lipid-based delivery reagents that are preferentially used for cytosolic delivery—following endosomal release of ssRNAs (Forsbach and others 2011). Because they are preferentially targeted to the endosome, DOTAP-duplexed RNAs induce higher immune responses than Dharmafect and HiPerfect-duplexed RNAs (Forsbach and others 2011). Polymer-based reagents such as poly-L-lysine (Sigma) can also elicit immune responses, through endosomal retention of the RNAs (Ablasser and others 2009; Forsbach and others 2011). The dendromer-based transfection reagent Superfect (Qiagen) can also promote immune activation (Forsbach and others 2011). Interestingly, ssRNA delivery with ESCORT (which contains a mix of DOTAP and DOPE—dioleoyl phosphatidylethanolamine) in human PBMCs selectively resulted in strong TNF-α production, but not IFN-α production, while both cytokines were induced by the same ssRNA sequence complexed with DOTAP (Forsbach and others 2011). Although warranting further validation, these results suggest that DOPE may inhibit TLR7-driven innate immune activation through limited pDC targeting, and preferential monocyte uptake.
In addition to the protective effect of transfection reagents, the presence of specific motifs in the RNA sequence can modulate its vulnerability to degradation by ubiquitous ribonucleases. For example, the presence of UA and CA dinucleotides in the sequence can increase susceptibility to endonuclease degradation (Lan and others 2007). This process is initiated by 3′ exonucleases. One strategy proposed to overcome this nuclease activity involves the linkage of two phosphorothioate RNA sequences at their 3′-end through a glycerol linker (Lan and others 2007). Such molecules can specifically activate TLR8 and were shown to induce transient immune stimulation when injected naked into nonhuman primates (Lan and others 2007; Kandimalla and others 2011).
Activation of TLR8 by its ligands
Although TLR8 is present in both the endoplasmic reticulum and early endosome, it is activated only in the early endosome, in a pH-dependent process (Gibbard and others 2006; Itoh and others 2011). As such, TLR8 activation by its ligands occurs by acidification of the endosomal compartment following phagocytosis of the ligands (ribonucleic acids and chemical agonists). This is thought to lead to conformational changes in the TLR8 ectodomain and receptor homodimerization, presumably through the ionization of histidine side chains (Gibbard and others 2006). Similar to TLR3, ligand-induced TLR8 dimerization is thought to result in TLR8–TIR/MyD88 interaction and downstream signal transduction (Govindaraj and others 2011). Whether RNA and chemical agonists directly bind to TLR8 to activate it remains poorly defined. It is possible that TLR8 recognition of its ligands is facilitated by other proteins, such as high-mobility group box proteins (Yanai and others 2009). Noteworthy, the endogenous level of TLR8 is likely to be a critical factor in its dimerization. Indeed, overexpression of TLR8 leads to TLR8 complexes that appear to self-associate, independent of stimulation (Zhu and others 2009; Liu and others 2010; Gantier and others 2010a). In addition to homodimerization, there is evidence that TLR8 can bind to TLR7 to form heterogenous complexes (Wang and others 2006). This association between TLR7 and TLR8 was shown to repress TLR7 sensing of its specific ligand in HEK293 cell overexpression studies (Wang and others 2006), suggesting a functional regulation by TLR8 of TLR7 sensing. In agreement with this, cytokine-induced modulation of the ratio of TLR7 to TLR8 in human THP-1 cells resulted in the sensing of different ssRNA sequences (Gantier and others 2008). Furthermore, these studies revealed the requirement for TLR7 in the detection of all ssRNAs studied, including those with a preference for TLR8 (Gantier and others 2008), thus highlighting a complex functional interplay between both receptors.
TLR8 Function in Mice
Early studies with imidazoquinolines demonstrated that mice lacking Tlr7/Myd88 did not respond to immiquimod or resiquimod (R-848) (Hemmi and others 2002), suggesting common sensing of these two chemical compounds in mice—through Tlr7. However, overexpression studies in human embryonic kidney cells (HEK293) showed that R848 could also activate human TLR8, but not its murine homolog (Jurk and others 2002), indicating that species-specific differences existed between human and mouse TLR8. Critically, these findings also suggested that the mouse Tlr8 did not respond to the same ligands as human TLR8.
In accord with this, further studies in Tlr7 −/− mice demonstrated that mouse Tlr8 was not directly required to sense RNA (Diebold and others 2004; Heil and others 2004; Hornung and others 2005). Indeed, while uridine-rich short synthetic RNAs elicited a strong immunostimulation in mouse pDCs and macrophages and in vivo, this response relied entirely on Tlr7 (Diebold and others 2004; Heil and others 2004; Hornung and others 2005). Furthermore, ssRNAs specifically activating human TLR8 over TLR7 did not activate downstream cytokine production in mouse macrophages or in HEK293 cells overexpressing mouse Tlr8 (Forsbach and others 2008; Gantier and others 2008). Taken together, these findings led to the conclusion that TLR8 was not functional in mice.
