
Abstract
Hydroxychloroquine (HCQ) is an antimalarial drug also used in treating autoimmune diseases. Its antiviral activity was demonstrated in restricting HIV infection in vitro; however, the clinical implications remain controversial. Infection with dengue virus (DENV) is a global public health problem, and we lack an antiviral drug for DENV. Here, we evaluated the anti-DENV potential of treatment with HCQ. Immunofluorescence assays demonstrated that HCQ could inhibit DENV serotype 1–4 infection in vitro. RT-qPCR analysis of HCQ-treated cells showed induced expression of interferon (IFN)-related antiviral proteins and certain inflammatory cytokines. Mechanistic study suggested that HCQ activated the innate immune signaling pathways of IFN-β, AP-1, and NFκB. Knocking down mitochondrial antiviral signaling protein (MAVS), inhibiting TANK binding kinase 1 (TBK1)/inhibitor-κB kinase ɛ (IKKɛ), and blocking type I IFN receptor reduced the efficiency of HCQ against DENV-2 infection. Furthermore, HCQ significantly induced cellular production of reactive oxygen species (ROS), which was involved in the host defense system. Suppression of ROS production attenuated the innate immune activation and anti-DENV-2 effect of HCQ. In summary, HCQ triggers the host defense machinery by inducing ROS- and MAVS-mediated innate immune activation against DENV infection and may be a candidate drug for DENV infection.
Introduction
F
Mosquitoes transmitting DENV in humans have been the main cause of dengue diseases globally, with 50 million people infected per year worldwide (Guzman and others 2010). DENV-infected people show typical syndromes of self-limited febrile dengue fever and dengue hemorrhagic fever (DHF). Life-threatening dengue shock syndrome (DSS) is more likely to occur after a second DENV infection (Kyle and Harris 2008; Martina and others 2009). A recent case surveillance revealed that in particular older adults are at increased risk of DHF/DSS and death (Lin and others 2012). Other than supportive treatments, no specific therapy or vaccine is available (Guzman and others 2010).
Hydroxychloroquine (HCQ) and chloroquine are safe and well-tolerated antimalarial drugs. Their modulation of inflammation also improves survival and reduces disease activity in autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and Sjögren syndrome (Lee and others 2011). Several possible mechanisms of action of HCQ and chloroquine were revealed from in vitro or in vivo studies and included suppression of autoantigen presentation, reduced prostaglandin synthesis, antiproliferative effects, photoprotection, and decreased metalloproteinase activity and leukocyte activation (Lee and others 2011; Ben-Zvi and others 2012).
HCQ and chloroquine are cellular autophagy modulators that interfere with the pH-dependent steps of endosome-mediated viral entry and late stages of replication of enveloped viruses such as retroviruses, flaviviruses, and coronaviruses (Savarino and others 2003; Vincent and others 2005). The anti-HIV-1 effect of HCQ or chloroquine was demonstrated with the combination of antiretroviral drugs such as zidovudine, hydroxyurea, and didanosine in vitro or in patients (Savarino and others 2001b; Paton and Aboulhab 2005). However, HIV-infected patients receiving HCQ but not other antiretroviral therapy showed increased viral replication (Paton and others 2012). HCQ and chloroquine were reported to interfere in flavivirus infection at JEV internalization, YFV replication, and DENV maturation (Brandriss and Schlesinger 1984; Randolph and others 1990; Zhu and others 2012). However, in a randomized controlled trial, chloroquine failed to reduce viremia in patients with dengue disease (Tricou and others 2010), so despite the known antiviral mechanism of antimalarial drugs, other signaling pathways remain to be explored.
Innate immunity is a fast response of type I interferon (IFN) and inflammatory cytokine secretion that can be triggered by nucleic acids of virus replication products and toll-like receptor (TLR) signaling (Kumar and others 2009). During virus infection, double-stranded RNA can be detected by a group of cellular sensors such as melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I). The signal is transferred to mitochondrial antiviral signaling protein (MAVS), then TANK binding kinase 1 (TBK1) and IKKɛ for activation of IFN regulatory factor 3 (IRF3), IRF7, and NFκB, thus leading to expression of type I IFN and various cytokines (Kumar and others 2009). These cytokines inhibit virus replication in infected cells and regulate the induction of adaptive immunity, for swift eradication of viruses.
