
Abstract
Natural killer T (NKT) cells constitute an important subset of T cells that can both directly and indirectly mediate antitumor immunity. However, we and others have reported that cancer patients have a reduction in both NKT cell number and function. NKT cells can be stimulated and expanded with α-GalCer and cytokines and these expanded NKT cells retain their phenotype, remain responsive to antigenic stimulation, and display cytotoxic function against tumor cell lines. These data strongly favor the use of ex vivo expanded NKT cells in adoptive immunotherapy. NKT cell based-immunotherapy has been limited by the use of autologous antigen-presenting cells, which can vary substantially in their quantity and quality. A standardized system that relies on artificial antigen-presenting cells (aAPCs) could produce the stimulating effects of dendritic cell (DC) without the pitfalls of allo- or xenogeneic cells. In this review, we discuss the progress that has been made using CD1d-based aAPC and how this acellular antigen presenting system can be used in the future to enhance our understanding of NKT cell biology and to develop NKT cell-specific adoptive immunotherapeutic strategies.
Natural Killer T Cells
N
There are functionally and phenotypically distinct subtypes of NKT cells. Type I NKT cells (also known as invariant or semi-invariant NKT cells -iNKT) express an invariant Vα14Jα18 T cell receptor (TCR) in mice and Vα24Jα18 TCR in humans. Type II NKT cells are CD1d restricted T cells that express a more diverse set of α chains in their TCR and recognize different lipid antigens. It is thought that type I NKT cells exert potent anti-tumor effects, whereas type II NKT cells generally suppress tumor immunity through the production of interleukin (IL)-4 and IL-13 (Ambrosino and others 2007). This review will focus mainly on canonical, type I iNKT cells. Type I NKT cells are further classified based on their expression of classic TCR cell surface markers. In mice, iNKT cells are CD4+ or CD4−CD8− double-negative (DN) (Terabe and Berzofsky 2008), whereas human NKT cells are either CD4+, CD8+, or DN (Gumperz and others 2002; Kim and others 2002; Lee and others 2002; Lin and others 2006). In addition, these subsets have distinct Th1 and Th2 cytokine profiles. CD4+ NKT cells produce Th1 and Th2 cytokines, whereas the CD4− subset, which includes both CD8+ & DN, primarily produces Th1 cytokines (Kim and others 2002). The majority of human NKT cells express CD161, CD56 as well as NKG2D. Interestingly, NKT cells are phenotypically similar to effector T cells because they express nonlymphoid tissue-homing chemokine receptors, such as CCR2, CCR5, and CXCR3. Given that NKT cells can rapidly produce Th1, Th2, and Th17- type cytokines, and activate cells of both the innate and adaptive immune systems, they are thought to be important modulators of the immune response. It is clear that NKT cells can also play a pivotal role in maintaining immune homeostasis (Godfrey and others 2004), see schematic shown in Fig. 1. In fact, NKT cells have been implicated in the regulation of autoimmunity (Illes and others 2000), tumor surveillance (Terabe and Berzofsky 2008; Swann and others 2009), hematological cancers (Neparidze and Dhodapkar 2009) as well as infectious disease (Kinjo and others 2005).
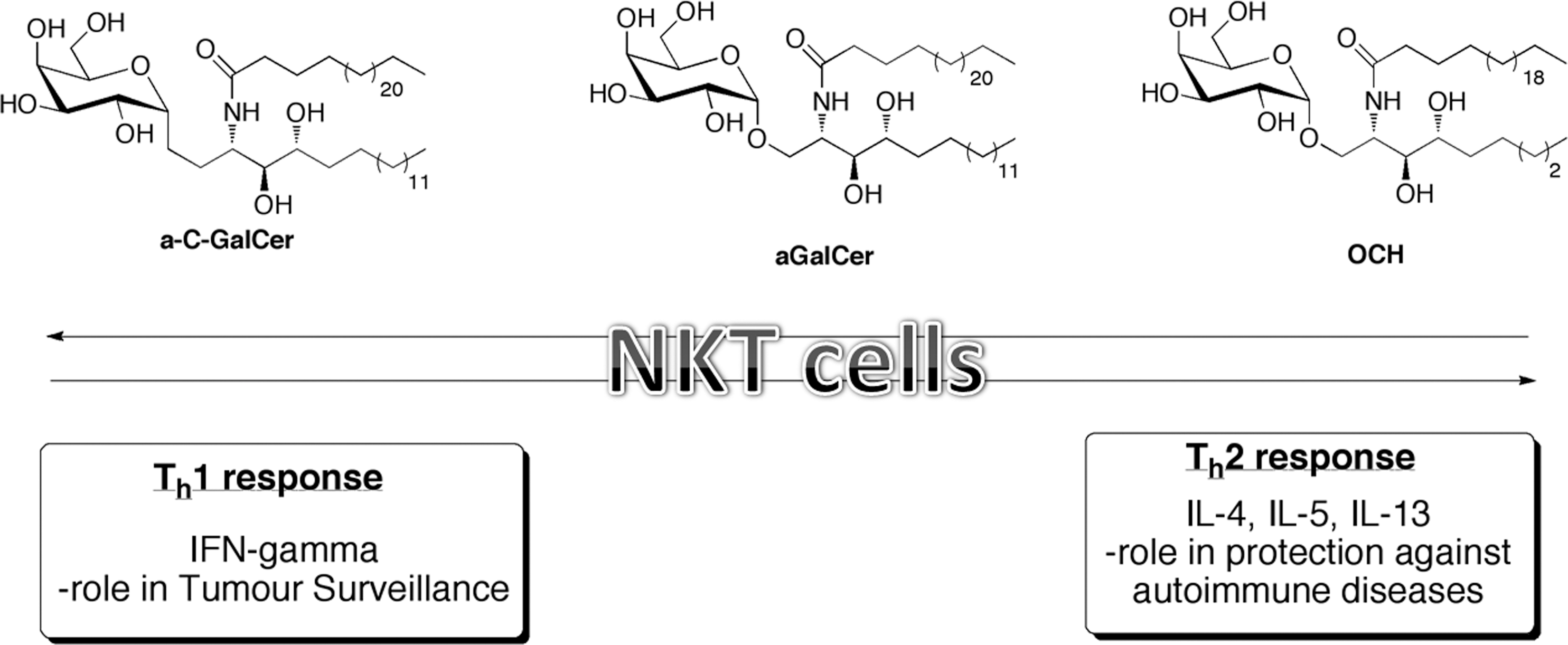
NKT cells are potent immunoregulatory cells that can drive both antitumor immune responses and protect against autoimmunity. NKT cells can produce both Th1 & Th2 cytokines. Classic NKT cell agonists are highlighted. α-GalCer induces the production of both Th1 & Th2 cytokine profiles, while–OCH results in primarily Th2 and c-GalCer stimulates Th1 responses. NKT, natural killer T.