Conversely, however, following the report that poly(dT) oligodeoxynucleotides (ODNs) could favor sensing of imidazoquinolines by human TLR8 (Gorden and others 2006a), Gorden and others (2006b) demonstrated that conjugation of poly(dT) ODNs with TLR7/8 (3M-003) or TLR8 (3M-002) chemical agonists resulted in immune activation in HEK293 cells overexpressing mouse Tlr8 or in mouse PBMCs derived from Tlr7 −/− mice. Similar observations were also made with poly(dT) ODNs conjugated to TLR8-specific ssRNA (Forsbach and others 2008), indicating that mouse Tlr8, although not directly responsive to ssRNA, could mediate downstream signaling when the ssRNA is combined to poly(dT) ODNs. It has recently been proposed that residues (RQSYA) located in the region directly following the leucine-rich repeat 14 of TLR8, which are lacking in mouse and rat TLR8, are critically required for human TLR8 responses and could explain these species differences (Liu and others 2010).
These findings raise the question as to what is the biological significance of Tlr8 in mouse—and that of the poly(dT) ODN-mediated immune response. One possible explanation is that mouse Tlr8 may be involved in the detection of DNA viruses—which would combine both DNA and RNA agonists. In support of this, a recent study has shown that the dsDNA vaccinia virus (VV), which is rich in poly A/T regions, can activate murine Tlr8 (Martinez and others 2010). Tlr8-expressing HEK293 cells responded to VV in a Tlr8-specific fashion. Moreover, primary murine pDCs failed to promote IFN-α production with both VV and its purified DNA when Tlr8 was downregulated by RNAi (Martinez and others 2010). Conversely, sensing of human adenovirus type 5, which is not enriched in A/T regions, was not affected by Tlr8 RNAi in murine pDCs, but rather relied on Tlr9 (Martinez and others 2010). While these results suggest that murine Tlr8 might be functional in the context of A/T-rich DNA viruses such as VV, this remains controversial (Bauer and others 2010), and further work in Tlr8 −/− mice will be required to tease out the possible direct involvement of mouse Tlr8 in VV sensing.
As mentioned above, unlike mouse Tlr7, mouse Tlr8 does not respond to ssRNA or its associated chemical agonists alone. Nevertheless, there is evidence that mouse Tlr8 directly modulates Tlr7 function, similarly to human TLR8. Indeed, mouse Tlr8 was found to inhibit Tlr7 sensing of the chemical Tlr7 agonist 3M-001 in HEK293 overexpression studies (Wang and others 2006). This is in agreement with recent studies in Tlr8 −/− mice, suggesting that Tlr8 closely modulates the activity and expression of Tlr7 (Demaria and others 2010). Bone marrow-derived DCs from Tlr8 −/− mice displayed increased TNF-α/IL-12/IL-6 production following Tlr7 activation by R848, suggesting enhanced responsiveness to Tlr7 ligand in the absence of Tlr8. Whether such a role for TLR8 is important in humans remains to be determined, but the coexpression (although not in a 1:1 ratio) of both receptors in several cell types suggests that it could also be an important aspect of TLR8 function.
TLR8 in Viral Detection
Localization of TLR8 in the endosomal compartment safeguards against the sensing of self-cellular RNA [which can also activate TLR8 as seen in human monocyte-derived DCs (Kariko and others 2005)]. In the context of viral infection, some viral RNA can reach the endosomal compartments via clathrin-coated vesicles and thus activate TLR8, as seen with human parechovirus 1 (Joki-Korpela and others 2001; Triantafilou and others 2005a). Initial studies of influenza infection established that TLR7 was critical in the pDC-driven production of IFN-α (Diebold and others 2004). In addition, a GU-rich synthetic oligoribonucleotide mimicking the U5 region of the HIV-1 RNA genome resulted in TLR8 activation (Heil and others 2004). Several additional viruses have now been shown to be sensed by TLR7 and TLR8, including human parechovirus 1 (Triantafilou and others 2005a), Sendai virus (Melchjorsen and others 2005), influenza A virus (Wang and others 2008), coxsackievirus (Triantafilou and others 2005b), VV (Martinez and others 2010), measles virus (Clifford and others 2011), and respiratory syncytial virus (RSV) (Bendelja and others 2010). Nevertheless, most of these studies did not selectively delineate between the contributions of TLR7 and TLR8 in the innate immune response to these viruses. The recent finding that TLR8 sensing is usurped by HIV-1 for its own replication purposes directly demonstrates that TLR8, independently of TLR7, can play a critical role in viral sensing (Gringhuis and others 2010). As previously mentioned, there is also evidence that VV could activate TLR8 independently of TLR7 in mouse (Martinez and others 2010).