Autophagy has been described as a regulatory mechanism of both innate and adaptive immunity (Deretic and Levine 2009; Levine and others 2011). Pharmacological study indicated that suppression of autophagy with inhibitors such as chloroquine and Bafilomycin A1 (Baf A1) activated innate immunity (Ke and Chen 2011a, 2011b).
In this study, we investigated the efficiency of the multipurpose drug HCQ against DENV infection and its ability to activate the host defense machinery.
Materials and Methods
Virus, cell lines, and chemicals
We used local Taiwanese strains of DENV-1 766733A and DENV-2 PL046 (Genbank accession no. AJ968413.1) isolated from patients with dengue fever and DENV-4 466088A isolated from a patient with DHF. DENV-3 H87 strain was kindly provided by D. J. Gubler (Lin and others 1998). These viruses were propagated in mosquito cell line C6/36 (ATCC: CRL-1660) grown in RPMI 1640 medium containing 5% fetal bovine serum (FBS). A549 human lung epithelial carcinoma cells (ATCC: CCL-185), Hepa1-6 hepatoma cells (BCRC: 60051), WS1 human fetal skin normal fibroblasts (BCRC: 60300), J774A.1 mouse macrophages (BCRC: 60140), and HEK-293T cells (ATCC: CRL-3216) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Invitrogen). The reagents HCQ (Sigma-Aldrich; H0915), Baf A1 (Calbiochem; #196000), and recombinant human IFN-α 2a (Prospec; CYT-204) were used. TBK1/IKKɛ inhibitors BX795 (InvivoGen; tlrl-bx7) and Amlexanox (Sigma-Aldrich; SML0517) (Reilly and others 2013) and reactive oxygen species (ROS) inhibitor N-tert-Butyl-(-phenylnitrone (PBN) (Sigma-Aldrich; B7263) (Fidanboylu and others 2011) were used.
Plaque-forming assay
To determine virus titers, culture medium from DENV-2–infected cells was harvested for plaque-forming assays. Various virus dilutions were added to 80% confluent BHK-21 cells (BCRC: 60041) and incubated at 37°C for 2 h. After adsorption, cells were washed and overlaid with 1% agarose (SeaPlaque; FMC BioProducts) containing RPMI 1640 with 1% FBS for 7 days, then fixed with 10% formaldehyde, and stained with 0.5% crystal violet.
Cell proliferation and viability assay
Cells were assayed with 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT; Roche) at 1 mg/ml for 4 h, then the cell supernatant was replaced with DMSO to completely resolve the purple formazon crystals. The spectrophotometric absorbance of samples was measured by use of a microplate reader (Anthos). The net absorbance at OD570 indicated the enzymatic activity of mitochondria and cell viability.
