
Abstract
The family of Interleukin-17 (IL-17) cytokine is the essential inflammatory mediator that influences the pathophysiology of various inflammatory diseases. Many studies focused on investigating the expression, signaling, and biological impacts of IL-17A and IL-17F, and the neutralization of these cytokines exhibited some promising results in clinical trials. In contrast, the expression resources and physiological relevance of IL-17C remained to be studied. In this study, through a microarray approach conducted with nontransformed human colonic epithelial cells (NCM460), we found that bacterial flagellin stimulation elicited potent IL-17C mRNA expression. We also confirmed that IL-17C protein production was strongly induced by flagellin in these cells. Flagellin-induced IL-17C expression was also observed in human colon adenocarcinoma cells such as DLD-1 and HT-29, indicating that IL-17C could be a signature inflammatory cytokine from intestinal epithelial cells in response to flagellin. Since inhibited in TLR5-, or MyD88- or TRIF-silenced cells, flagellin-induced IL-17C expression was specifically mediated by TLR5 and, subsequently, MyD88 and TRIF adaptor molecules. Furthermore, in line with inflammatory nature of IL-17, we found that IL-17C expression was substantially enhanced in the intestinal tissues from Ulcerative colitis patients. Given the facts that TLR5 is a key pattern recognition receptor which mediates microbial recognition in the intestinal epithelium and IL-17C turned out to be a unique member of the IL-17 family expressed in intestinal epithelial cells on TLR5 activation, our study may provide an important clue on understanding how intestinal microbes would contribute to an inflammatory program in the gut.
Introduction
T
Although the biochemical features and biological impacts of each IL-17 family member still remain to be intensively studied, IL-17A and F have mesmerized many to study their intracellular signaling and inflammatory impacts for years. Intriguingly, IL-17A and F were known to collaborate with inflammatory cytokines such as tumor necrosis factor-α (TNF-α) or interferon-γ or IL-1β to synergistically derive potent inflammatory responses, although the molecular mechanism of the synergy is not yet clear (Pappu and others 2011). In line with the pro-inflammatory propensity of IL-17, indeed, elevated IL-17A and F were observed in various autoimmune diseases (Pappu and others 2011).
As for IL-17B, C, D, and E, their expression resources, targeting cells, cellular signaling, and biological impacts have been poorly studied. Very recent studies, however, showed that IL-17C mRNA is expressed in human colon adenocarcinoma cells SW480 and HCT-15 stimulated with bacterial products (peptidoglycan, lipopolysaccharide, or flagellin) or inflammatory cytokines (IL-1β, TNF-α) (Ramirez-Carrozzi and others 2011; Song and others 2011). These studies also suggested that IL-17C utilizes a heterodimeric receptor complex of IL-17RA and IL-17RE with a higher affinity to IL-17RE than IL-17RA. Since IL-17RE and IL-17RA are preferentially expressed in epithelial cells, IL-17C appears to play an essential role in host mucosal defense against microbial infection and inflammation in the intestine (Ramirez-Carrozzi and others 2011; Song and others 2011).
TLR5 is a pattern recognition receptor that specifically recognizes bacterial flagellin at the plasma membrane and is abundantly present in many epithelial cell types from various organs (Rhee and others 2004; Schaefer and others 2004; Blohmke and others 2008). We demonstrated that TLR5 utilizes the adaptor molecules MyD88 and TRIF, but not TRAM, to mediate flagellin-induced NF-κB and AP-1 transcription factor activation and corresponding cytokine expression in intestinal epithelial cells (Choi and others 2010a). Given the facts that intestinal epithelial cells are at the front line of microbial recognition and these cells are strongly responsive to flagellin via TLR5 to induce potent inflammatory and innate immune responses, it would be of interest to study whether TLR5 engagement elicits the expression of IL-17 family members in intestinal epithelial cells.
In this study, using the microarray approach, we found that TLR5 activation by flagellin elicited both IL-17C protein production and IL-17C mRNA expression in nontransformed human colonic epithelial cells (NCM460). We also showed that the adaptor molecule MyD88 and TRIF mediated TLR5-induced IL-17C expression. In light of the inflammatory impact of IL-17, we examined the expression of IL-17 family members in the intestinal tissues from inflammatory bowel disease (IBD) patients represented by Crohn's disease (CD) and Ulcerative colitis (UC), and observed that IL-17C expression was significantly augmented in UC patients' tissues compared with normal tissues. Thus, our data suggest that IL-17C expression by flagellin-TLR5 engagement at the surface of epithelial cells may confer a crucial impact on the inflammatory pathophysiology of intestinal mucosa.
Materials and Methods
Materials
Nontransformed human colonic epithelial (NCM460) cells developed from normal human colon (Moyer and others 1996; Kumar and others 1997) were cultivated as previously described (Rhee and others 2004). MyD88-knock down NCM460 (NCM460-MyD88-KD), NCM460-TRIF-KD, and its control NCM460-control cells were previously described (Choi and others 2010a). NCM460-TLR5-KD and its control cells were previously described (Rhee and others 2006). Human colon carcinoma DLD-1 cells, DLD-1-TLR5-KD, and DLD-1-control cells were previously described (Rhee and others 2008). Human colon carcinoma HT-29 cells were obtained from ATCC (Manassas, VA) and maintained in McCoy's 5A complete media containing fetal bovine serum to a final concentration of 10%. Flagellin purified from Salmonella typhimurium (<0.1 Endotoxin Unit/μg purified protein determined by Limulus Amebocyte Lysate test) was purchased from “Enzo Life Sciences” (Farmingdale, NY) and dissolved in endotoxin-free water. Human IL-17C and IL-8 ELISA were from “MyBioSource” (San Diego, CA) and “Invitrogen” (Grand Island, NY), respectively.