Viral evasion of antiviral pathways underlines the importance of such pathways in the immune response to viral infections. In accord with a direct role for TLR8 in antiviral responses, TLR8 downregulation was observed in PBMCs following RSV infection, which is responsible for lower respiratory tract infection in infants (Bendelja and others 2010). This was concurrent with lower TNF-α levels in the blood of these patients following TLR8 stimulation (Bendelja and others 2010). Depletion of macrophages is associated with increased RSV infection, indicating that the macrophage activity limits RSV infection (Soukup and Becker 2003; Pribul and others 2008). Furthermore, there is a strong negative correlation between the TLR8 expression level in monocytes and severity of RSV infection (Bendelja and others 2010). Based on these observations, it is tempting to speculate that TLR8 might be an essential component of the antiviral response to RSV, and is targeted for inhibition by the virus.
Detection of Bacterial RNA
Although originally thought to be restricted to the detection of viral RNA, there is now growing evidence that TLR7 and 8 also play an important role in the detection of bacterial RNA. Both TLR7 and 8 can be activated by bacterial RNA much more potently than by human RNA (Kariko and others 2005). This relates to the fact that human RNA contains more frequent nucleoside modifications than bacterial RNA. For instance, human ribosomal RNA (which represents ∼80% of human total RNA) contains 25 times more 2′-O-methylated (2′-Ome) nucleosides and 10 times more pseudouridines than bacterial ribosomal RNA (Kariko and others 2005). Such modified nucleosides dampen TLR7 and 8 sensing of RNA, thereby limiting sensing of phagocytosed self-RNA (Kariko and others 2005).
By reason of its specific high expression in monocytes/macrophages, TLR8 activation—like that of TLR2 or TLR4—predominantly results in the induction of proinflammatory cytokines (TNF-α, IL-6, and IL-12) to favor a cell-mediated antimicrobial immune response (Gorden and others 2005). TLR8 would therefore be expected to be a critical component of bacterial detection by the immune system. This was first suggested by a genetic study analyzing 149 polymorphisms across all known TLRs and associated adaptor proteins in patients with active tuberculosis (TB) due to Mycobacterium tuberculosis (Davila and others 2008). TLR8 polymorphisms were the only variations significantly associated with disease progression of TB (Davila and others 2008). In addition, TLR8 mRNA was upregulated in patients with active TB (Davila and others 2008). Crucially, M. tuberculosis parasitizes the macrophage phagosomal compartment by blocking the phagosome–lysosome maturation (Vergne and others 2004). This property is not exclusive to M. tuberculosis and many other bacteria are able to survive intracellularly when phagocytosed by macrophages, by impeding phagosomal maturation (Gantier and others 2010a). For example, we showed that TLR8 signaling is induced in response to Helicobacter pylori phagocytosis by human monocytic THP-1 cells (Gantier and others 2010a).
These findings suggest that TLR8 contributes to the sensing of phagocytosed bacteria. However, given the intracellular localization of TLR8, it could be argued that such a contribution of TLR8 to bacterial sensing is only secondary given that extracellular bacterial sensors such as TLR2 or TLR4 would be first recruited. In a recent publication, TLR2 and TLR8 were found to cooperate in the monocytic detection of the pathogen Borrelia burgdorferi, the spirochetal agent of Lyme disease (Cervantes and others 2011). Cell surface TLR2 initially detected B. burgdorferi, resulting in phagocytosis of the bacterium together with translocation of TLR2 into the phagosomal compartment of the monocyte. TLR8 was subsequently recruited and colocalized with TLR2 and the bacterium in the phagosomal vacuole, where the bacterium was rapidly degraded. Critically, TLR8 directly contributed to the NF-κB-driven response induced by TLR2, and was required for the production of IFN-β by the monocytes (Cervantes and others 2011). Both receptors were induced by the bacterium at the mRNA level, resulting in a positive feedback. These findings clearly establish an important role for the phagosome compartment in the detection of bacteria, where both cell surface and endosomal TLRs (here TLR2 and 8), can cooperate in the detection of the degrading pathogen.