Real-time quantitative PCR
TRIzol reagent (Invitrogen) was used for total RNA extraction, and cDNA was synthesized from 0.5 μg total RNA by use of Superscript III reverse transcriptase (Invitrogen). qPCR amplification involved 3 ng cDNA in 10 μL SYBR Green PCR master mix (Applied Biosystems) with 3 μM primers in ABI StepONE Plus Real-Time PCR system (Applied Biosystems). Transcript levels were normalized to that of hypoxanthine phosphoribosyltransferase. The primer sequences for gene detection are in Supplementary Tables S1 and S2 (Supplementary Data are available online at
Luciferase reporter assay
TurboFect transfection reagent (Thermo Scientific) was used for transient transfection following the manufacturer's protocol. Cells cultured in 12-well plates were transfected with NFκB-, AP-1-, ISRE-, or IFN-β-Luc reporter plasmids (Chang and others 2009). pRL-TK (Promega), encoding Renilla luciferase under an herpes simplex virus thymidine kinase promoter, was an internal control. In some cases, pcDNA3.1 V5-tagged-MAVS was used (Yu and others 2010). Cell lysates were collected for dual-luciferase assay (Promega). Firefly luciferase activity was normalized relative to that of Renilla luciferase.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30 min, then permeabilized with 0.5% Triton X-100 for 10 min. After 2 washes with phosphate-buffered saline (PBS), cells were blocked with 10% skim milk in PBS. DENV-2 NS3 was detected by incubation with a monoclonal antibody against NS3 (#YH3304, 1:500 dilution; Yao-Hong Biotechnology) (Chang and others 2012), plus Alexa Fluor-488-conjugated goat anti-mouse IgG antibody (Invitrogen). Fluorescence signals were observed by fluorescence microscopy (ZEISS Observer. A1). The 50% inhibition concentration (IC50) of HCQ against DENV-2 in cells was estimated by immunofluorescence intensity measured with use of a microplate reader (Fluroskan Ascent FL; Thermo Scientific). Anti-E antibody (#YH3304, 1:100 dilution; Yao-Hong Biotechnology) was used for antibody-dependent enhancement (ADE) of DENV-2 infection in J774A.1 cells.
IRF3 and NFκB p65 nuclear translocation assay was described previously (Chang and others 2006). IRF3 and NFκB p65 location was detected on incubation with the primary antibody anti-IRF3 (#sc-9082) and anti-NFκB p65 (#sc-372; both Santa Cruz Biotechnology), respectively, then Alex 568-conjugated anti-rabbit IgG antibody (Invitrogen). Nuclei were stained with DAPI.
Immunoblot analysis
Cells were lyzed in RIPA buffer (150 mM NaCl, 0.5% sodium deoxycholate, 1% NP40, 0.1% SDS, and 50 mM Tris-HCl [pH 8.0]) containing protease inhibitor and phosphotase inhibitor cocktail (Roche). Harvested extracts were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes, which were incubated with primary antibody, then horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratory) and visualized by enhanced chemiluminescence (Thermo Scientific). Images were acquired by use of a digital image system, BioSpectrum Image System (UVP). The following primary antibodies were used: anti-phospho-c-Jun (Ser73) (#3270, D47G9), anti-c-Jun (#9165, 60A8), anti-MAVS (#3993), and anti-IκBα (#4814S; all Cell Signaling); anti-phospho-IRF3 (pS386) (#2562-1; Epitomics); anti-IRF3 (sc-9082) and anti-NFκB p65 (sc-372; both Santa Cruz Biotechnology); and anti-V5 (Invitrogen). Endoplasmic reticulum (ER) stress was measured by immunoblotting with use of an ER-stress antibody kit (#9956; Cell Signaling), which includes antibodies against immunoglobulin heavy chain-binding protein/glucose-regulated protein of molecular (BiP/GRP78), Calnexin, endoplasmic oxidoreductin1α (Ero1α), inositol-requiring protein 1α (IRE1α), C/EBP homologous protein (CHOP), PKR-like ER protein kinase (PERK), and protein disulfide isomerase (PDI).
MAVS knockdown and type I IFN receptor signaling blocking
A549 cells were transfected with MAVS knockdown siRNA (siMAVS) and negative control siRNA (siNC) (35 pmol, Dharmacon; Thermo Scientific) by use of DharmaFECT (Thermo Scientific) for 24 h, then treated with HCQ for 24 h. MAVS knockdown efficiency was analyzed by immunoblotting with anti-MAVS antibody. A549 cells with shMAVS or control shLacZ stable expression were previously established (Yu and others 2010). For blocking type I IFN receptor signaling, A549 cells were incubated with type I IFN receptor neutralization monoclonal antibody (Millipore; #MAB1155, MMHAR-2) at 1:100 dilution for 8 h before HCQ treatment and virus infection.