Microarray analysis
NCM460-control, NCM460-MyD88-KD, and NCM460-TRIF-KD cells were cultivated in 100 mm dishes in quadruplicate and treated with or without flagellin (100 ng/mL) for 3 h. Total RNA was isolated using the miRNeasy Mini kit (Qiagen, Valencia, CA). Microarray analysis was performed by the Clinical Microarray Core at UCLA. Briefly, an equal amount of total RNA (200 ng) was processed using the Low Input Quick Amp Labeling Kit One-Color (Agilent Technologies, Santa Clara, CA), and Cyanine-3-labeled cRNA was purified by RNeasy Mini Kit (Qiagen). Cy-3-labeled cRNA (600 ng) was then fragmented for 30 min at 60°C before loading onto Agilent Whole Human Genome arrays (Agilent Technologies). The samples were hybridized for 17 h at 65°C with constant rotation. Arrays were washed, dried, and scanned on an Agilent DNA Microarray Scanner at a resolution at 3 μm by following the manufacturer's protocol. Microarray images were analyzed using the Agilent's Feature Extraction software.
Array normalization, comparison analysis, and hierarchical clustering were performed with the dChip software (Li and Wong 2001) (
Quantitative real-time PCR
As previously described (Im and others 2009; Choi and others 2010b), total RNA was prepared from cultivated cells, using the “RNeasy Plus Mini Kit” (QIAGEN, Valencia, CA), and an equal amount of RNA (2 μg) was transcribed into cDNA, using “High Capacity Reverse Transcription Kit” obtained from “Applied Biosystems” (Foster City, CA) by following the manufacturer's instruction. To evaluate the cytokine gene expression, quantitative real-time PCR (qPCR) was performed on “Applied Biosystems 7500 Fast Real-Time PCR System” with the TaqMan Universal Master Mix, using the standard condition from “Applied Biosystems”. After the sample had been incubated for 2 min at 50°C followed by AmpliTaq Gold activation for 10 min at 95°C, 40 cycles were run with a denaturing temperature of 95°C (15 s) and an annealing/extension temperature 60°C (1 min). The primer pairs and FAM™ dye-labeled TaqMan® MGB (minor groove binding) probes for human TRIF, MyD88, IL-8, IL-17(A, B, C, D, and F), IL-1β, and the internal control GAPDH gene were purchased from “Applied Biosystems.” The level of expression was calculated based on the PCR cycle number (C T) at which the exponential growth in fluorescence from the probe passes a certain threshold value (C T). Relative gene expression was determined by the difference in the C T values of the target genes after normalization to the RNA input level, using a C T value of GAPDH. Relative quantification was represented by standard 2−ΔCT calculations. ΔC T=(C T-target gene−C T-GAPDH). Each reaction was performed in triplicate.
Measuring IL-17C protein production
An enzyme-linked immunosorbent assay (ELISA) was performed to measure the level of human IL-17C protein production using the appropriate kits from “MyBioSource” (San Diego, CA) by following the manufacturer's instructions. All assays were performed in triplicate, and data were shown as mean±SD.
IPA network analysis
Excel files exhibiting fold change values were obtained from Significance Analysis of Microarrays analyses using a 10% false discovery rate, followed by including the gene name and symbol. The microarray data were analyzed by the Ingenuity Pathway Analysis (IPA) 7.5 software (Ingenuity Systems, Inc., Mountain View, CA) (Li and others 2005; Dorsam and others 2010). Network scores and p-values were assigned by IPA software; thus, according to the degree of interconnectedness between the molecules, IPA software determined a higher or lower IPA network score. The most significant networks of each dataset were imported into the path designer, and networks were graphically displayed to describe the relationships between genes and their neighboring genes.
Quantitative real-time PCR with human UC CD and normal intestinal tissues
We used a commercially available cDNA panel (TissueScan™ qPCR Arrays, Origene Technologies, Inc., Rockville, MD) made from tissues of CD and UC patients and normal controls, which were selected from mixed ages, gender, and ethnic groups. The panels are of normalized cDNA prepared from pathologist-verified human CD and UC tissues. qPCR was performed as described earlier (Im and others 2009; Choi and others 2010b).
Statistical analysis
Statistical analyses were conducted with GraphPad Prism (GraphPad Software, La Jolla, CA), and detailed information of statistical analysis was presented in the corresponding figure legends.
