
Abstract
The endoribonuclease RNase-L is the terminal component of an interferon-regulated RNA decay pathway known as the 2′-5′-oligoadenylate (2–5A) system, whose established functions include antimicrobial and tumor suppressive activities. RNase-L activity requires binding of the small molecule 2–5A, leading to RNase-L dimerization and cleavage of single-stranded RNA. RNase-L expression is controlled post-transcriptionally by its 3′-untranslated region (3′ UTR), which exerts a strong negative effect on RNase-L levels. MicroRNAs (miRNAs) are a class of small noncoding RNAs that repress expression of target genes by binding to regions of complementarity often in the 3′ UTR. The miR-29 family acts as a tumor suppressor in several cancers, including acute and chronic myelogenous leukemia (CML), and has many oncogenic targets. We report that the miR-29 family represses RNase-L protein expression across several cell types. Using a luciferase reporter, we showed that miR-29 acts via 4 target sites within the RNASEL 3′ UTR. Mutation of all sites is required for abrogation of miR-29 repression. In light of the reported tumor suppressive role of miR-29 in K562 CML cells and miR-29 repression of RNase-L in these cells, we generated K562 cells with stable RNase-L knockdown and demonstrated that loss of RNase-L inhibits proliferation in vitro as well as tumor growth in a xenograft model. Our findings identify a previously unknown miRNA regulator of RNase-L expression and support a novel oncogenic role for RNase-L in CML and potentially other hematopoietic malignancies.
Introduction
MicroRNAs (miRNAs) are small noncoding RNAs that bind to regions of partial complementarity in target mRNAs to inhibit translation or enhance transcript turnover (Bartel 2004). A single miRNA can regulate multiple targets, and multiple miRNAs may regulate a single mRNA; thus, modulation of the cellular miRNA/target profile provides a potent mechanism to post-transcriptionally alter the gene expression program in distinct biologic settings. Indeed, miRNAs have emerged as critical regulators of virtually all physiologic and pathologic processes, including cancer (Bartel 2004; Croce 2012). The miR-29 family has been extensively studied in the context of human cancer, where it was shown to mediate either tumor suppressive or oncogenic functions in distinct malignancies (Pekarsky and Croce 2010; Kriegel and others 2012). The miR-29 family is comprised of 3 isoforms arranged in 2 clusters: the miR-29b-1/miR-29a cluster located at chromosome 7q32 and the miR-29b-2/miR-29c cluster at chromosome 1q23 (Kriegel and others 2012). Decreased expression of miR-29 members has been reported in many cancers, including rhabdomyosarcoma, cholangiocarcinoma, acute myelogenous leukemia (AML), lung cancer, nasopharyngeal carcinoma, and the aggressive form of chronic lymphocytic leukemia characterized by high ZAP-70 expression and unmutated IgH V(H) (Pekarsky and others 2006; Fabbri and others 2007; Mott and others 2007; Sengupta and others 2008; Wang and others 2008; Garzon and others 2009). Restoration of miR-29 sensitized cholangiocarcinoma and AML cells to apoptotic stimuli (Mott and others 2007; Garzon and others 2009) and inhibited growth of rhabdomyosarcoma, lung cancer, and chronic myelogenous leukemia (CML) xenografts in nude mice (Fabbri and others 2007; Wang and others 2008; Garzon and others 2009). Consistent with a tumor suppressor role for miR-29, confirmed targets of miR-29 repression include prosurvival and proproliferation factors such as the antiapoptotic protein myeloid cell leukemia sequence 1 (Mcl-1) (Mott and others 2007), the oncogenic protein T-cell leukemia/lymphoma 1 (Tcl1) (Pekarsky and others 2006), and cyclin-dependent kinase 6 (CDK6) (Zhao and others 2010), as well as other targets relevant to tumorigenesis, such as the DNA methyltransferases 3A and 3B (DNMT3A and B) (Fabbri and others 2007). In contrast, miR-29 upregulation is observed in a subset of malignancies, including breast cancer, colorectal cancer, prostate cancer, pancreatic cancer, and the indolent form of chronic lymphocytic leukemia characterized by low ZAP-70 expression and mutated IgH V(H) (Volinia and others 2006; Gebeshuber and others 2009; Santanam and others 2010). In this context, miR29 may exhibit oncogenic functions such as promotion of epithelial-to-mesenchymal transition in breast cancer cells (Gebeshuber and others 2009). Thus, understanding the roles of miR-29 in specific cancers and identifying the relevant mRNA targets that mediate its tumor suppressor or oncogenic activities are essential to develop miR-29 as a therapeutic target.