CD1 Molecules
In humans, 5 genes (CD1A–E) located within 200-kb on chromosome 1q22-q23 encodes CD1a, CD1b, CD1c, CD1d, and CD1e lipid antigen-presenting molecules. The CD1 proteins are classified into 2 groups based on amino acid sequence homology: group 1 is composed of CD1a, b, c, and e; and group 2 contains CD1d. Like MHC Class I molecules, CD1 isoforms (CD1a, b, c, and d) are assembled in the ER and transported to the cell surface. However, in contrast to MHC molecules, CD1 complexes are then reinternalized into specific endocytic compartments where they can bind lipid antigens. These include a broad scope of both self and foreign molecules that range from simple fatty acids or phospholipids, to more complex glycolipids, isoprenoids, mycolates, and gangliosides (De Libero and Mori 2012). Lipid-loaded CD1 molecules are then delivered to the cell surface and can be surveyed by CD1-restricted T cells expressing αβ or γδ TCR. CD1a-, b-, and c-restricted T cells have been found to recognize a number of lipid antigens from Mycobacterium tuberculosis. Mice only express group 2 CD1d molecules-CD1d1 and CD1d2. iNKT cells are CD1d1-restricted and the focus of this review. Notably, the crystal structures of both mouse and human CD1d have been described (Zeng and others 1997; Koch and others 2005). These studies have shown that the antigen-binding groove is deeper, narrower, and much more hydrophobic compared to MHC class I and class II molecules. The hydrophobic nature of the antigen-binding groove is ideal for binding lipid antigens. In vitro binding studies have determined the molecular mechanism for lipid antigen presentation by CD1 molecules: the alkyl chains of a lipid–ligand bind within a highly hydrophobic groove inside the CD1 protein, while the polar head group remains exposed on top of the extracellular domain, thereby allowing direct contact with the TCR, leading to NKT cell activation (Moody and others 2005).
NKT Cell-Based Immunotherapy
Numerous studies have reported that circulating NKT cells are reduced in cancer patients (Kawano and others 1999; Tahir and others 2001; Fujii and others 2003a). In fact, Molling and others (2005) examined a large cohort of cancer patients and healthy controls and found that NKT cell numbers were 47% lower in cancer patients compared to age and gender matched healthy controls. This reduction in NKT cell numbers was independent of tumor type or stage/grade. Clinical trials for the activation of endogenous NKT cells have focused on direct intravenous (i.v.) injection of α-GalCer or infusion of α-GalCer-pulsed DC. In two phase I clinical trials, patients injected with either α-GalCer (Giaccone and others 2002) or α-GalCer-loaded immature dendritic cells (Nieda and others 2004), showed strong immune responses as assessed by serum interferon (IFN)-γ levels, but this was true only in patients with detectable NKT cell numbers. Chang and others (2005) showed that multiple injections of α-GalCer-loaded mature dendritic cells lead to sustained expansion of NKT cells and antigen-specific T cells. However, these expanded NKT cells from cancer patients still exhibited reduced capacity for IFN-γ secretion compared to NKT cells from healthy controls. In contrast to the direct injection of α-GalCer, Nakayama's group has carried out a few phase I/I-II studies to evaluate the immunological response and clinical outcome and safety by using α-GalCer-pulsed DC in lung cancer patients (Ishikawa and others 2005a, 2005b). They reported that vaccination was well-tolerated by all of the patients, with no severe adverse effects. In addition, there was a dramatic increase in NKT cell numbers in the peripheral blood and augmentation of IFN-γ mRNA from circulating NKT cells. Several other groups observed similar results in different cancer types, such as multiple myeloma, head and neck squamous cell carcinoma, and other type of solid cancers (Chang and others 2005; Uchida and others 2008).
Several groups have investigated the in vivo efficacy of in vitro-activated NKT cells. Motohashi and others (2006) performed the adoptive transfer of in vitro-activated NKT cells in patients with refractory lung cancer. They found an increase in both the number of circulating NKT cells and the number of IFN-γ producing cells in the peripheral blood, but the objective antitumor response rate remained low. Collectively, these studies show that cancer patients have a deficiency in both NKT cell number and function, which suggests that in vivo NKT cell modulation may be ineffective in patients with low NKT cells and further suggests that adoptive immunotherapy of ex vivo-expanded NKT cells may be a more productive strategy for these patients.
Adoptive Immunotherapy
Adoptive immunotherapy involves stimulation of tumor-specific T cells leading to their ex vivo expansion, followed by transfer of these expanded autologous T cells back into patients. The most successful efforts in cancer immunotherapy have been focused on the expansion of melanoma-specific T cells from surgically resected tumor samples or ex vivo expansion of melanoma-reactive T cells from the peripheral blood patients. Studies by Rosenberg and others (1988) were the first to demonstrate that ex vivo-expanded autologous tumor-specific cells are able to traffic to tumor sites and directly induce tumor shrinkage, in vivo.
For classical CD4+ and CD8+ T cell subsets, effective adoptive immunotherapy requires the expansion of large numbers of tumor antigen-specific T cells for adoptive immunotherapy. Then, the expanded T cells need to migrate to the site of the tumor and mediate their effector functions. A limitation of this approach is that patients must have pre-existing tumor reactive cells that are difficult to identify in nonmelanoma malignancies. To overcome this limitation, TCR gene transfer (Zhao and others 2007), MHC streptamers, or chimeric antigen receptors (Morgan and others 2010) can be utilized. It has been hypothesized that the effectiveness of T cells after the TCR gene transfer is reduced due, at least in part, to the pairing of the newly introduced TCR α and β chains with the endogenous TCR proteins. MHC streptamers are multimerized MHC-peptide complexes that reversibly bind in an antigen-specific manner to the TCR. Thus, a benefit to using these molecules is that secondary enrichment steps can be performed in which one could select for additional phenotypic markers to obtain an optimal effector T cell subpopulation. A disadvantage is that these molecules are only available for a select few antigens, primarily viral antigens. As mentioned above, tumor reactive T cells have been engineered to express tumor-associated antigens or chimeric antigen receptors, which make them tumor-specific rather than depending on tedious isolation and expansion steps. However, the transduced TCRα or β chains can mispair with endogenous TCR and can potentially result in autoreactive cells and toxicity from cytokine overproduction. Therefore, all of these current methodologies have major limitations, indicating an urgent need for the development of cell-based and noncellular artificial antigen-presenting cells (aAPCs) for the activation and expansion of tumor-specific T lymphocytes (Turtle and Riddell 2010).
Artificial Antigen-Presenting Cells
Cellular aAPCs derived from primary, transformed human, or xenogenic cells have been developed (Paulos and others 2008). These cells are generated using retroviral or lentiviral transduction to introduce molecules that provide the necessary TCR, costimulatory, and adhesion molecules necessary for synapse formation. One key example is the human erythroleukemia cell line- K562, which does not express endogenous HLA A, B, or DR. Upon transduction with the human Fc receptors CD32 and CD64, K562 can bind and present anti-CD3 and anti-CD28. Recently, K562 cells were transduced with HLA-A*201 and HLA-DR*0401, which permits the presentation of exogenous peptide antigen or endogenously processed antigens (Maus and others 2002).
The use of MHC streptamers, chimeric antigen receptors, and cellular aAPC all have major limitations to their use as effective therapy in patients. This has led to the development of aAPCs. The artificial or noncellular aAPC can be utilized both for their potential clinical value in ex vivo T cell expansion as well as to investigate the basic requirements for T cell activation. It has been reported that soluble forms of recombinant HLA-A2 molecules loaded with antigen-associated peptides are able to directly target cognate CD8+ T cells in vitro (Groh and others 2002). Tham and others described one of the first bead-based systems, where they coupled biotinylated murine MHC class I–peptide–single-chain constructs together with biotinylated costimulatory molecules B7.1 and B7.2 via streptavidin to the surface of latex microspheres (Tham and others 2001; Goldberg and others 2003). The limitation of this approach is that it uses a single-chain MHC-peptide complex to ensure homogenous loading of the MHC molecules. Consequently, each target peptide antigen requires a new transfection for expression of the desired single-chain MHC-peptide complex.