TLR8 in Autoimmune Diseases
TLR7 and TLR8 are the only two TLRs expressed on chromosome X. Most autoimmune diseases are more prevalent in females than in males, with the extreme example of systemic lupus erythematosus (SLE) being nine times more abundant in females than males (Voskuhl 2011). Critically, the incidence of SLE directly relates to a chromosome X dosage effect, as evidenced by the report that XXY men are as prone as women to developing SLE (Sawalha and others 2009; Voskuhl 2011). These observations point to a direct role for X-inactivated gene expression in autoimmune disorders such as SLE, and thus it is possible that X-linked TLR7 and TLR8 may play a role in the aberrant detection of self-ribonucleic acids. TLR7 mRNA levels are elevated in PBMCs from SLE patients (Komatsuda and others 2008), and PBMCs from females produced more IFN-α following R848 stimulation, indicating a role for TLR7 in human SLE (Berghofer and others 2006), consistent with animal models of SLE where TLR7 is critical (Groom and others 2007; Anders and others 2008). While no direct role in SLE for TLR8 has been established to date, elevated TLR8 expression levels may play a role in this disease through the modulation of TLR7 function, as described above. This is supported by the recent observation that Tlr8 −/− mice displayed lupus-like increased autoantigens to ribonucleoproteins, dsDNA, and glomerulonephritis, and this was directly dependent upon Tlr7 expression (Demaria and others 2010). In addition to SLE, TLR8 could be at play in other autoimmune disorders. For example, there is evidence that TLR8 is an X-linked inflammatory bowel disease (IBD) susceptibility gene, and is highly upregulated in colonic epithelium from patients with active IBD (Saruta and others 2009; Steenholdt and others 2009). TLR8 has also been implicated in rheumatoid arthritis (Sacre and others 2008), psoriasis (Ganguly and others 2009), and type I diabetes (Cooper and others 2009).
Therapeutic Applications of TLR8 sensing
TLR8 and RNAi
The RNAi machinery is an evolutionarily conserved pathway allowing for specific control of gene expression. Introduction of a synthetic short RNA of ∼21-base pair duplex (siRNA) in mammalian cells can recruit the RNAi machinery and promote the degradation of perfectly complementary cognate sequences. Because RNAi can be directed against any gene of interest, it is a formidable tool for controlling gene expression and presents great therapeutic potential. However, initial in vivo studies demonstrated that injection of siRNAs could activate TLR7/8 signaling pathways and induce flu-like symptoms (Hornung and others 2005; Judge and others 2005; Gantier and Williams 2010). As previously mentioned, this response was sequence dependent and varied greatly depending on the siRNA administered. Because siRNA design itself is constrained by the efficacy of target sites in mRNAs (which vary greatly among siRNAs), the selection of sequences that do not activate the immune system, but retain excellent RNAi properties requires extensive testing. Relying on natural mammalian nucleoside modifications that inhibit TLR7/8 sensing, it was shown that 2′-Ome incorporation in the antisense strand of the siRNA could ablate immune activation of siRNAs in vitro and in vivo, while retaining most of the RNAi efficacy (Morrissey and others 2005; Judge and others 2006; Sioud 2006; Zamanian-Daryoush and others 2008).
In fact, 2′-Ome nucleoside-modified RNAs were found to act as potent antagonists to TLR7 and 8 (Judge and others 2006; Robbins and others 2007), and have been proposed to have a greater affinity for TLR7/8 than normal RNA (Robbins and others 2007; Hamm and others 2010). As such, 2′-Ome-modified ribonucleic acids could present therapeutic avenues for the modulation of aberrant TLR7/8 activation [in diseases such as lupus or asthma (Moller-Larsen and others 2008; Garcia-Ortiz and others 2010)].
On the other hand, we and others have proposed that activation of the innate immune system by siRNAs could potentiate their effect in specific disease contexts such as cancer or viral infections (Gantier and Williams 2009; Gantier and others 2010b). There is growing evidence that concurrent recruitment of both siRNA-driven innate immunity and gene-specific silencing can have synergistic effects in antitumor treatments (Poeck and others 2008; Gantier and Williams 2009; Kortylewski and others 2009; Khairuddin and others 2011) or antiviral treatments (Gantier and others 2010b; Stewart and others 2011). Advancing this concept, we recently demonstrated that Dicer-substrate siRNAs (D-siRNA), which require processing by Dicer to enter the RNAi pathway, can easily be modified to increase their ability to activate TLR7/8, with a predominant activation of TLR8 (Gantier and others 2010b). Such modification relies on the addition of nonpairing uridines in the nontargeting strand of the D-siRNA, and specifically increases sensing by TLR7/8 without affecting the gene-silencing efficacy of these D-siRNAs (Gantier and others 2010b). This increase in TLR7/8 sensing presumably relates to the structural distortion introduced within the RNA duplex together with the additional uridine residues (Gantier and others 2010b; Gantier 2012).