Our previous study demonstrating that RNase-L expression is post-transcriptionally downregulated via elements in the 3′ UTR of its mRNA (Li and others 2007) and the established role of miRNAs in repressing gene expression through sites in the 3′ UTR of target mRNAs suggested that miRNAs may negatively regulate RNase-L expression and modulate its biologic activities. Analysis of the RNase-L 3′ UTR with multiple miRNA target prediction algorithms identified several potential miRNA target sites. Among these, miR-29 has 4 predicted target sites in the RNase-L 3′ UTR and represented the best-candidate miRNA. Ectopic expression of miR-29 downregulated RNase-L, miR-29 knockdown upregulated RNase-L, and RNase-L was inversely correlated with miR-29 expression in several cell types indicating that endogenous RNase-L is regulated by miR-29. A luciferase reporter containing the RNase-L 3′ UTR exhibited miR-29-dependent downregulation, and mutation of miR-29 sites rescued luciferase expression, demonstrating functional regulation of the RNase-L 3′ UTR by miR-29. Consistent with our identification of RNase-L as a miR-29 target, RNase-L was identified as a candidate miR-29-regulated transcript in a study of miR-29-mediated tumor suppression in myelogenous leukemias (Garzon and others 2009), and we validated this regulation in K562 CML cells. The miR-29-dependent repression of RNase-L expression in the context of a tumor suppressor phenotype suggested a previously undescribed oncogenic role for RNase-L. To determine if RNase-L represented an important target of miR-29 tumor suppressor activity independent of other miR-29 targets, we stably knocked down RNase-L expression in K562 cells and analyzed the proliferative phenotype. Remarkably, RNase-L knockdown reduced K562 proliferation in culture, and more strikingly, inhibited tumorigenesis in nude mouse xenografts. Thus, RNase-L knockdown phenocopied the tumor suppressive activity of miR-29 in K562 xenografts, revealing a novel tumorigenic role for RNase-L in this setting. These findings suggest that RNase-L functions to promote tumorigenesis in some malignancies, and thus RNase-L inhibition may represent a viable strategy for therapeutic intervention.
Materials and Methods
Plasmids and miRNA precursors and inhibitors
The ZC5 and ZC5+ RNase-L constructs have been previously described (Zhou and others 1993; Li and others 2007). To generate the RNase-L 3′ UTR reporter, the firefly luciferase gene was cloned into the NheI and XhoI sites of the pcDNA3.1(+) plasmid after digestion with the appropriate restriction enzymes. The RNase-L ZC5+ construct was then partially digested with EcoRI, and the 3′ UTR fragment was gel-purified and cloned into the EcoRI site downstream of firefly luciferase. The resulting chimeric transcript contains both luciferase and the RNase-L 3′ UTR and utilizes the pcDNA3.1-encoded polyadenylation signal. All restriction enzymes were purchased from New England Biolabs. The Renilla luciferase plasmid was generously provided by Dr. Myriam Gorospe (National Institute on Aging, Baltimore). miR-29-site mutations were created using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) using primers that deleted the 7 nucleotides predicted to bind the seed region (nt2–8) of the miR-29 family. Primers used for mutagenesis were as follows: site A (forward 5′-GCACTTTATAAATTTATGATTGGTACCTCTCATTTGGGC-3′, reverse 5′-GCCCAAATGAGAGGTACCAATCATAAATTTATAAAGTGC-3′); site B (forward 5′-CCAGACAAAAATATCAAGAGGTTGAGAAAACCTGAC-3′, reverse 5′-GTCAGGTTTTCTCAACCTCTTGATATTTTTGTCTGG-3′); site C (forward 5′-CTGTCTTACGTTTTTCTTATAATGTATACATTACATCTGAG-3′, reverse 5′-CTCAGATGTAATGTATACATTATAAGAAAAACGTAAGACAG-3′); site D (forward 5′-CTTGATTTGAACAAATTTTCAAGTCTGATGTTCTTTCCATG-3′, reverse 5′-CATGGAAAGAACATCAGACTTGAAAATTTGTTCAAATCAAG-3′). Constructs were verified by sequencing (Biopolymer/Genomics Core Facility, University of Maryland, Baltimore). pGIPZ-encoded nonspecific and RNase-L short-hairpin RNAs (shRNAs) were purchased from Open Biosystems. miR-29 Pre-miR miRNA precursors and mirVana miRNA inhibitors were purchased from Ambion.
Cell culture, transfection, and transduction
293T, HeLa, and MDA-MB-231 cells were passaged in the Dulbecco's modified Eagle's medium (Cellgro) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals), antibiotic/antimycotic (Invitrogen), and 2.5 μg/mL Plasmocin (InvivoGen). K562 cells were passaged in RPMI 1640 (Cellgro) with
Antibodies and Western blotting
Cell lysates were prepared using a radioimmunoprecipitation assay buffer (Millipore) plus a protease inhibitor cocktail (Sigma). Protein concentrations were determined using the Bradford protein assay (Bio-Rad), and equal amounts of protein per sample were separated on 10% SDS-PAGE gels (Bio-Rad). Proteins were then electrotransferred to Immobilon-P membranes (Millipore). Membranes were blocked for 1 h at room temperature in a TBST buffer (10 mM Tris, pH 8.0, 150 mM NaCl, and 0.1% Tween 20) plus 10% FBS (RNase-L antibody only) or 5% nonfat milk (all other antibodies). Anti-human RNase-L mouse monoclonal antibody (clone 2E9; Alexis Biochemicals) was used at a 1:1,000 dilution in TBS-T plus 5% bovine serum albumin. All other antibodies were diluted in TBS-T plus 5% nonfat milk. Anti-human MCL-1 mouse monoclonal antibody (clone 22; BD Biosciences) was used at 2 μg/mL. Anti-human DNMT3A rabbit polyclonal antibody (Cell Signaling) was used at 1:1,000. Anti-human β-actin (clone AC15) and α-tubulin (clone B-5-1-2) mouse monoclonal antibodies (Sigma) were used at 1:10,000 dilutions. Membranes were incubated with a primary antibody at 4°C overnight and then incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch) at 1:10,000 dilution in TBST plus 5% nonfat milk for 1 h at room temperature. Membranes were washed again and visualized using a SuperSignal West Pico Chemiluminescent Substrate (Pierce) and Hyblot CL autoradiography film (Denville Scientific).