ROS detection
Cellular ROS was determined by dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay (Fluska-35845; Sigma-Aldrich). A549 cells were washed with PBS, then 10 μM DCFH-DA was added for 30 min. The dye was discarded and cells were washed with PBS; DCF fluorescence was observed under a fluorescence microscope and quantified by use of a microplate reader (Fluroskan Ascent FL; Thermo Scientific). DCF excitation and emission wavelength were 488 nm and 529 nm, respectively.
Statistical analysis
Quantitative data are presented as mean±SD. Student's t test was used to determine the significance between treatment groups. P<0.05 was considered statistically significant.
Results
HCQ inhibits DENV infection in vitro
We evaluated the use of HCQ against DENV-2 infection by immunofluorescence assay (IFA) with anti-NS3 antibody in A549 lung carcinoma cells, murine hepatocellular carcinoma Hepa1-6 cells, and normal human fibroblast WS-1 cells. HCQ dose-dependently reduced DENV-2 infection (Fig. 1A). The 50% inhibition concentration (IC50) of HCQ against DENV-2 infection was estimated at 10.1±1.6 μM for A549 cells, 12.9±4.2 μM for Hepa1-6 cells, and 12.9±1.9 μM for WS-1 cells. Plaque assay indicated that 50–80 μM HCQ decreased DENV-2 titers by ∼100-fold in A549 cells (Fig. 1B). Because cell viability could be a factor in virus replication, we monitored the cytotoxicity of HCQ in A549, Hepa1-6, and WS-1 cells by MTT assay. The results indicated that equitable HCQ doses were used in our system (Fig. 1C). Thus, HCQ-restricted DENV2 replication was not due to impaired cell proliferation. Moreover, HCQ inhibited DENV-2 infection in J774A.1 macrophages (IC50=9.7±1.3 μM) (Supplementary Fig. S1A). MTT assay clarified that the dose of HCQ did not affect macrophage viability (Supplementary Fig. S1B). Consistent results were shown in HEK-293T cells, with DENV-2 infection, viral protein NS3 expression, 5′-UTR (untranslated region) viral gene expression, and virion production attenuated by HCQ (Supplementary Fig. S1C–F).