Studies from Schneck's group pioneered the noncellular, bead-based aAPC, made by coupling HLA-Ig (signal) and anti-CD28 (signal 2) onto magnetic beads. His group initially developed HLA-Ig, a unique multimeric form of HLA fused to an immunoglobulin molecular scaffold (Dal Porto and others 1993; Greten and others 1998). HLA-Ig as a molecular scaffold for expressing soluble HLA molecules takes advantage of the intrinsic flexibility associated with Ig proteins for expression of a dimeric HLA complex. Based on their valence, these molecules can be used to stimulate antigen-specific T cells and can be easily derivatized to solid supports, such as beads based on the Ig domains. Subsequently, this group developed MHC-Ig-based aAPCs, which have been shown to effectively expand CMV and MART-1-specific CTL (Oelke and others 2003). To determine the effectiveness of this approach on NKT cell propagation and activation, CD1d-Ig-based aAPCs have been generated, see schematic shown in Fig. 2 (Shiratsuchi and others 2009; Webb and others 2009).
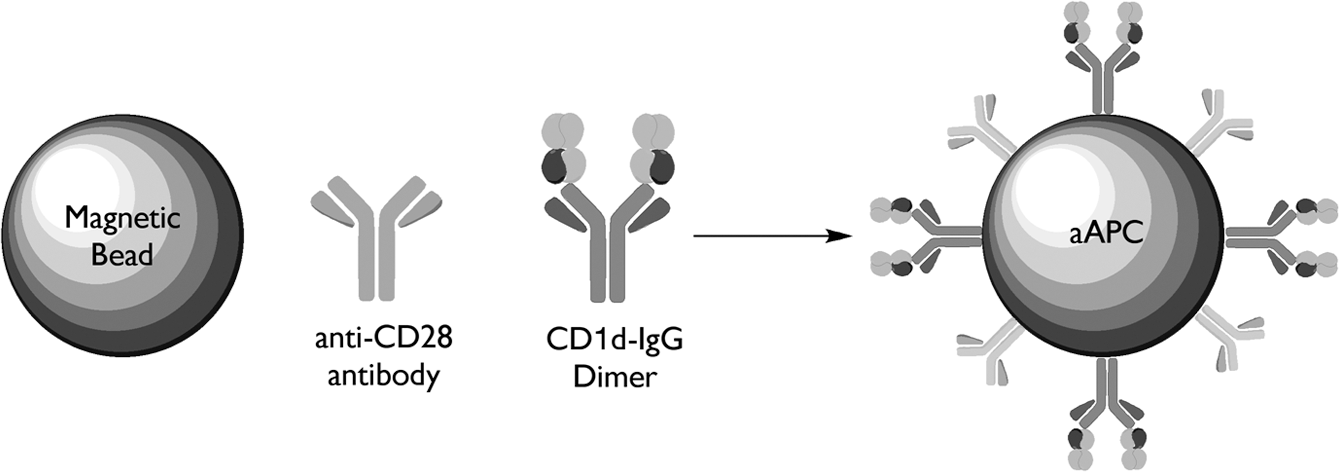
Schematic diagram of CD1d-based aAPCs. aAPCs can be made by coupling CD1d-Ig and various costimulatory Abs to magnetic beads. These noncellular aAPCs can then be loaded with lipid antigen of choice. In this system, CD1d-Ig is used to provide the cognate antigen-specific signal through the TCR and anti-CD28, CD44, CD161 Abs will provide the costimulatory signal. The B7–CD28 interaction was one of the first costimulatory pairs defined at a molecular level. As such, this has served as a cornerstone for studies involving costimulation. However, these molecules may not be the major sources of costimulation for NKT cells. In fact, a variety of additional costimulatory complexes have been defined over the past few years, including CD161 and CD44, which will offer interesting alternatives to the B7–CD28 interaction. aAPCs, artificial antigen presenting cells; TCR, T cell receptors.
CD1d-Based aAPC
To date, immunotherapy utilizing the NKT/CD1d system has been limited by the use of autologous antigen-presenting cells (APCs) in the presence or absence of α-GalCer. The quantity and quality of these stimulator cells can vary substantially. For example, it has been shown that monocyte-derived DC from cancer patients, express reduced levels of costimulatory molecules, and produce less inflammatory cytokines (Onishi and others 2002; Bella and others 2003). Therefore, Shimizu and others (2006) recently reported using murine DC rather than autologous APC to test the function of NKT cells from CML patients. However, this system can only be used for in vitro testing, since NKT cells cannot be expanded by murine DC and then given back to patients. A standardized system that relies on aAPCs could produce the stimulating effects of DC without the pitfalls of allo- or xenogeneic cells.
Importantly, CD1d-Ig-based aAPC can be used for both the activation and expansion of mouse and human NKT cells. Since the engagement of the TCR by the CD1d-antigen complexes is a fundamental requirement of NKT cell activation, antigen:CD1d-Ig complexes, possibly along with appropriate costimulatory molecules, potentially offer a reliable method to isolate, activate, and expand effector NKT cell populations. In addition, we have demonstrated that CD1d-Ig-based aAPCs, made by covalent coupling of CD1d-Ig and potential costimulatory molecules to magnetic beads can be used as a standardized method for the propagation of NKT cells. Importantly, this system can be used to expand the NKT cell population from the peripheral blood mononuclear cells (PBMC) of cancer patients (Webb and others 2009).
The culture conditions have been optimized so that relatively large numbers of Vα24+Vβ11+ NKT cells can be obtained and functional studies can be performed to assess cytokine profiles and cytotoxic activity. Remarkably, when compared to the standard protocol of Vα24+ NKT cell purification using magnetic beads, followed by expansion using α-GalCer pulsed, irradiated autologous PBMC, the CD1d-based aAPC-expansion method results in similar NKT cell expansion (Fig. 3) in donors with higher starting numbers of NKT cells. For example, if cultures are initiated with 2 million CD161+CD3+ T cells, 2–3 weeks following aAPC culture one can expect to obtain beween 7–28 million cells, of which 30%–70% are Vα24+Vβ11+ NKT cells. Notably, in samples from donors with lower starting percentages of NKT cells, high levels of proliferation still occur using the CD1d-Ig aAPC platform. This is important because the adoptive transfer of 50 million NKT cells has been reported in patients (Yamasaki and others 2011), thus, increasing the starting cell number should permit the expansion of clinically relevant numbers. The CD1d-based aAPC system was established using a medium supplemented with human AB serum and a monocyte-derived cytokine cocktail. In an effort to move this system toward clinical standards, the protocol has been modified to aAPC-mediated NKT cell expansion using clinical grade IL-2 alone, specifically in the absence of the cytokine cocktail.