Quantitative real-time polymerase chain reaction
Total RNA was prepared using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Concentration and 260/280 ratios were determined on a Tecan Infinite 200 Promultimode reader. mRNA quantitative real-time polymerase chain reaction (qRT-PCR) analysis was carried out on a CFX96 Touch Real-Time PCR Detection System using the iScript One-Step RT-PCR Kit with SYBR Green (Bio-Rad) and 0.5μg total RNA per triplicate reaction. Primers for human DICER were as follows: forward 5′-AGTGCCAGTCCTGCAGTAGTTGAT-3′; reverse 5′-ATGTGATTCACCAACATGCCAGCC-3′. Primers for human ribosomal protein L13a (rpl13a) have been previously described (Li and others 2007). Primers for human RNase-L were as follows: forward 5′-CAGGATCTGCAACCACAAAA-3′; reverse 5′-CCCACTTGATGCTCTTATCAAA-3′. Primers for human hydroxymethylbilane synthase were designed by Vandesompele and others (2002): forward 5′-GGCAATGCGGCTGCAA-3′; reverse 5′-GGGTACCCACGCGAATCAC-3′. Cycling conditions were as follows: 50°C for 5 min; 95°C for 10 min; and 40 cycles of 95°C for 10 s and 58°C for 30 s. miRNAs and control U6 small RNAs were detected by qRT-PCR using TaqMan miRNA Assays (Applied Biosystems) according to the manufacturer's instructions.
Luciferase assays
293T cells were seeded in 12-well plates at 350,000 cells per well and allowed to adhere overnight. Cells were then transfected with a combination of firefly luciferase reporter (0.5 μg per well) and Renilla internal control (5 ng per well) plasmids, along with the indicated miRNA mimetics at 25 nM. Cells were harvested 24 h later and analyzed for luciferase activity on a PerkinElmer Victor X3 Multilabel Plate Reader using the Dual Luciferase Reporter Assay (Promega) according to the manufacturer's instructions. Within each sample, firefly was normalized to Renilla luciferase, and for each reporter construct, the firefly/Renilla ratio of the nonspecific control-transfected sample was set to 1.
Proliferation studies
K562 cell proliferation was measured using the Promega Cell Titer 96 Nonradioactive Cell Proliferation MTT assay according to the manufacturer's instructions. For proliferation studies, 1000 K562 cells were seeded in triplicate in 100 μL complete RPMI per well in 96-well plates. MTT dye was added to a new plate and absorbance measured at 570 nM daily over 4 days. For plates measured on later days, cells were spun down, and 25 μL of media was replaced daily.