Results
Gene array analysis reveals IL-17C expression in flagellin-treated human colonic epithelial (NCM460) cells
Intestinal epithelial cells are in continuous contact with microbial factors released from commensal and pathogenic microbes in the gut. Therefore, the gene expression in the intestinal epithelial cells is significantly influenced by these microbial factors that are recognized by pattern recognition receptors such as TLRs. Bacterial flagellin is an abundant microbial pattern molecule that is released by microbes in the gut, and the level of serum antibody against flagellin is elevated in IBD patients. Therefore, flagellin is considered one of the biomarkers for IBD (Lodes and others 2004; Yu and others 2011). Accordingly, it is reasonable to speculate that an array of gene expression elicited by flagellin in intestinal epithelial cells would be related to the development and progress of IBD. Thus, using a microarray analysis, we examined the flagellin-induced gene expression profile in nontransformed human colonic epithelial cells (NCM460). In order to perform a microarray analysis, we used MyD88-knock-down NCM460 (NCM460-MyD88-KD) cells and TRIF-knock-down NCM460 (NCM460-TRIF-KD) cells, which were stably transfected with MyD88 and TRIF shRNA encoding constructs, respectively, as previously described (Choi and others 2010a). In parallel, we also used NCM460-control cells, which were also stably transfected with scramble shRNA constructs (Choi and others 2010a).
To conduct a microarray analysis, NCM460-control, NCM460-MyD88-KD, and NCM460-TRIF-KD cells were stimulated with flagellin or a vehicle, and total RNA was isolated. To confirm whether the knock-down cells have genuinely silenced MyD88 or TRIF gene expression, we first checked MyD88 and TRIF expression. Our qPCR data clearly showed that MyD88 expression was dramatically silenced in NCM460-MyD88-KD cells, compared with the intact MyD88 expression in both vehicle- and flagellin-treated NCM460-control cells (Fig. 1A). We also confirmed that MyD88 expression was preserved in flagellin-treated NCM460-TRIF-KD cells. In addition, we examined TRIF expression in these cells, and as expected, TRIF expression was significantly diminished in NCM460-TRIF-KD cells (Fig. 1B). It is of interest to note that flagellin stimulation resulted in increased TRIF expression in NCM460-control cells, compared with vehicle-treated NCM460-control cells and flagellin-treated NCM460-MyD88-KD cells. As previously shown, inflammatory stimulation such as TNF-α enhanced TRIF expression in NCM460 cells (Choi and others 2010b), and flagellin stimulation appeared to up-regulate TRIF expression. Nonetheless, these data (Fig. 1A, B) demonstrated that MyD88 and TRIF gene expressions were successfully silenced in NCM460-MyD88-KD and NCM460-TRIF-KD cells, respectively, and NCM460-control cells have intact MyD88 and TRIF gene expression.
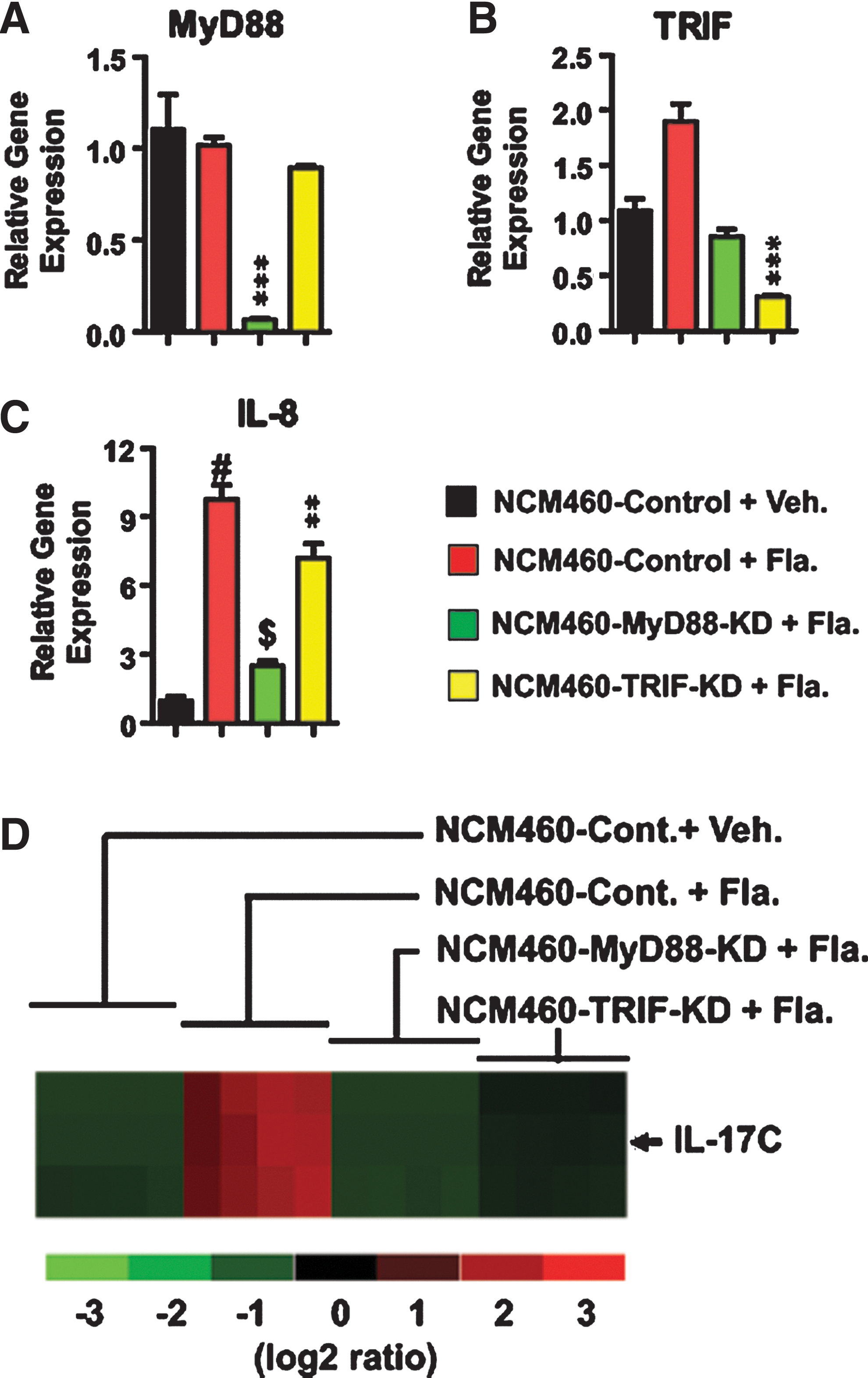
Microarray analysis reveals that flagellin stimulation elicits IL-17C expression, mediated by adaptor molecules MyD88 and TRIF. MyD88-knock down (NCM460-MyD88-KD), TRIF-knock down (NCK460-TRIF-KD), and its control (NCM460-control) cells were stimulated with vehicle or flagellin (100 ng/mL) for 3 h.