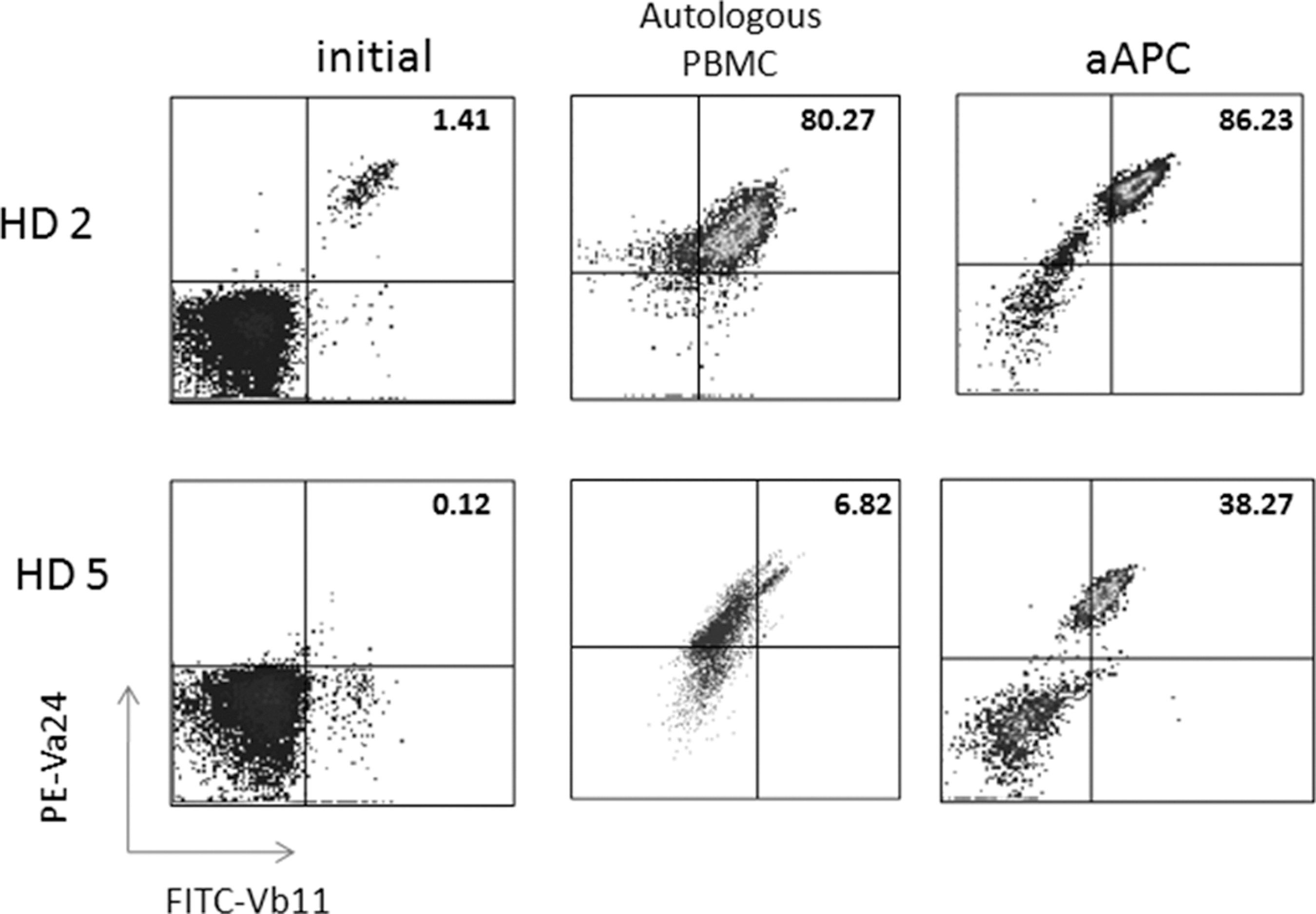
CD1d-based aAPCs can be used to expand human NKT cells. CD161+ T cells were isolated from PBMC using magnetic bead separation. The cells were stimulated biweekly with irradiated PBMC pulsed with 100 ng/mL α-GalCer (10:1 ratio) or a-GalCer-loaded aAPC+αCD28 (1:1 ratio). Data shown are after 2 rounds of stimulation. PBMC, peripheral blood mononuclear cells.
Generation of NKT Cells Using aAPC and CD34+ Cells
Despite the importance of NKT cells in regulating immune responses, their low frequency significantly restricts their potential for clinical application. It has been previously shown that OP9 stromal cells transduced with Notch ligand delta-like 1 (OP-DL1) can be used for the directed differentiation of embryonic stem cells into T cell lineages (Schmitt and Zúñiga-Pflücker 2002; de Pooter and others 2003). The induction of Notch signals directs stem cells to differentiate into immature double-positive T cells and inhibits B cell development, demonstrating that Notch signaling is required as a proximal event in T cell commitment from progenitors (Robey and others 1996; Pui and others 1999). While the OP9-DL1 system has been shown to generate functionally mature human CD4, CD8, regulatory T cells (Schmitt and others 2004; La Motte-Mohs and others 2005; Hutton and others 2009; Van Coppernolle and others 2009; Awong and others 2011) and murine NKT cells from transduced embryonic stem cells or fetal liver hematopoietic progenitors (Nunez-Cruz and others 2008; Watarai and others 2010), it is unclear whether this system could be utilized to generate functional human NKT cells from adult stem cells. To address this question, studies performed by our group have recently demonstrated that functionally mature human NKT cells can be generated in vitro from CD34+ cells utilizing the OP9-DL1 system in combination with aAPCs (manuscript submitted).
Our findings demonstrating that OP9-DL1 cultures in combination with aAPCs support the differentiation of functionally mature NKT cells are highly encouraging because the propagation of human NKT cells to date has been technically challenging due to the low frequency of human circulating NKT cells and no other culture system has been previously reported to generate human NKT cells from adult progenitor stem cells. In the context of adoptive immunotherapy, the generation of NKT cells from CD34+ cells isolated from granulocyte-macrophage colony-stimulating factor immobilized donors, bone marrow, or cord blood could be utilized in combination with other tumor-specific T cell types, as well as in the setting of transplantation to boost host immune responses. Moreover, the availability of technologies and clinical grade reagents required for the selective expansion of NKT cells coupled with the cost–effectiveness and versatility of the bead based-aAPC system highlights the feasibility of this therapeutic strategy.
The Function of Costimulatory Molecules Can Be Assessed Using aAPCs
There has been great interest in the modulation of antitumor T cell responses by engaging the TCR or by stimulation through costimulatory pathways. While the intracellular signaling for conventional T cells is well understood, this information for NKT cells is as yet unavailable. Using aAPCs to stimulate NKT cells, a thorough investigation of the intracellular-signaling cascades using various different NKT cell ligands can be performed. These factors can yield other targets to boost NKT cells responses along with administration of activating ligands. CD1d-based aAPCs can also be redesigned to include various costimulatory molecules. Since aAPCs can be designed to express any costimulatory molecule, they can lead to immediate identification of potential activating signals that can be included during patient antitumor therapy. The use of aAPCs for the activation of NKT cells facilitates the study of cellular proteins and signaling molecules. It also allows researchers to mimic various conditions of NKT cell activation, since the precise mechanisms that drive different types of responses are not completely understood.
Initial studies have been conducted to examine the impact of varying costimulatory ligand complex on aAPC stimulation-mediated expansion of NKT cells. T cell activation requires delivery of a combination of signals through the TCR (signal 1) and through costimulatory molecules (signal 2), such as engagement of CD28 by B7. One of the parameters that one can vary easily using aAPCs is the ratio of signal 1 to signal 2 as well as the type of signal 2 delivered. As depicted in Fig. 4, NKT cells express the majority of costimulatory molecules found on classic CD4+ and CD8+ T lymphocytes.