Xenograft studies
All mouse experiments were carried out by the University of Maryland Translational Core Laboratory (Baltimore, MD). Mice were maintained in accordance with the protocols approved by the Institutional Animal Care and Use Committee at the University of Maryland. For xenograft studies, 6-week-old female nude (nu/nu) mice were subcutaneously injected in the flank with K562 cells. Cells were washed and reconstituted in phosphate-buffered saline (PBS; Cellgro), and 100 μL of PBS containing 10 million cells was mixed with an equal amount of Matrigel (BD Biosciences) before injection. Each mouse was injected with a matched pair of K562 cell lines, the shNS cells in the left flank and the shRNL cells in the right flank. Each pair of cells was tested in 10 mice (n=10). Mice were monitored every 2 to 3 days for weight loss and tumor size. Tumor length and width measurements were taken with calipers, and the tumor volume was calculated according to the following formula: (volume=width2×length/2). Mice were euthanized and tumors were weighed and frozen for protein analysis when tumor volume on either side exceeded 1,500 mm3.
Results
miRNA-dependent repression of RNase-L by the miR-29 family
Our previous report of the post-transcriptional, 3′ UTR-dependent repression of RNase-L expression suggested that this regulation may be mediated by miRNAs. To test the hypothesis that miRNAs are involved in RNase-L repression, we used shRNAs to knockdown the essential miRNA-processing enzyme DICER in 293T cells. DICER knockdown was confirmed by qRT-PCR and resulted in an increase in RNase-L protein (Fig. 1A), indicating that production of mature miRNAs is necessary for RNase-L repression. This approach does not rule out the possibility of indirect regulation (i.e., miRNAs targeting a regulator of RNase-L); therefore, we used miRNA target prediction algorithms to identify miRNAs that may directly interact with the RNase-L message. Analysis of the RNase-L 3′ UTR (NM_021133) using TargetScan (version 5.1) (Lewis and others 2003) and
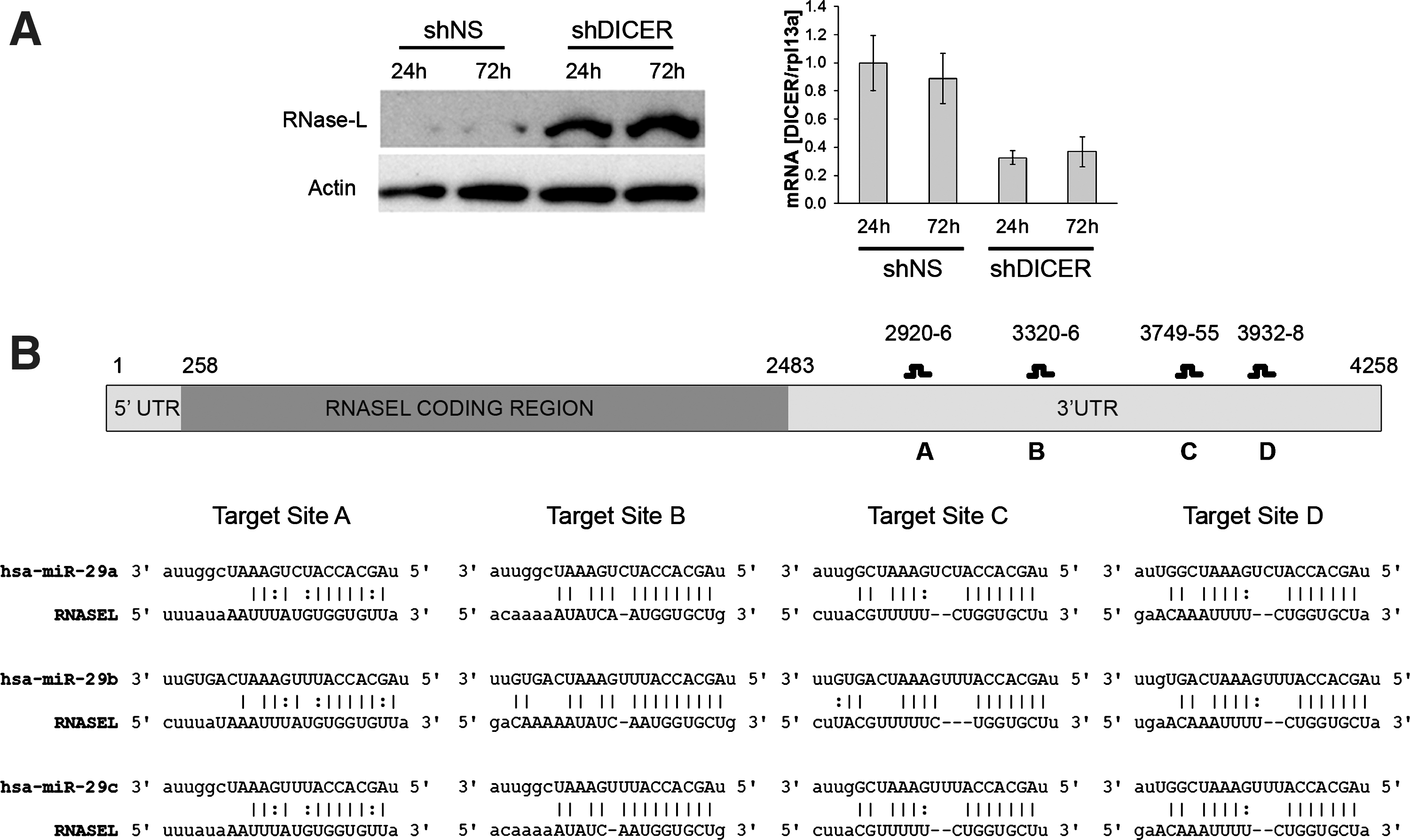
miRNA-mediated repression of RNase-L and identification of miR-29 family target sites in the RNase-L 3′ UTR.
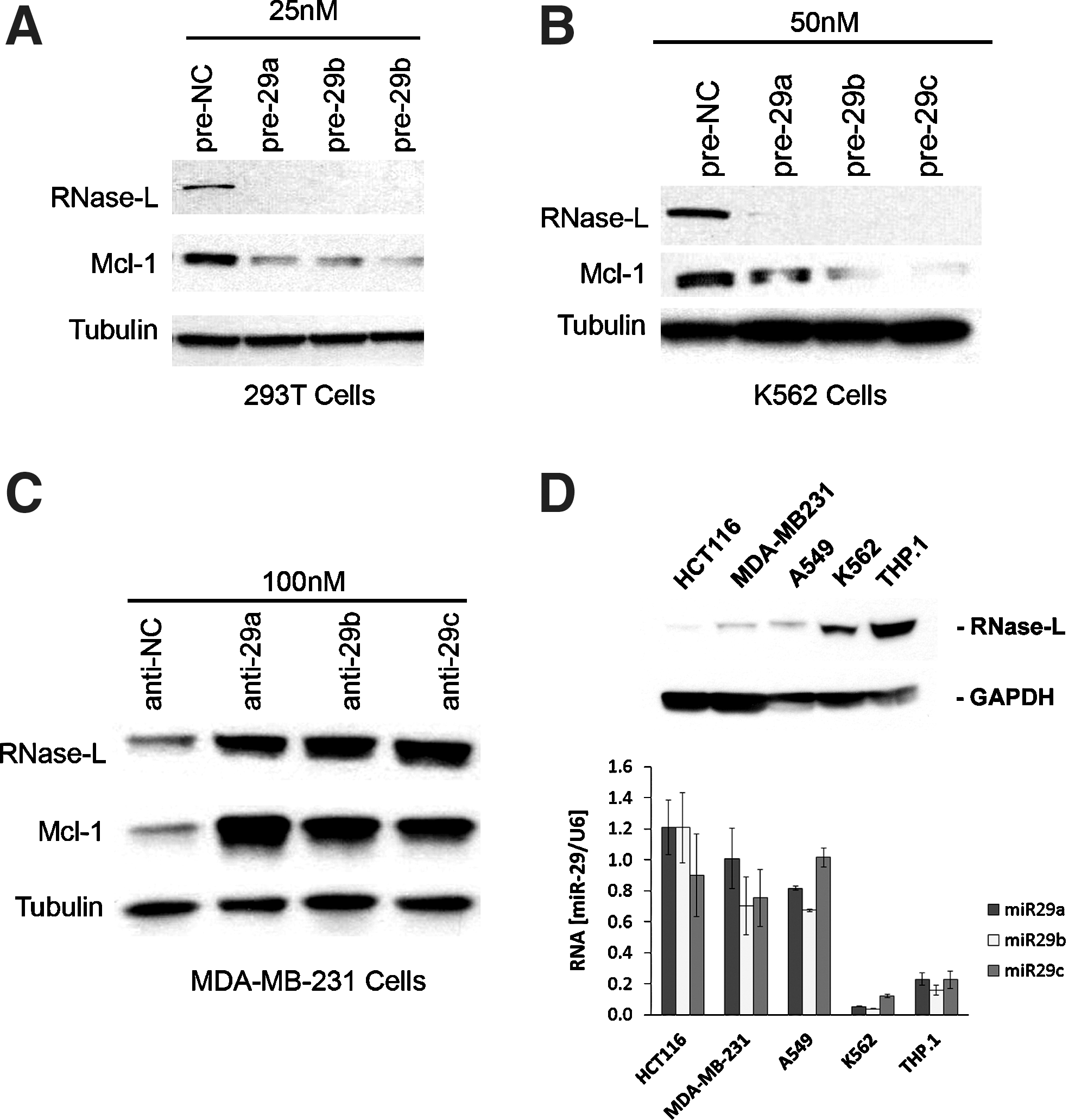
RNase-L is regulated by the miR-29 family in multiple cell types.