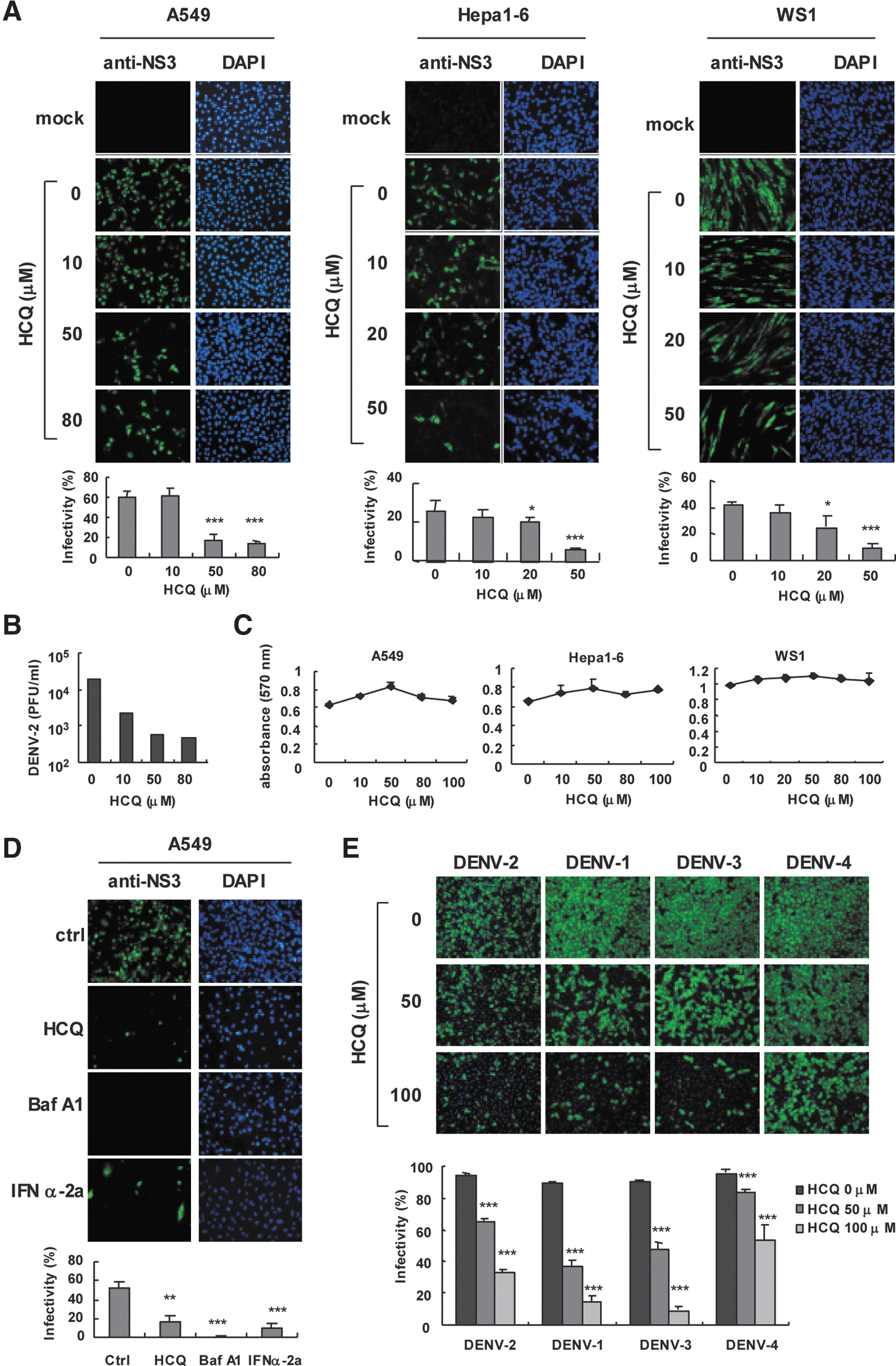
Hydroxychloroquine (HCQ) attenuates dengue virus infection in vitro.
Baf A1 is an autophagosome–lysosome fusion modulator with a similar pharmacological effect as HCQ (Rubinsztein and others 2012) and has been found to inhibit influenza A and B virus infection (Ochiai and others 1995). Here, we noted an anti-DENV-2 activity of Baf A1 similar to HCQ and the positive control IFN-α (Fig. 1D); so, DENV-2 infection requires cellular activity of autophagosome–lysosome fusion. Besides blocking DENV-2 infection, HCQ also had antiviral effects against DENV-1, -3, and -4 serotypes (Fig. 1E); however, a higher dose of HCQ might be required to suppress DENV-4 efficiently. Overall, our data show that HCQ interferes with DENV infection in vitro.
To address the antiviral mechanism of HCQ in DENV-2 infection, HCQ was added to cells before, during or after viral adsorption (Fig. 2A, upper panel). IFA of DENV-2 infectivity revealed that HCQ pretreatment suppressed DENV-2 infection, whereas DENV-2 replication was only partially reduced with HCQ added during virus adsorption or after viral adsorption (c–e in Fig. 2A). DENV-2 infection was greatly inhibited when cells were incubated with HCQ before and during viral adsorption or during all times (f, g in Fig. 2A). Thus, long-term HCQ treatment may have better inhibition effect on DENV-2 replication. Infectivity estimated by IFA gave similar results (Fig. 2B) and plaque-forming assay of DENV-2 titration confirmed the HCQ-mediated restriction of DENV-2 infection (Fig. 2C). Thus, HCQ pretreatment may change the host cell physical condition or trigger the host defense system against DENV infection.