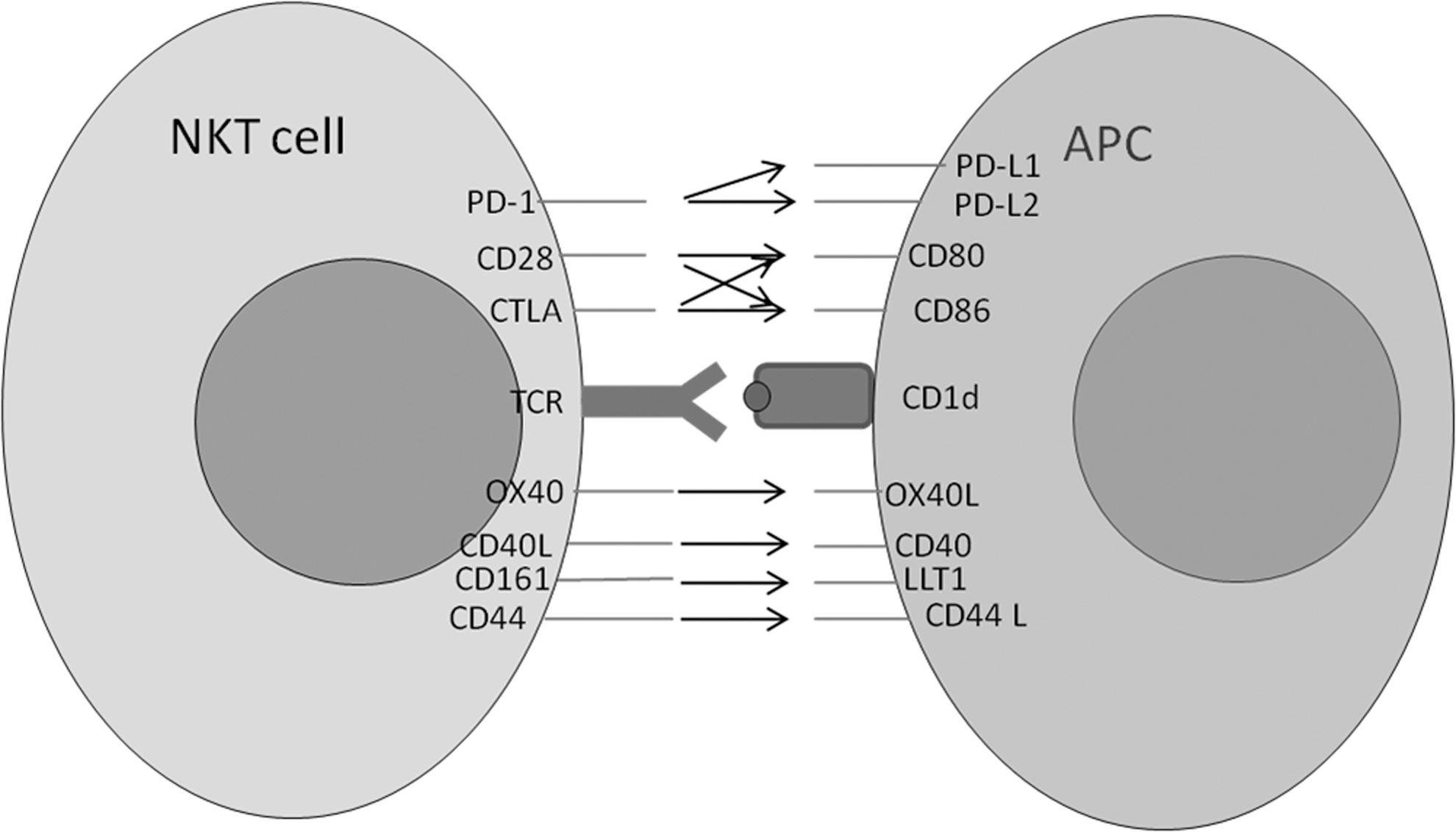
Potential costimulatory ligand-receptor interactions between NKT cells and APC. A selection of the key costimulatory molecule pairs involved in NKT cell activation is depicted. The CD28 co-stimulatory families are shown above the CD1d:TCR synapse and the CD40 families are shown below. The arrow indicates cross-reaction between the receptor-ligand pair. APC, antigen-presenting cells.
Given the relatively high expression of CD44 and CD69 on murine liver NKT cells, and the study by Larkin and others (2006) identifying CD44 as a costimulatory molecule, we generated CD1d-Ig aAPC to compared the costimulatory potential of these molecules (Webb and others 2009), in the context of 2 different NKT cell antigens, α-GalCer and-OCH stimulation. We found that activation by α-GalCer-loaded aAPCs was enhanced by the addition of CD44. A modest increase was observed in the presence of anti-CD28, while little or no effect was observed with anti-CD69. When we examined the stimulatory capacity of OCH-loaded aAPCs, the addition of anti-CD44 and CD28 resulted in a slight increase cytokine production; however, anti-CD69 abrogated activation in NKT cell hybridomas. We also characterized the stimulatory potential of the aAPC using freshly isolated liver NKT cells from C57BL/6 mice and found that anti-CD28 and CD44 significantly increased NKT cell activation. Thus, CD1d-Ig based aAPCs can be used to investigate the efficacy of potential costimulatory molecules on NKT cell function.
To determine the impact of using different costimulatory ligands (i.e., CD28, CD44, and CD161) (Exley and others 1998) on primary human NKT cell expansion, CD161+CD3+ T cells were isolated from the peripheral blood and stimulated with human CD1d-based aAPCs in the presence or absence of costimulatory molecules every 10–14 days. Table 1 shows that following 2 rounds of stimulation, α−GalCer loaded aAPCs were able to expand the NKT cells population in the presence of all costimulatory molecules examined. Notably, the expansion rate was highly donor dependent. As expected, the higher the initial population of Vα24+ cells, the greater the percentage of expansion. In addition, stimulation with anti-CD28 in the presence of a cytokine panel results in a significantly higher level of expansion of canonical NKT cells compared to the other costimulatory molecules examined. Ex vivo-expanded NKT cells remain responsive to α-GalCer stimulation and are potent producers of IFN-γ, tumor necrosis factor (TNF)-α, and IL-17A (Fig. 5). These data demonstrate that costimulation enhances NKT cell proliferation as well as function, and that signaling through CD28 is important for proliferation and cytokine production. When the function of this ex vivo-expanded population was assessed by intracellular cytokine/cytotoxicity staining, it was found that different costimulatory elicited functional differences in the iNKT cell population, as determined by CD107a, IFN-γ, and IL-4 expression (data not shown). In addition, expansion of different subsets of NKT cells (CD4+, CD8+, DN) appear to be dependent upon the type of costimulatory molecule used (data not shown). Thus, these data suggest that costimulatory molecule expression can be used to modulate NKT cell proliferation and activation; consequently, a combination of costimulatory signals may be necessary to expand best effector NKT cell subset. Such studies facilitated by aAPCs can provide valuable insights into the NKT cell activation process with respect to the activating ligand as well as the APC and lead to the development of improved cancer immunotherapy.