miR-29 represses RNase-L via multiple target sites in its 3′ UTR
Given that multiple miR-29 sites were identified in the RNase-L 3′ UTR, we expected that deletion of the 3′ UTR would abrogate miR-29-mediated regulation. Accordingly, a combination of pre-miR-29a/b/c was cotransfected with an RNase-L expression construct that contained (ZC5+) or lacked (ZC5) the 3′ UTR (Zhou and others 1993; Li and others 2007). Consistent with the presence of miR-29-binding sites in the RNase-L 3′ UTR, miR-29 potently repressed RNase-L expression from the ZC5+ construct, but did not markedly alter expression from ZC5 (Fig. 3A). To quantify the contributions of individual target sites to miR-29-mediated RNase-L repression, the human RNase-L 3′ UTR was cloned downstream of the firefly luciferase reporter gene. Transfection of the reporter construct and pre-miR-29 family members into 293T cells resulted in a significant inhibition of luciferase activity that was not observed in the absence of the 3′ UTR; thus, the RNase-L 3′ UTR conferred miR-29-dependent regulation by all 3 family members (Fig. 3B). Mutation of miR-29 target sites that are critical for RNase-L repression is predicted to rescue expression and luciferase activity. Therefore, we generated mutations in which the seed regions (nt2–8) of predicted miR-29 target sites (designated A–D) were deleted (Fig. 1B). Mutation of all 4 target sites (mutABCD) resulted in the complete loss of miR-29-mediated repression and full rescue of luciferase activity (Fig. 3B), while deletion of between 1 and 3 target sites led to partial repression that was proportional to the number of intact target sites (Supplementary Fig. S2). Target site A was the weakest, as it was neither necessary for complete repression (mutBCD), nor capable of mediating repression on its own (mutA). Nonetheless, site A exhibited some functionality, as its presence enhanced repression by other more robust target sites (e.g., mutABD versus mutBD). Together, these findings identify miR-29-mediated repression as a novel mechanism regulating RNase-L expression.
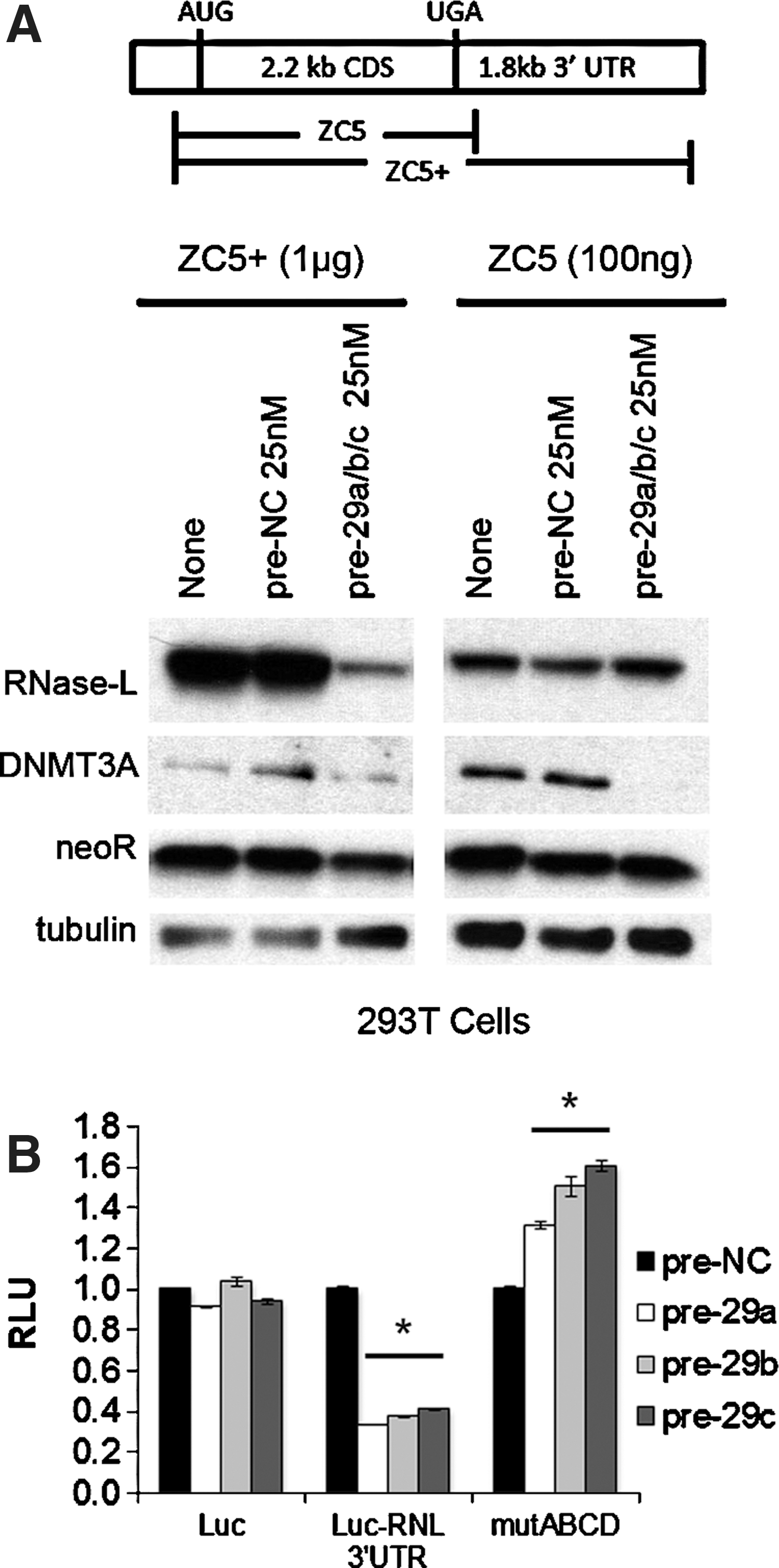
miR-29 represses RNase-L via multiple target sites in the 3′ UTR.
miR-29-dependent regulation reveals a novel role for RNase-L in tumorigenesis
Our data demonstrating the miR-29-dependent regulation of RNase-L in cell culture systems suggested that RNase-L is an important miR-29 target and contributes to its biologic functions in physiologic and pathologic settings. Consistent with this prediction, a study of miR-29 tumor suppressor activity in myelogenous leukemias identified RNase-L as the most highly repressed transcript after miR-29 transfection of K562 CML cells (Garzon and others 2009). We demonstrated that miR-29 transfection also downregulated RNase-L protein, validating their inverse relationship in K562 cells (Fig. 2B). The role of RNase-L in antiproliferative and tumor suppressor activities is well established (Hassel and others 1993; Castelli and others 1997; Andersen and others 2007), so its identification as a target of miR-29 repression and tumor suppressor activity was surprising, and suggested that RNase-L may serve a novel oncogenic function in this setting. miR-29 regulates the expression of many transcripts that may contribute to its tumor suppressor activity (e.g., Mcl-1, Tcl1, CDK6, DNMT3A, and 3B) (Pekarsky and others 2006; Fabbri and others 2007; Mott and others 2007; Zhao and others 2010); to examine the role of RNase-L in miR-29-mediated tumor suppressor activities independent of other miR-29 targets, we stably knocked down RNase-L in K562 cells (Fig. 4A). An 80-kDa immunoreactive band that migrates just below the authentic 83-kDa RNase-L is observed in some conditions and cell types, but is not consistently repressed by shRNA directed against RNase-L, thus demonstrating its nonspecific nature. RNase-L knockdown resulted in a modest reduction in the proliferation of cultured cells (42% in the pools and 31% in the clones, Fig. 4B), suggesting that RNase-L functions to stimulate proliferation in K562 cells. Similar results were observed in all knockdown clones tested (data not shown). More strikingly, RNase-L knockdown dramatically inhibited tumorigenesis in nude mouse xenografts, whereas nonspecific shRNA-transfected control cells exhibited robust tumor growth (Fig. 4C). All control shRNA K562 cells formed detectable tumors by 11 days postinjection, whereas RNase-L knockdown cells failed to produce tumors in the majority of mice even at 26 days postinjection. Furthermore, RNase-L knockdown cells failed to produce tumors in several mice that were monitored through day 46 postinjection (data not shown). In the few small tumors generated from RNase-L knockdown cells, functional knockdown of RNase-L expression was maintained at the time of tumor harvest (Supplementary Fig. S3A). Thus, RNase-L knockdown phenocopied the tumor suppressive activity of miR-29 in K562 xenografts, revealing a novel tumorigenic role for RNase-L in this setting. This remarkable phenotype provides the first evidence of a tumorigenic role for RNase-L, and suggests that RNase-L mRNA is an important target of miR-29 tumor suppressor activity. Consistent with this finding, RNase-L activity is increased in CML patient samples (Hubbell and others 1994), and was among the top 10% of upregulated genes in a large microarray analysis of multiple leukemias, including CML and B-cell acute lymphocytic leukemia (Rhodes and others 2004; Haferlach and others 2010). However, further studies are required to determine the specific settings in which RNase-L mediates oncogenic functions and the molecular mechanisms involved in these activities.