Given that these cells were stimulated with flagellin, before proceeding with a microarray analysis, we checked whether these cells were properly stimulated with flagellin. To confirm this, we tested whether these cells exhibited differential IL-8 expression in response to flagellin, because a previous study had already shown that IL-8 expression was induced by flagellin, and blocking MyD88 or TRIF expression reduced IL-8 expression in intestinal epithelial cells (Rhee and others 2006; Choi and others 2010a). Indeed, flagellin stimulation dramatically induced IL-8 expression in NCM460-control cells, compared with vehicle-treated NCK460-control cells (Fig. 1C). In contrast, flagellin-induced IL-8 expression was significantly reduced in NCM460-MyD88-KD and NCM460-TRIF-KD cells, although MyD88 knock down resulted in more potent inhibition than TRIF knock down. Collectively, these data assured that MyD88 or TRIF gene expression was successfully silenced, and flagellin stimulation in these cells were successfully achieved.
From total RNA isolated from these cells, we subsequently performed a microarray analysis. Among the various genes whose expressions were differentially altered in flagellin-treated NCM460 cells, intriguingly, IL-17C expression was dramatically elevated in flagellin-treated NCM460-control cells compared with vehicle-treated NCM460-control cells (Fig. 1D). Moreover, our microarray analysis also showed that flagellin-induced IL-17C expression was dramatically diminished in both NCM460-MyD88-KD and NCM460-TRIF-KD cells (Fig. 1D). These data imply that flagellin stimulation in nontransformed human colonic epithelial cells elicits IL-17C gene expression, for which the TLR-associated adaptor molecules, MyD88 and TRIF, are required as a regulatory adaptor molecule.
Silencing TLR5 expression inhibits flagellin-induced IL-17C expression
Having found that flagellin stimulation elicited IL-17C expression in NCM460 cells determined by a microarray analysis, we subsequently carried out independent qPCR experiments to confirm whether flagellin stimulation could induce IL-17C expression. Indeed, we observed that IL-17C expression was robustly up-regulated by flagellin stimulation in NCM460-control cells relative to vehicle-treated NCM460-control cells. In contrast, flagellin-induced IL-17C expression was dramatically inhibited in both NCM460-MyD88-KD and NCM460-TRIF-KD cells (Fig. 2A). In addition to evaluating IL-17C mRNA expression, we furthermore measured IL-17C protein production in these cells by ELISA. Similarly, IL-17C protein production was strongly induced by flagellin stimulation in NCM460-control cells (Fig. 2B, C), compared with vehicle-treated NCM460-control cells. Again, both NCM460-MyD88-KD and NCM460-TRIF-KD cells exhibited almost completely inhibited IL-17C protein production in response to flagellin (Fig. 2B). Together, these data clearly showed that flagellin stimulation induced both IL-17C mRNA expression and IL-17C protein production in nontransformed human colonic epithelial cells.

Flagellin-induced IL-17C expression is specifically mediated by TLR5.
Despite the fact that TLR5 is a specific pattern recognition receptor to bacterial flagellin at the plasma membrane, NLRC4 (known as Ipaf) is also known to recognize flagellin in the cytosolic region of myeloid cells (e.g., macrophages)(Franchi and others 2006; Miao and others 2007). Although we believe that TLR5 is an exclusive receptor that specifically recognizes flagellin in NCM460 cells, it may be necessary to show that flagellin-elicited IL-17C expression is specifically mediated by TLR5 in these cells. To address this, TLR5-knock-down NCM460 (NCM460-TLR5-KD)(Rhee and others 2006) and its control cells were stimulated with flagellin, followed by evaluating IL-17C expression. Notably, silencing TLR5 completely blocked flagellin-induced IL-17C mRNA and protein expression in NCM460-TLR5-KD cells, while control cells strongly induced IL-17C gene expression on flagellin (Fig. 2D, E).
Taken together, these data demonstrate that flagellin stimulation elicits IL-17C expression in a TLR5-specific manner, and moreover, TLR5-interacting adaptor molecules, MyD88 and TRIF, participate in mediating TLR5-elcited IL-17C expression.