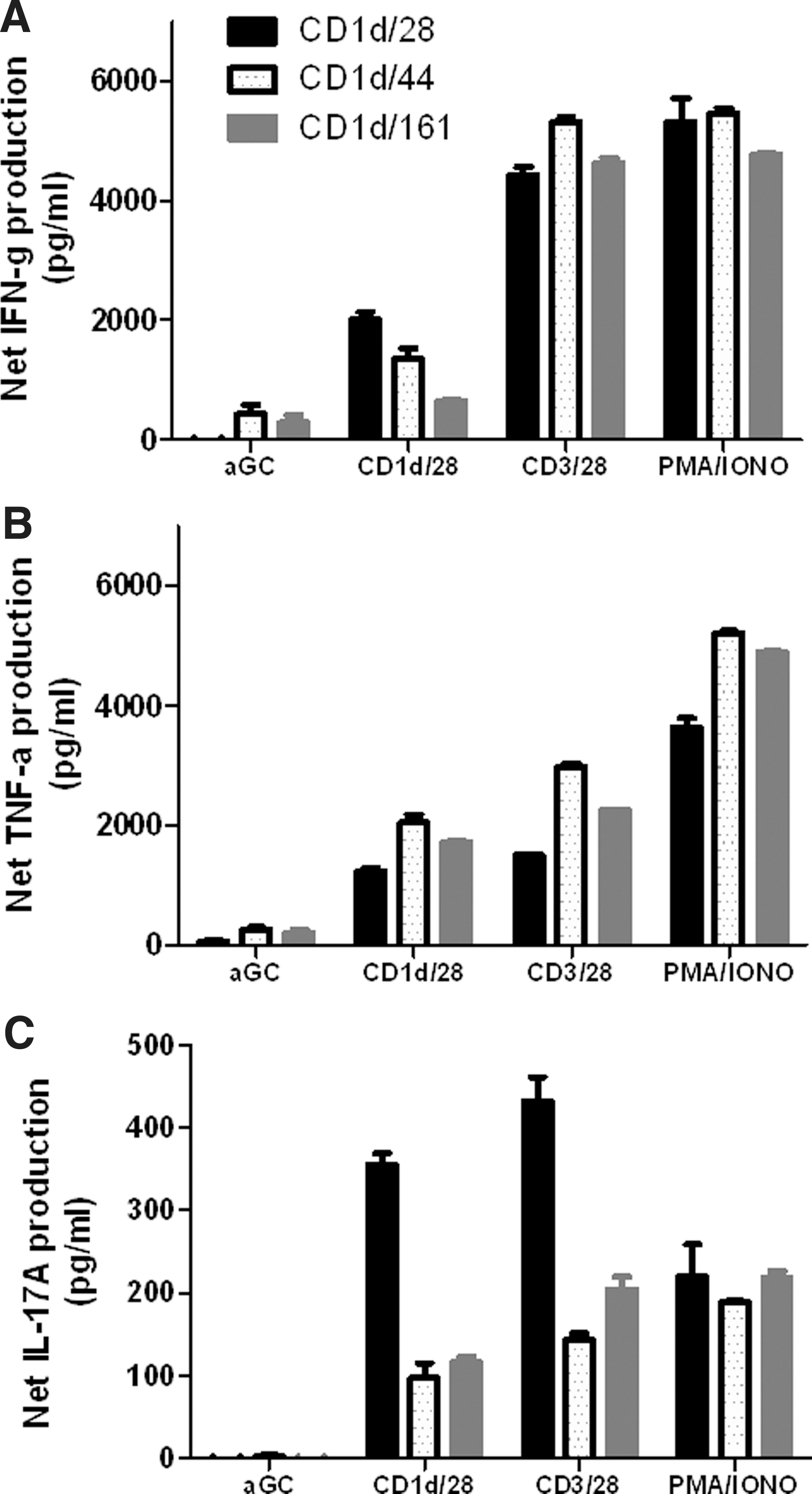
Cytokine profiles of aAPC-expanded NKT cells. After stimulation with α-GalCer-loaded aAPCs for 2 weeks, the expanded NKT cells (1×105/well) were cocultured with soluble α-GalCer, PMA/ionomycin, αCD3/28 microbeads, or α-GalCer-loaded aAPCs (2×105/well) for 48 h.
a
aAPC, artificial antigen-presenting cells.
Different costimulatory molecules may modulate diverse downstream signaling cascades. Collectively, these data are very intriguing and have lead us to ask what are the molecular “triggers” that modulate NKT cell effector function. Bessloes and others (2008) found that activation of IL-2R on NKT cells leads to the induction of PI3K, ERK, p38MAPK, and STAT 3, 4, 5, and 6, as well as IFN-γ and IL-4. We hypothesize that both environment and subset (CD4, CD8, DN) play a role in determining the effector function of NKT cells. Our data suggest that the expression of specific costimulatory molecules alters the cytokine profile, cytotoxic function, and the expansion of specific subsets of the NKT cell population (Fig. 5, data not shown). In addition, propagation of NKT cells in IL-2 alone compared to a monocyte-derived cytokine cocktail had an effect on proliferation rates (data not shown). It is unclear whether the kinetics alone are affected or if different costimulatory signals will activate or downregulate specific signaling cascades. We do know that the data will significantly enhance our understanding of NKT cell biology.
NKT-Specific aAPCs as Adjuvants
Considering the unique adjuvant effect that occurs following NKT cell activation and its modulation, other immune effectors, developing combination therapeutic regimens that activate NKT cells as well as promote adaptive immunity may be very useful. Hermans and others (2003) found that activation of NKT cells by administration of α-GalCer along with i.v. delivery of soluble OVA protein resulted in the significant enhancement of OVA-specific T cell response, including CTL response upon cross-presentation ex vivo. They also found a significant resistance to tumor challenge when the mice were treated with OVA together with α-GalCer before tumor challenge in their EG7.OVA mouse cancer model. Fujii and others (2003b) also demonstrated that a single i.v. injection of α-GalCer can rapidly mature DC in vivo through activation of NKT cells and showed increased CD4+ and CD8+ T cell immunity and high resistance to OVA-expressing tumors.
Cytokine-Mediated Modulation of NKT Cell Function
NKT cells are known to rapidly produce large amounts of cytokines after activation. NKT cells are inherently capable of producing Th1 as well as Th2 cytokines (Yoshimoto and others 1995; Kawakami and others 2001; Miyamoto and others 2001; Niemeyer and others 2008). The precise mechanism for cytokine bias still remains somewhat elusive although several groups have cited one or more mechanisms (Goff and others 2004). Previously, it was believed that the cytokine response from NKT cells depends mainly on the interaction between the glycolipid antigen and TCR or the CD1d-binding properties of the antigen. This was supported by the observation that ligands like OCH, which are known to bind less stably as compared to α-GalCer, elicit a Th2 response (Velmourougane and others 2009). On the other hand, compounds like α-C-GalCer, which have better binding capabilities, stimulated a stronger Th1 response. However, with the development of newer compounds such as α-GalCer acC8 and α-GalCer acC20:2, the differences in antigen–TCR interaction have proved insufficient to explain the Th2 bias of particular compounds (Yu and others 2005). A recent report has suggested distinct APC as the mechanism driving different immune responses (Bai and others 2012). This was done using a CD1d conditional knockout mouse model focusing on 3 major types of APC- DCs, macrophages, and B cells. This study showed that Th1-biased ligands are more likely to be presented by DCs as compared to other APC. On the contrary, Th2-biased ligands were more likely to be presented on APC other than DCs. This study further underscores the importance of APC in eliciting appropriate NKT cell responses. The natural antitumor immune response is typically characterized by Th1 cytokines, such as IFN-γ (Molling and others 2005). This leads to the rapid recruitment of CD8+ cytotoxic T lymphocytes, which can mount a strong antitumor cytolytic response (Schofield and others 1987; Shibolet and others 2003). This IFN-γ response is fortified by a feedback loop involving NK cells (Carnaud and others 1999). When DCs present glycolipid antigen to activate NKT cells, they are also activated reciprocally due to CD40L expression on the NKT cell, which engages the CD40 on the DC. This stimulation leads to secretion of IL-12 by the DC. This IL-12 further activates NK cells, which produce a second wave of IFN-γ. Thus, the primary Th1 response from the NKT cells can be boosted and/or sustained due to DC IL-12 production and NK cell activation. IL-12 and IL-18 produced by DCs are also known to activate NKT cells in an antigen-independent manner. Pathogens that do not bear NKT cell-activating glycolipid antigens can thus activate NKT cells. For example, viral pathogens bearing CpG DNA can activate TLR9 on DCs and lead to the production of IL-12 and IL-18. These cytokines can cause activation of NKT cells and lead to IFN-γ production (Tyznik and others 2008). Similarly, bacterial lipopolysaccharides can activate DCs in a TLR4-dependent manner and lead to CD1d-independent activation of NKT cells (Nagarajan and Kronenberg 2007). Administering cytokines along with aAPCs can thus be a useful method to achieve robust NKT cell responses by engaging both the antigen-dependent and the antigen-independent routes of NKT cell activation (Fig. 6). aAPC can allow fine tuning of the immune response through administration of cytokines, such as IFN-γ to directly boost the Th1 response, or indirectly using IL-12 to activate NK cells as well as NKT cells.