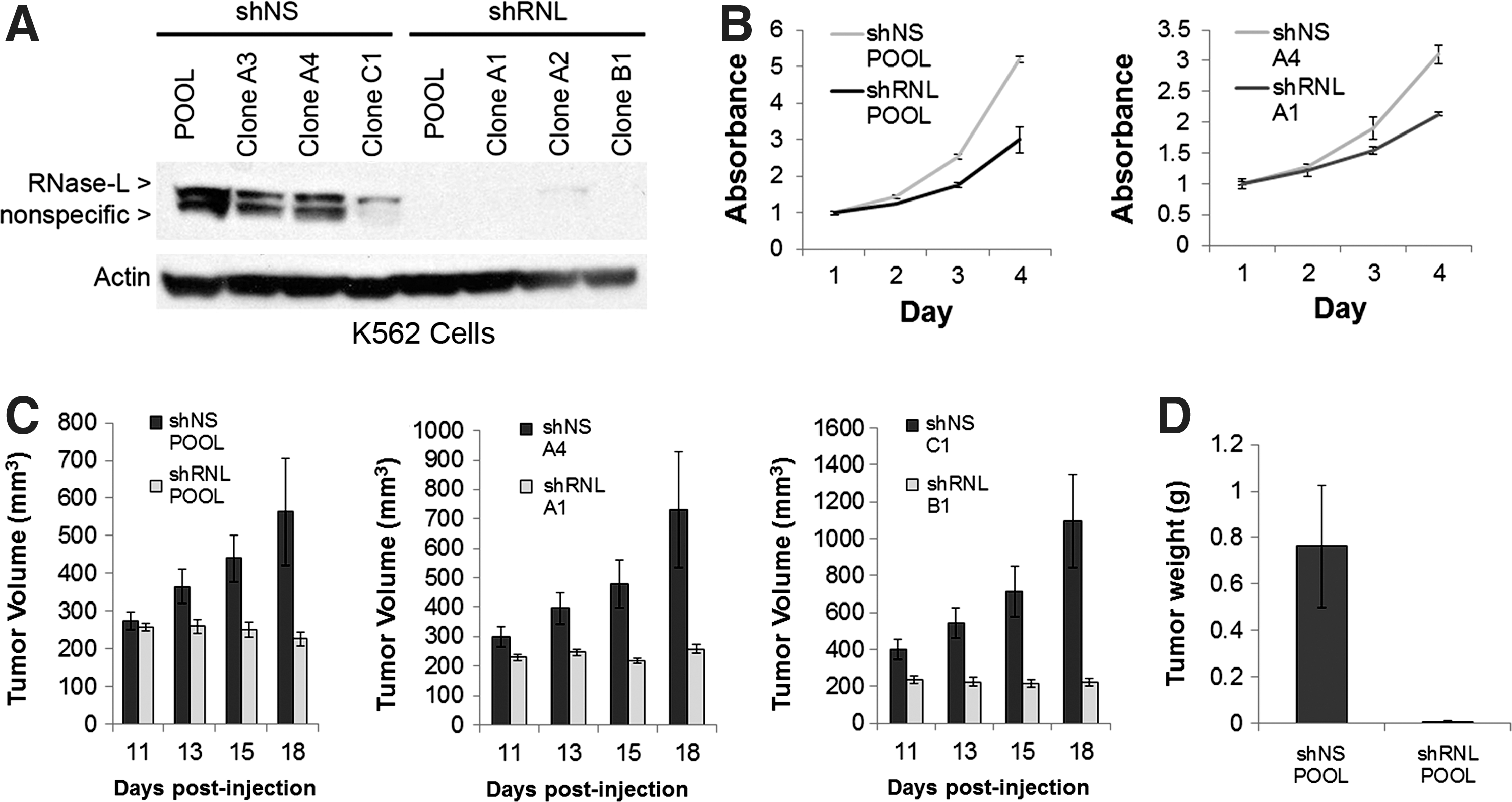
RNase-L promotes K562 proliferation and xenograft growth.
Discussion
In this study, we demonstrate that RNase-L expression is post-transcriptionally regulated by the miR-29 family of miRNAs via 4 target sites in its 3′ UTR. The RNase-L 3′ UTR-mediated repression of a luciferase reporter required multiple miR-29 sites, as deletion of at least 3 sites was necessary to rescue expression (Fig. 3 and Supplementary Fig. S2). Consistent with this finding, 3 of the miR-29 sites occur in a region that we previously reported to negatively regulate RNase-L expression (Li and others 2007). At the same time, binding of miR-29 to the RNase-L 3′ UTR may impact the activities of additional 3′ UTR regulatory elements and their cognate binding factors (van Kouwenhove and others 2011). Indeed, our prior study identified AU-rich elements (AREs) in the RNase-L 3′ UTR that interact with ARE-binding proteins (AREBPs) to modulate mRNA stability and translation (Li and others 2007). Interestingly, the fourth miR-29 site in the RNase-L 3′ UTR is adjacent to a binding site for the AREBP HuR that enhances RNase-L expression in response to stress stimuli; therefore, miR-29 and HuR may mediate antagonistic effects on RNase-L expression in conditions where they are expressed together. In addition to the several miR-29-binding sites, other potential miR-binding sites were identified in the RNase-L 3′ UTR; the presence of multiple candidate miRNA- and RNABP-binding sites in the RNase-L 3′ UTR provides a platform for complex regulatory interactions to mediate rapid modulation of RNase-L expression. Identification of the specific cis-elements and trans-acting regulators that control RNase-L expression in distinct biologic settings may reveal strategies to modulate its expression for therapeutic applications.