Flagellin-induced IL-17C expression is extended to other intestinal epithelial cells, DLD-1 and HT-29
Given that robust IL-17C expression by TLR5 activation was observed in NCM460 cells, which originated from normal human colon (Moyer and others 1996; Kumar and others 1997), we next tested whether TLR5-induced IL-17C expression could be observed in other types of human intestinal epithelial cells. Since it was recently suggested that IL-17C mRNA expression was enhanced in human colon adenocarcinoma SW480 and HCT-15 cells stimulated with flagellin, we tested DLD-1 cells which are human colon adenocarcinoma cells, and TLR5-knock-down DLD-1 (DLD-1-TLR5-KD) and its control (DLD-1-control) cells (Rhee and others 2008) were stimulated with or without flagellin. Notably, TLR5 activation by flagellin strongly induced IL-17C expression in DLD-1-control cells relative to vehicle-treated DLD-1-control cells. However, flagellin-induced IL-17C expression was completely inhibited in DLD-1-TLR5-KD cells (Fig. 3A).
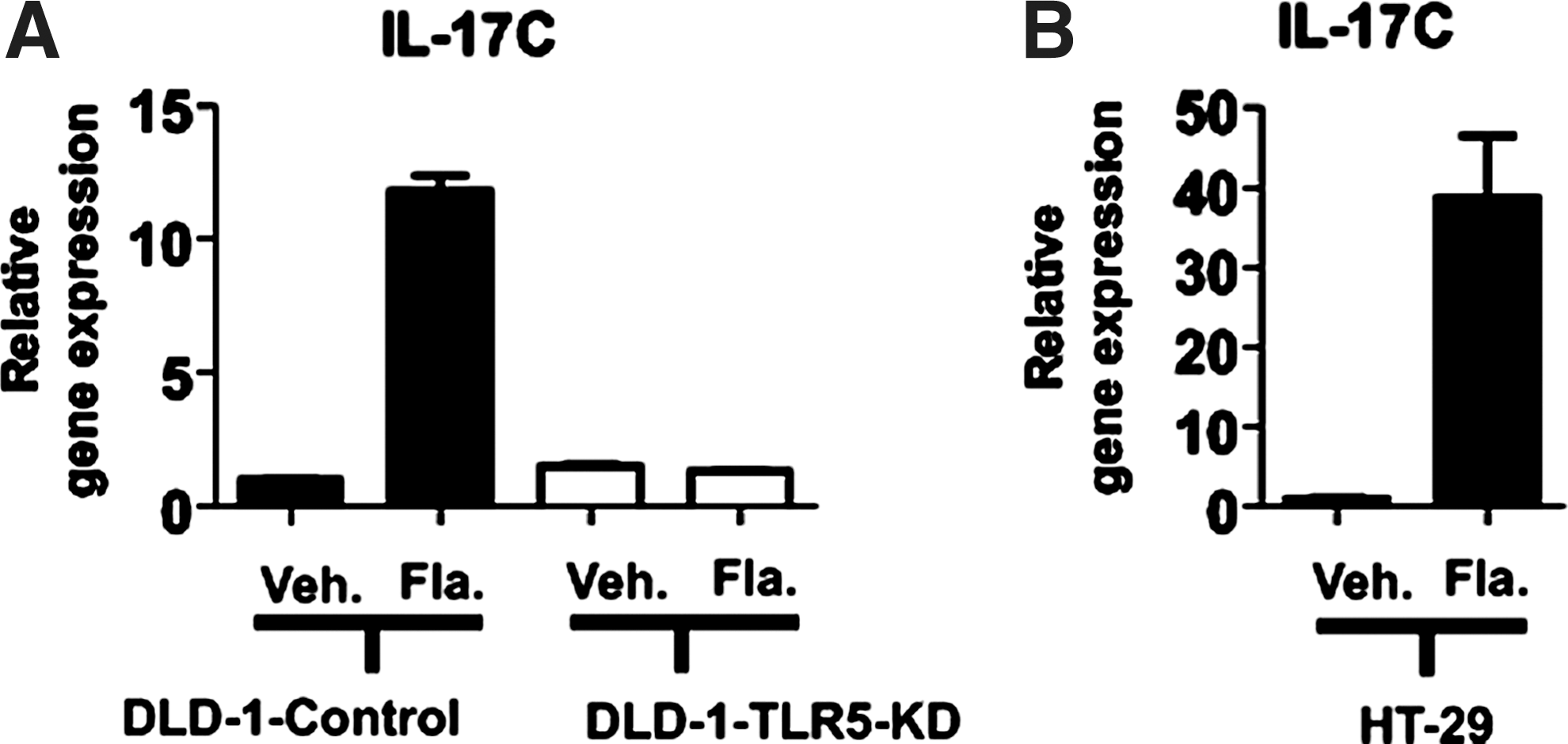
Flagellin stimulation elicits IL-17C expression in DLD-1 and HT-29 cells.
Furthermore, we tested whether another type of human colon adenocarcinoma cell line, HT-29 cells, could also express IL-17C in response to flagellin. Similar to DLD-1 and NCM460 cells, strong IL-17C expression was measured in HT-29 cells on flagellin stimulation compared with vehicle-treated HT-29 cells (Fig. 3B).
Together, these data show that, in addition to NCM460 cells, TLR5 activation induces IL-17C expression in other types of human intestinal epithelial cells, DLD-1 and HT-29. Thus, TLR5-induced IL-17C expression appears to be one of the general outcomes of TLR5 activation at least in human intestinal epithelial cells. It should be noted that TLR5-silenced DLD-1 cells failed to induce IL-17C expression on flagellin. Together with the data (Fig. 2C), this result further demonstrates that flagellin-induced IL-17C expression is specifically mediated by TLR5.