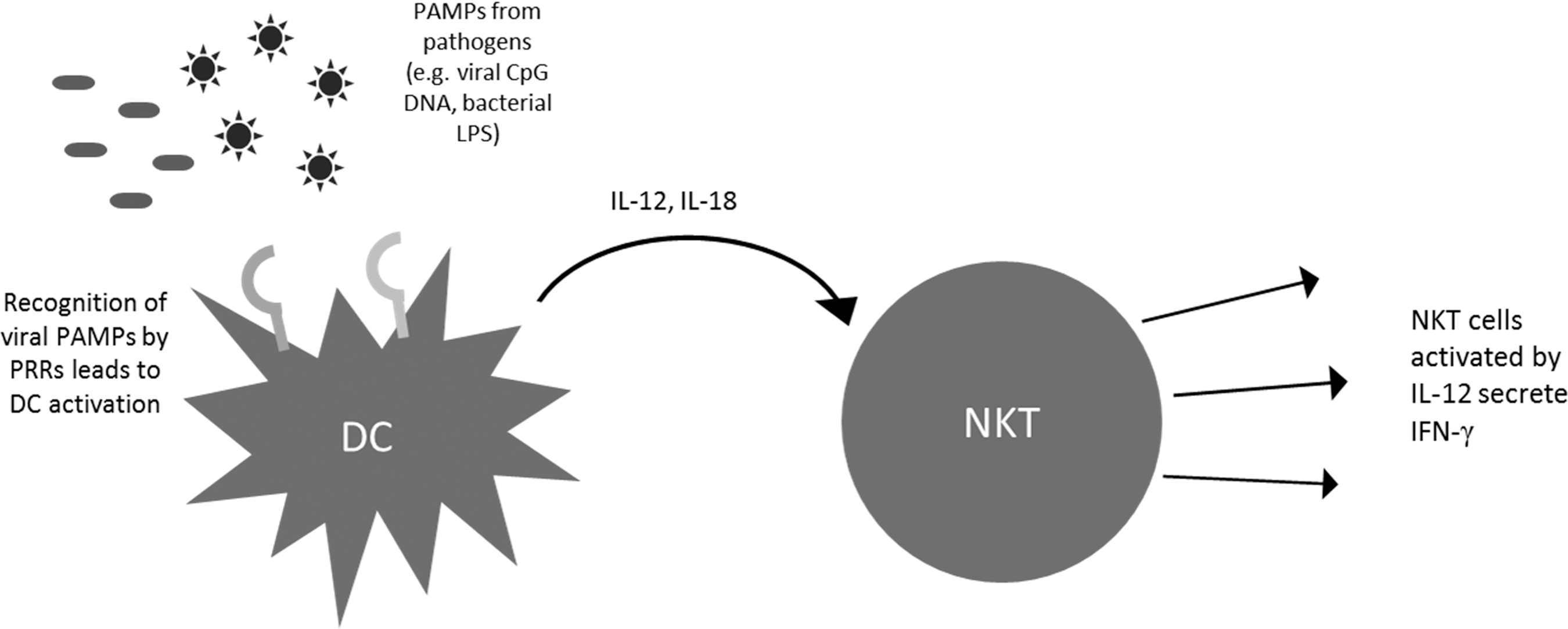
Cytokine driven NKT cell activation. PAMPs from pathogens, such as viral CpG DNA or bacterial LPS, can be recognized by PRRs, such as TLR9 or TLR4, respectively. This leads to the activation of APCs, such as DCs, and results in rapid IL-12 and IL-18 production. This cytokine signal can then activate NKT cells in a CD1d-independent manner. Thus, pathogens that do not bear CD1d-binding antigens can also activate NKT cells in this manner. DC, dendritic cells; PAMPs, pathogen-associated molecular patterns; LPS, lipopolysaccharides; PRR, pattern recognition receptors.
In an interesting study by Steenblock and others (2011), biodegradable polymer-based aAPCs were generated, which also released IL-2. Specifically, these avidin-coated aAPCs contained biotinylated anti-mouse CD3, anti-mouse CD28, and IL-2 biotin. Thus, the authors were able to investigate the importance of the paracrine delivery of IL-2 on the activation and proliferation of CD8+ and CD4+ T cells. It was found that paracrine IL-2 delivery by aAPCs result in enhanced CD8+ T cells proliferation, but caused activation-induced apoptosis in CD4+ T cells. These studies also demonstrated the importance of the temporal and spatial context in which the cytokine is presented. The dose of aAPCs and cytokine can thus be precisely controlled to achieve maximum possible antitumor responses, while preventing detrimental immunopathology. CD1d-Ig-based aAPCs thus provide a valuable tool for stimulating NKT cell responses and afford a greater level of control over the NKT cell responses generated by such anti-cancer therapy (Webb and others 2009). There has been great interest in the development of α-GalCer analogs to achieve stronger Th1 responses (Lu and others 2006). However, the role of APC in the modulation and sustenance of Th1 responses is now being appreciated, thereby making aAPCs a valuable tool in the regulation of NKT cell responses elicited by the administration of NKT cell antigens.
Polarizing NKT Cell Responses Based on Antigen
The types of responses seen by effector cells can be greatly altered depending on the type of antigen encountered by the effector cell as well as the antigen-presenting molecule. In the case of NKT cells, different analogues of α-GalCer [PBS57 (Courtney and others 2011), OCH (Velmourougane and others 2009), and C-GalCer (Fujii and others 2006)] can elicit Th1 (Wu and others 2011) or Th2 (Chang and others 2007) responses (see Fig. 1). In addition to effector functions, these lipid antigens can be used to initiate proliferation of NKT cells. Typically, autologous dendritic cells are pulsed with different lipid antigens, and then incubated with NKT cells to determine which types of cytokines are secreted. This process is laborious and time consuming. aAPCs can be incubated with desired antigen and provide a constant source of stimulation for the expansion and activation of NKT cells (East and Sun, manuscript submitted). Our group has shown that aAPCs are capable of enriching the NKT cell population in PMBCs isolated from humans, while still retaining their functionality (Webb and others 2009).
A number of different methods of delivery of α-GalCer have been demonstrated to effectively stimulate NKT cells, seen most often in the context of using these cells as vaccine adjuvants. α-GalCer is considered a viable vaccine adjuvant because of its ability to stimulate NKT-mediated cytokine release and its related biological functions (Subrahmanyam and Webb 2012). α-GalCer along with BSA, monophosphoryl lipid A, polyinosinic–polycytidylic acid, and alginate in poly(lactic-co-glycolic acid) (PLGA) microspheres has been used to stimulate an increased peptide release in vitro and a more balanced IgG1/IgG2 response versus soluble antigen in vivo (Salvador and others 2012) Poly-lactic acid-based nanoparticles have been used to present α-GalCer to NKT cells via dendritic cells and macrophages, but not B cells (Thapa and others 2009) This delivery method stimulates NKT cell growth and proliferation over multiple administrations without inducing anergy. Similar methods have been successful in stimulating NKT cells, including α−GalCer analogue KRN7000 encapsulated in PLGA microspheres (Macho Fernandez and others 2012).