The strong miR-29-dependent regulation of RNase-L involving multiple target sites suggests that it is an important target in miR-29-mediated biologic functions. Consistent with this prediction, miR-29 family members and RNase-L are implicated in an overlapping set of physiologic and pathologic activities, including antiviral immunity (Silverman 2007b; Ahluwalia and others 2008; Bandyopadhyay and others 2011), tumorigenesis (Casey and others 2002; Pekarsky and others 2006; Volinia and others 2006; Fabbri and others 2007; Liu and others 2007; Mott and others 2007; Madsen and others 2008; Sengupta and others 2008; Wang and others 2008), and myogenesis (Wang and others 2008; Salehzada and others 2009; Winbanks and others 2011). In this study, we focused on the regulation of RNase-L in the context of a tumor suppressor function for miR-29 in myelogenous leukemias, as RNase-L was identified as a candidate miR-29 target in this system (Garzon and others 2009). miR-29 transfection of K562 CML cells reduced proliferation, enhanced apoptosis, and inhibited tumorigenesis; therefore, our validation of RNase-L as a miR-29-repressed transcript in K562 cells suggested that RNase-L serves a novel oncogenic function in this setting. Consistent with this interpretation, stable knockdown of RNase-L, independent of other miR-29 targets, decreased K562 proliferation and inhibited tumorigenesis in a xenograft model (Fig. 4). However, RNase-L knockdown did not phenocopy the enhanced sensitivity to proapoptotic chemotherapeutic agents observed after ectopic expression of miR-29 (Mott and others 2007; Garzon and others 2009), indicating that other miR-29 targets (e.g., Mcl-1) also contribute to its tumor suppressor phenotype (Supplementary Fig. S2B). Importantly, endogenous RNase-L is functional in K562 cells (Castelli and others 1998); therefore, a mutation in RNase-L is unlikely to account for the tumorigenic phenotype. These findings provide the first evidence of a functional role for RNase-L in tumorigenesis and are supported by previous reports of cancer-associated increases in RNase-L activity and expression in CML and colon cancer (Hubbell and others 1994; Wang and others 1995). Furthermore, microarray analyses revealed that RNase-L is upregulated in diverse types of leukemia (Rhodes and others 2004; Haferlach and others 2010). RNase-L, however, was not altered in a separate analysis of AML (Garzon and others 2009), suggesting that it contributes to the oncogenic phenotype in a subset of hematopoietic malignancies. Further work is required to determine the extent to which RNase-L functions in proliferation and tumorigenesis in other types of cancer.
Our data indicating a tumorigenic role for RNase-L contrasts with the more-established antiproliferative and tumor suppressor activities of RNase-L (Hassel and others 1993; Castelli and others 1997; Andersen and others 2007) and suggest that it mediates context-specific tumor suppressor and oncogenic activities as has been reported for other proteins and regulatory RNAs (e.g., c-myc and miR-29) (Larsson and Henriksson 2010; Pekarsky and Croce 2010). This dual functionality is often observed for regulators of gene expression and is thought to reflect distinct profiles of targets or cofactors that promote the expression of tumor suppressor or oncogenic gene products. We hypothesize that RNABPs expressed in different cell types or cancers target RNase-L cleavage to specific RNAs and, in turn, dictate its function as an oncogene or tumor suppressor. Indeed, the dysregulation of RNABPs has been reported in multiple malignancies (Abdelmohsen and Gorospe 2010; Baou and others 2011); these and other cancer-associated alterations may lead to aberrant substrate recognition and cleavage by RNase-L. Consistent with this model, microarray analyses revealed distinct profiles of RNase-L-regulated RNAs in different cell types and conditions (Malathi and others 2005; Salehzada and others 2009; Ezelle and Hassel 2012). A comparison of RNase-L substrates in healthy and cancer tissues will provide insights into potential mechanisms by which RNase-L contributes to tumorigenic activity. In this regard, the diminished tumor growth in K562 RNase-L knockdown xenografts did not provide sufficient material for analysis of RNase-L targets in the context of tumorigenesis. Therefore, future studies will employ a conditional RNase-L knockdown approach in a mouse model of leukemogenesis to analyze the impact of RNase-L on gene expression and disease progression.
The discovery that RNase-L serves a novel oncogenic function as a target of miR-29 regulation has important clinical implications. Elevated levels of RNase-L may serve as a biomarker of miR-29 downregulation and compromised tumor suppressor activity. From a therapeutic standpoint, modulation of miRNA expression has shown efficacy in animal models and is an area of intense investigation (van Rooij and others 2012). Furthermore, the direct targeting of clinically relevant miRNA targets provides an alternate strategy that may increase specificity and potency. Therefore, as suggested by the antitumor activity of RNase-L knockdown, RNase-L inhibitors may represent a novel class of therapeutic agent for tumors in which RNase-L functions to promote malignancy. In this regard, sunitinib, an ATP-competitive inhibitor that is an FDA-approved chemotherapeutic agent, was recently shown to inhibit RNase-L activity (Jha and others 2011); thus, sunitinib may have novel indications as an RNase-L-targeting agent. Our findings demonstrate that RNase-L is an important target of miR-29 and identify a previously unknown role for RNase-L in tumorigenesis, opening a door for further investigations into the mechanisms behind its dual function.
Footnotes
Acknowledgments
The authors would like to thank Sarah Brennan-Laun, Tiha Long, and Kristina Pedersen for critical discussion of the manuscript. This study was funded by an NIH grant AI077556 (BAH) and a VA Merit Award (BAH). TYL is supported by an NIH grant T32 AI07540 (JBK) and the University of Maryland School of Medicine MSTP program.
Author Disclosure Statement
BAH is an inventor on patents relating to RNase-L licensed to Alios BioPharma. No competing financial interests exist for all other authors.