IL-17C expression is elevated in human IBD patients
A family of IL-17 consists of crucial inflammatory cytokines that regulate the pathophysiology of various inflammatory diseases. IBD represented by UC and CD is a representative inflammatory disease in a human bowel system in which intestinal epithelial cells are continuously in contact with gut microbes. Our data showed that flagellin recognition by TLR5 elicited robust IL-17C expression in intestinal epithelial cells. In this context, we hypothesized that IL-17C expression would be augmented in IBD patients' tissues. To test this, we evaluated the expression of IL-17 family members in the intestinal tissues from human IBD patients characterized into UC and CD. We found that IL-17C expression was remarkably enhanced in UC patients relative to normal control subjects (Fig. 4A). However, the expression level of IL-17C in CD patients was not significantly changed compared with normal subjects. In addition, the expression of IL-17A and IL-17F were substantially elevated in both UC and CD patients compared with control subjects (Fig. 4B, E), while the expression levels of IL-17B and IL-17D were similar in the tissues from IBD patients and normal subjects (Fig. 4C, D). In light of enhanced inflammatory responses in inflamed tissues, CD and UC tissues were featured with an augmented pro-inflammatory cytokine, IL-1β, expression, compared with normal tissues (Fig. 4F).
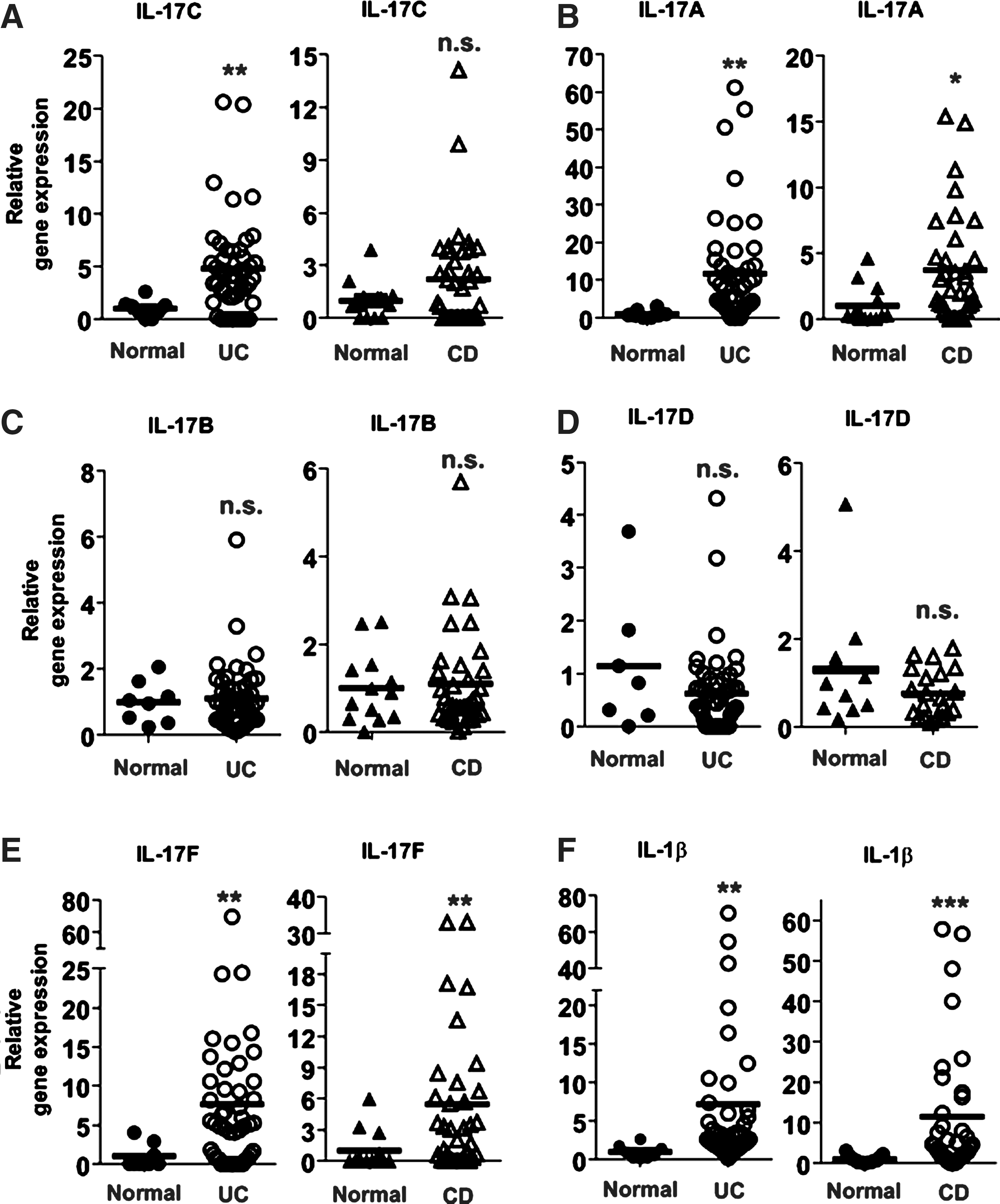
IL-17C expression is augmented in ulcerative colitis patients.