TLR8 agonists as therapeutics
TLR7/8 agonists, by virtue of their ability to induce IFN-α/TNF-α, present great potential as antiviral, anticancer, and vaccine adjuvants (Hennessy and others 2010). For instance, imiquimod (TLR7 chemical agonist) topical application is a current standard of care for nonmelanoma skin cancer (Desai and others 2011). Similarly, for their ability to modulate immune responses, TLR7/8 agonists present good therapeutic potential for the modulation of allergies such as seasonal and perennial allergic rhinitis—as seen with the nasal spray of VTX-1463, a chemical TLR8 agonist (Horak 2011). In addition, TLR7/8 agonists may help to improve the immunogenicity of vaccines when used in adjuvant formulations (Kasturi and others 2011).
Nevertheless, systemic administration of TLR7/8 chemical agonists such as R848 (resiquimod) in human trials resulted in adverse side effects similar to that of IFN-α treatment (flu-like symptoms) (Pockros and others 2007; Biffen and others 2011). To limit these effects, strategies are currently being devised to identify novel TLR7/8 ligands that are more rapidly metabolized into inactive products. For instance, AZ12441970, a novel TLR7 ligand, showed efficacy in a mouse allergic airway model, with dampened systemic cytokine production (Biffen and others 2011). Importantly, there are also concerns about the specificity of some chemical TLR8 agonists such as imidazoquinolines. For example, there is evidence that imiquimod does not exclusively regulate NF-κB through TLR7, but could also interact with receptors such as the human adenosine receptor (Schon and others 2006).
Alternatively, the use of modified oligonucleotides that are much less stable than the chemical ligands, but can be stabilized to avoid rapid ribonuclease degradation, could present avenues for the systemic administration of TLR7/8 agonists. As previously mentioned, stabilized phosphorothioate RNAs linked to their 3′-end through a glycerol linker were able to induce cytokine production in human nonprimates and showed antitumor effects in mice (Lan and others 2007; Wang and others 2010). Because the immunostimulatory intensity of such stabilized immunomodulatory RNAs can be fine-tuned by sequence modifications (Kandimalla and others 2011), oligonucleotides that activate TLR7/8 represent a new class of adjuvants that can be designed to suit best the therapeutic effect required.
Concluding Remarks
Several lines of evidence indicate the importance of human TLR8 in innate immunity: localization on chromosome X, which could explain the prevalence of autoimmune disorders in females (Saruta and others 2009; Voskuhl 2011); a complex regulation of genetic expression and correlation with protection against viral and bacterial infections (Gantier and others 2010a); a predominant expression in monocytes/macrophages and an associated ability to promote the production of proinflammatory cytokines such as TNF-α; a preferential inducibility by IFN-γ compared to all other TLRs (Hornung and others 2002; Zarember and Godowski 2002) through the recruitment of NK cells (Fig. 1); usurpation of TLR8 activation by pathogens such as HIV-1 (Gringhuis and others 2010); cooperation with cell-surface TLRs in the detection of phagocytosed bacteria (Qin and others 2006); and the ability to respond to RNA sequences that TLR7 cannot detect. There is a rapidly growing body of evidence to support the notion that TLR8 is a critical player in foreign RNA sensing in its own right, that is, independently of TLR7. Nevertheless, research to uncover the role of TLR8 in pathogen-driven diseases remains hampered by the lack of access to animal models that faithfully reproduce the role of this receptor. While in vitro studies with human PBMCs have been very informative on the role of TLR8 in agonist signaling, they do not account for a more physiological contact with pathogens. For example, it is likely that neutrophils play a more critical role in TLR8 function than currently appreciated, given their rapid migration to sites of infection/wounding. Studies relating to the genetic modulation of the TLR8 activity have so far been restricted to primates [with a similar TLR8 genomic context (Gantier and others 2010a)]. However, there is evidence that the contribution of TLR8 to the global immune response could be investigated further through the use of animal models such as sheep and cattle (Mikula and others 2010). Better characterization of the role played by TLR8 in the detection of pathogens such as M. tuberculosis and H. pylori will be important for the development of novel strategies to control pathogen-associated diseases worldwide.
Footnotes
Acknowledgments
We would like to thank Frances Cribbin for her editorial input in the creation of this article. Work in the authors' laboratory is supported by funding from the Australian NHMRC (1006590 and 1022144), a Monash University FMNHS Strategic Grant (2012), and the Victorian Government's Operational Infrastructure Support Program.
Author Disclosure Statement
No competing financial interests exist.