CD1d-Based Killer aAPC
Killer APC, like FasL-overexpressing DC, have been shown to induce antigen-specific apoptosis of T cells in both murine and human models. However, there is still a series of concerns, such as biosafety, time-consuming and batch-to batch variability. To overcome the limitations associated with cell-based APC, bead-based Killer artificial antigen presentation cells (KaAPC) have been developed and these KaAPC specifically eliminated antigen-specific T cell in vitro. Shen and others (2011) investigated the ability of KaAPC to eliminate antigen-specific T cells in vivo and found skin allograft survival was prolonged along with a decrease in antigen-alloreactive T cells in a murine model of alloskin transplantation. In addition, studies by Schütz and others (2008), showed that KaAPC, made by coupling HLA-A2 Ig and α-Fas IgM mAb covalently to the surface of beads successfully deleted antigen-specific CTL in vitro from a mixture of T cells with various specificities. Together, these studies highlight the therapeutic potential of KaAPC strategy for the treatment of autoimmune diseases and allograft rejection.
Type II NKT cells, also known as nonclassical NKT cells, have a diverse TCR repertoire and exhibit inhibitory activity, whereas type I NKT cells mount antitumor responses. Perhaps, CD1d-based KaAPC can be used to aid in the selective removal of type II NKT cells, so that one could restore the antitumor effects mediated by type I NKT cells. In this case, one could couple CD1d-Ig and anti-Fas L antibody to magnetic beads and load the CD1d molecules with sulfatide. This would potentially enable these KaAPC to specifically target the type II NKT and induce apoptosis of these NKT cells. This strategy would need to be tested rigorously by in vitro and in vivo experiments.
Conclusions
We have demonstrated that CD1d-Ig-based aAPCs, made by covalent coupling of CD1d-Ig and potential costimulatory molecules to magnetic beads, can be used as a standardized method for the propagation of NKT cells. We designed an aAPC, which is adaptable to any requirements we find necessary for optimal NKT cell proliferation. When aAPCs are compared to standard autologous PBMC-based induction and expansion, the current standard for NKT cell expansion, aAPCs compared favorably. As highlighted in Fig. 7, aAPCs represent a robust versatile technology useful for inducing and expanding NKT cells. Together, these studies demonstrate that the CD1d-Ig-based aAPC system has the potential to provide a better understanding of NKT cell biology, which may lead to new strategies to enhance current approaches in cancer immunotherapy.
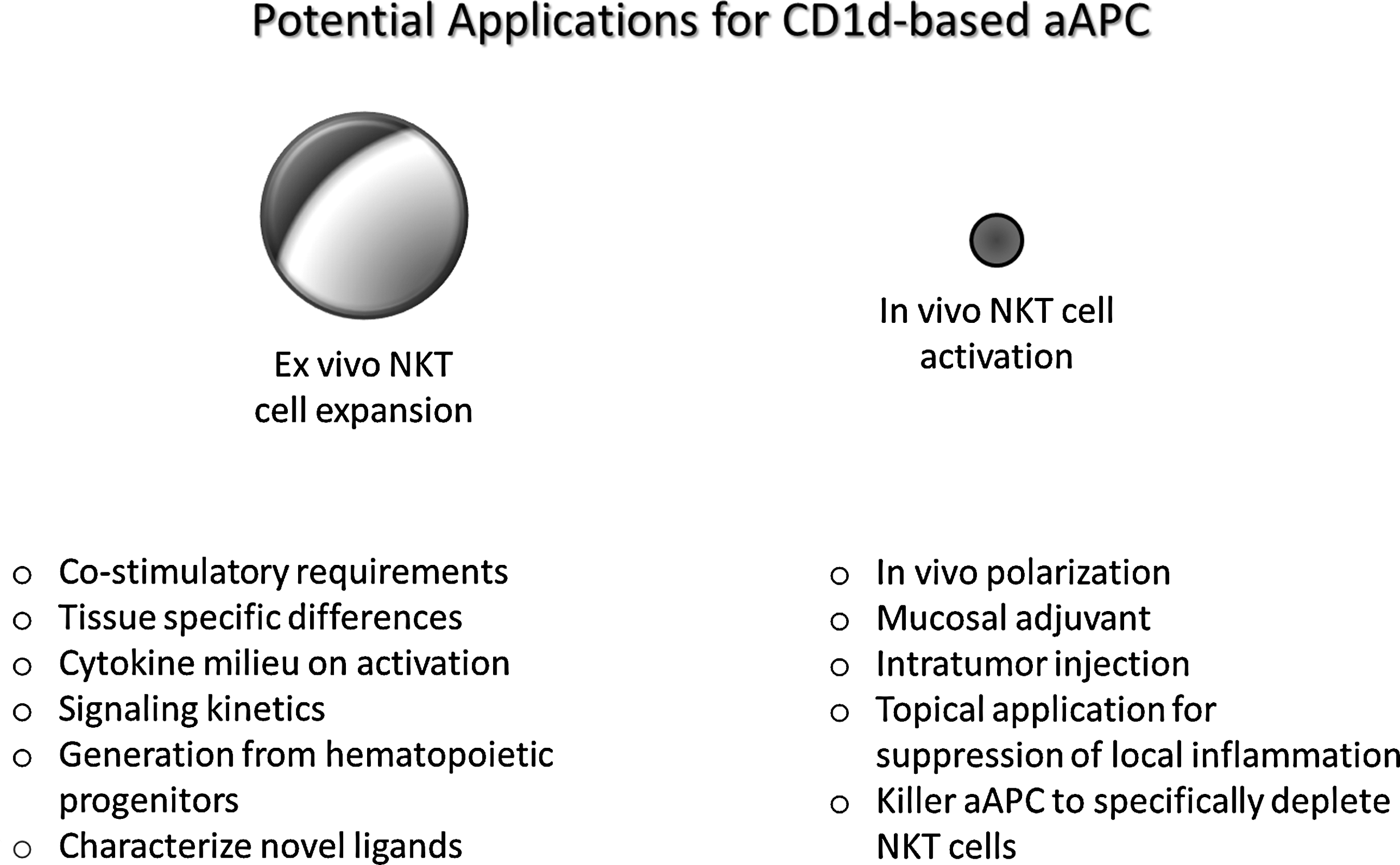
Utilization of CD1d-based aAPC mediated activation of NKT cells as a broad-based platform. CD1d-Ig-based aAPC is a unique system that can be used to characterize the requirements for NKT cell activation and proliferation both in vitro and in vivo. Given the feasibility and reproducibility of this system, it can help us connect the dots and assess the costimulatory requirements and tissue-specific differences in specific NKT cell subsets. In addition, NKT cells can be cultured with aAPCs in the presence of certain cytokines or putative ligands and following activation, their downstream signaling kinetics can be determined. aAPCs can be used to activate and expand NKT cells from humans as well different species of animals. Ongoing studies are focused on comparing different matrices for optimal in vivo use. Then, one could potentially use the CD1d-Ig-based aAPC to polarize NKT cell cytokine profiles in vivo and examine their efficacy in vaccination strategies to either enhance NKT cell activation or specifically deplete undesired NKT cell responses.
Footnotes
Acknowledgments
The authors have no competing financial interest. The authors would like to thank all of the donors who allowed their samples to be studied. PMBC were obtained from commercial vendors for all data provided in this manuscript. The authors also would like to thank the NIH Tetramer Core Facility for providing CD1d tetramers. This work was supported by grants from the American Cancer Society, NIH/NCI K01 CA131487, R21 CA162273, R21 CA162277, and P30 Tumor Immunology and Immunotherapy Program to T.J. Webb. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.