These data suggest that the expression of IL-17 family members could be associated with the development and progress of the intestinal inflammation. Given the facts that IL-17C expression is up-regulated by flagellin-TLR5 engagement in intestinal epithelial cells and the intestinal tissues from UC patients is characterized by enhanced IL-17C expression, our study suggests that, by enhancing IL-17C expression in intestinal epithelial cells, TLR5-mediated microbial recognition could play an important role in regulating the pathophysiology of intestinal inflammatory diseases.
Discussion
Through microarray analysis, we found that TLR5 activation by flagellin elicited potent IL-17C protein production and IL-17C mRNA expression in nontransformed human intestinal epithelial cells. Moreover, flagellin-induced IL-17C expression was also observed in other intestinal epithelial cells such as DLD-1 and HT-29 cells. It is noticeable that flagellin stimulation induced more potent IL-17C mRNA expression in NCM460 (fold change ≥150) cells than DLD-1 (fold change ≥11) or HT-29 (fold change ≥38) cells, compared with corresponding vehicle-treated control cells (Figs. 2 and 3). Similarly, while flagellin stimulation elicited robust IL-17C protein production in NCM460 cells, we could not measure IL-7C protein production in flagellin-treated DLD-1 and HT-29 cells. These data indicate that with regard to flagellin-induced IL-17C expression, NCM460 cells are more sensitive to flagellin stimulation than DLD-1 and HT-29 cells. Although originally developed from human colon tissues, DLD-1 and HT-29 cells are adenocarcinoma cells from terminal colon cancer patients and, therefore, harbor various oncogenic mutations such as K-Ras (Rhee and others 2008), p53(Rodrigues and others 1990; Ray and others 2001), and APC mutation (Rowan and others 2000), which may compound TLR5-induced responses. In contrast, NCM460 cells are developed from normal human colon (Moyer and others 1996; Kumar and others 1997), and, therefore, NCM460 cells have many advantages compared with adenocarcinoma cell lines in response to flagellin, resulting in potent cytokine gene expression. This might be the reason that we were able to measure TLR5-induced IL-17C protein production in NCM460 cells, but not in DLD-1 and HT-29 cells, while IL-17C mRNA expression by TLR5 activation was observed in all these cells.
The list of genes whose expression levels were altered (fold change ≥2, P<0.05) by flagellin stimulation was obtained by analyzing the microarray data after making a comparison of vehicle- and flagellin-treated NCM460-control cells. Then, to obtain IL-17C interactome profiling in the mammalian system, we performed IPA Core Analysis to interpret the microarray data in the context of biological functions, pathways, and networks. The most significant IPA-generated gene networks were presented in Fig. 5. The networks suggested NFκB and IRF1 as the transcription factors that would be possibly involved in regulating the expression of IL-17C by TLR5 activation. Moreover, biological pathways algorithmically based on their connectivity implied that granzyme B (GZMB) and an inflammatory cytokine IL-1β may have a significant biological relationship with IL-17C. Since recent studies suggested that IL-17C binds to a receptor complex of IL-1RA and IL17-RE (Ramirez-Carrozzi and others 2011; Song and others 2011), it would be worthwhile studying whether IL-17C stimulation alters the expression of GZMB or IL-1β. Thus, the network analysis confers a clue to speculate significant biological relationships between IL-17C and related molecules.
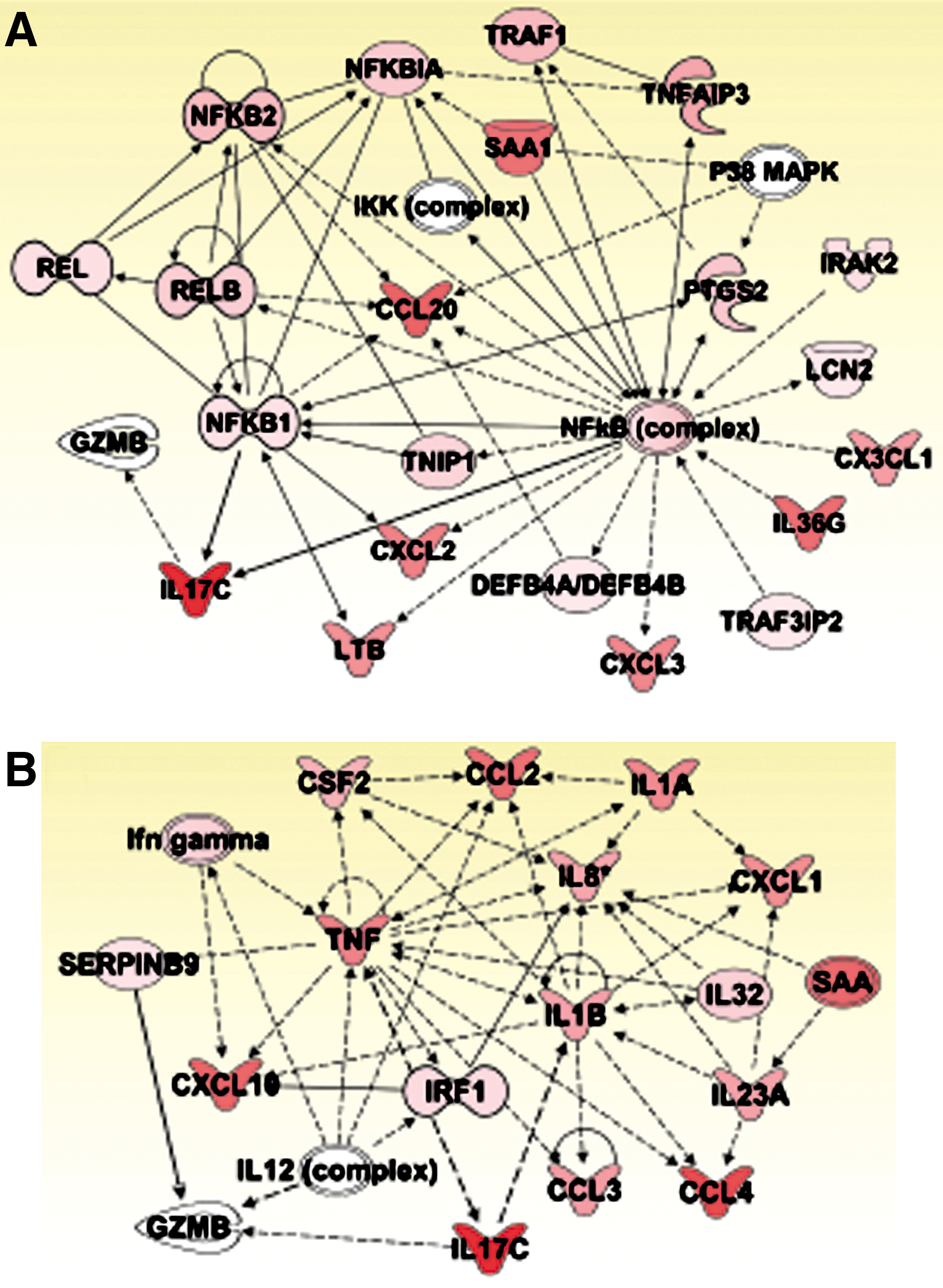
IPA generates IL-17C interactome.
Many studies indicated that a family of IL-17 cytokine plays an essential role in host defense and various inflammatory diseases, including psoriasis, RA, asthma, or IBD (Pappu and others 2011). While IL-17C expression in IBD remained to be examined, in agreement with our data (Fig. 4B, E), elevated IL-17A and F expressions were indeed previously shown in IBD patients (Fujino and others 2003; Seiderer and others 2008). Since clinical trials conducted with humanized IL-17A antibody showed that the neutralization of IL-17A could provide therapeutic benefits to psoriasis or RA or uveitis patients (Genovese and others 2010; Hueber and others 2010), investigating the expression of IL-17 in IBD would provide important information on a pro-inflammatory cytokine profile of the disease.
Since our study demonstrated that TLR5 activation by flagellin elicited strong IL-17C expression, which is a crucial cytokine in inflammatory disease activity, it would be reasonable to consider TLR5-induced events in pro-inflammatory responses. However, the impacts of TLR5-induced responses in the intestine appear to be controversial. Dr. Gewirtz's group reported that 10% of TLR5−/− mice (generated by Dr. Akira) exhibited spontaneous colitis in a TLR4-dependent manner (Vijay-Kumar and others 2010; Vijay-Kumar and others 2007), suggesting that TLR5 may protect intestinal mucosa from inflammation. However, spontaneous colitis in TLR5−/− mice was not observed in rederived TLR5−/− mice (Vijay-Kumar and others 2010), implying that the incidence of spontaneous colitis in TLR5−/− mice is caused by unique commensal microflora, which may be dependent on a housing environment. They also suggested that TLR5−/− mice had a metabolic syndrome, making the mice obese (Vijay-Kumar and others 2010). On the other hand, Dr. McSorley's group recently demonstrated that TLR5−/− mice developed neither spontaneous colitis nor metabolic defects (Letran and others 2011). In the study, TLR5−/− mice were simultaneously maintained in 2 independent animal facilities, and they concluded that TLR5−/− mice developed neither spontaneous colitis, nor metabolic defects. Moreover, the other line of TLR5−/− mice independently generated by Dr. Flavell's group has a normal phenotype without any obvious abnormalities (Feuillet and others 2006). Collectively, these studies demonstrate that spontaneous colitis and metabolic deficiency are not an obligatory phenotype of TLR5−/− mice (Feuillet and others 2006; Letran and others 2011). Meanwhile, several studies suggested that flagellin-TLR5 engagement were able to promote intestinal inflammation (Rhee and others 2005; Sitaraman and others 2005; Targan and others 2005; Gewirtz and others 2006; Carvalho and others 2008; Ivison and others 2009), and the level of serum antibody against flagellin is elevated in IBD patients (Lodes and others 2004; Yu and others 2011), while a few studies indicate that flagellin may protect mucosal epithelium against mucosal insults (e.g., radiation) (Burdelya and others 2008; Vijay-Kumar and others 2008). Therefore, it still remains to be studied whether TLR5-induced responses provoke inflammatory impacts in the intestine. Since our study shows that flagellin-TLR5 engagement elicits potent IL-17C expression in intestinal epithelial cells, however, our finding may suggest a possibility that TLR5 activation could participate in evoking inflammatory impacts in the gut. Thus, our study may provide an extensive insight into understanding an impact of microbial recognition by TLR5 in the gut.
Footnotes
Acknowledgments
This work was supported by the Pusan National University Research Grant, 2012 (E.I.) and the National Institutes of Health, DK079015 (S.H.R.). The authors have no conflicting financial interests pertaining to the current study.
Author Disclosure Statement
No competing financial interests exist.