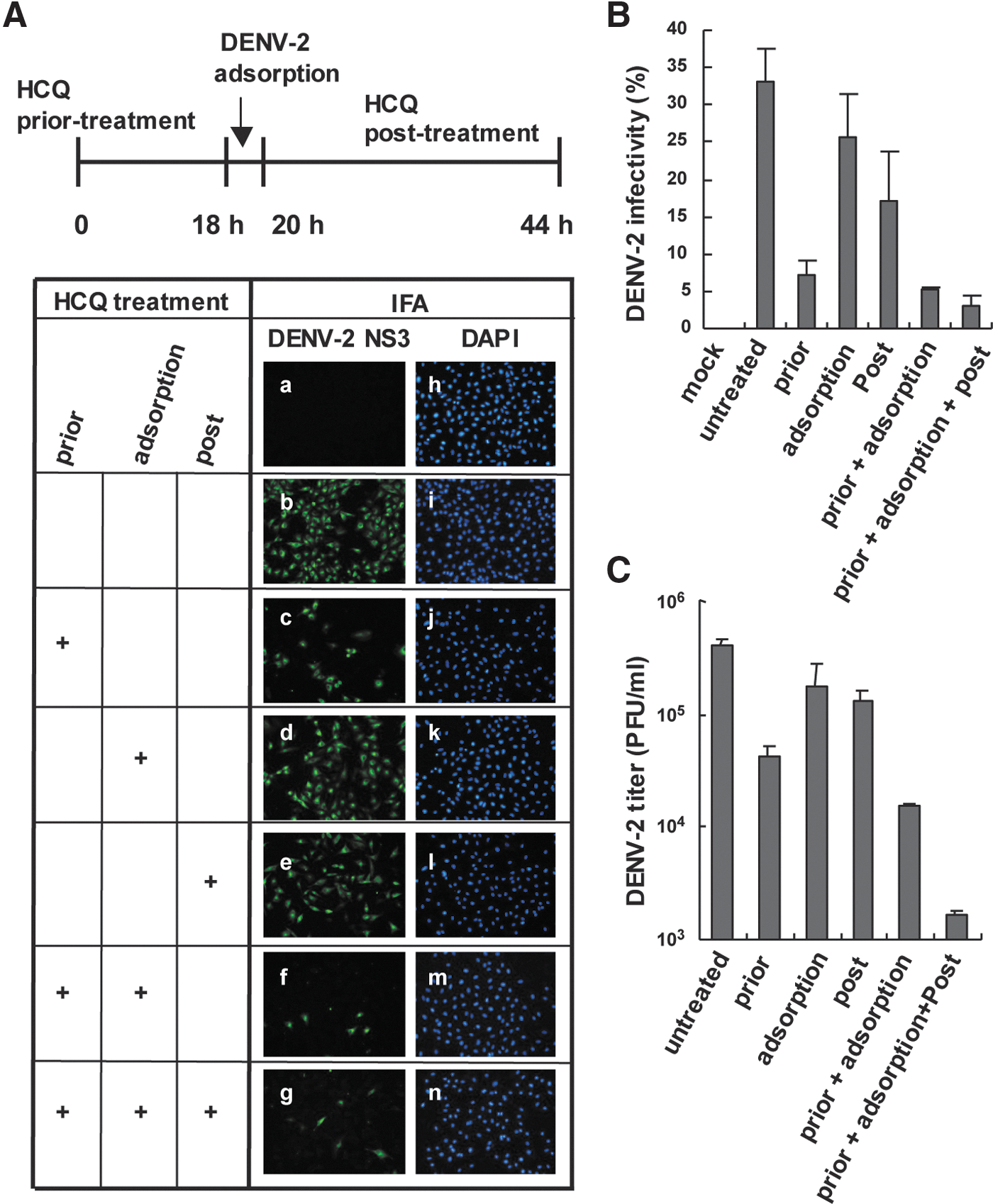
HCQ pretreatment reduces DENV-2 infectivity.
HCQ induces the expression of host antiviral genes
Type I IFN and associated antiviral proteins play crucial roles in host innate immunity. Because HCQ pretreatment efficiently inhibited DENV-2 infection, we investigated whether HCQ activates the host defense machinery. We found high induction of several innate immunity genes in human A549 cells treated with a high dose of HCQ (Table 1). The genes included IFN-β, IFN-induced protein with tetratricopeptide repeats 3 (IFIT3), C-X-C motif chemokine 10 (CXCL-10), MDA-5, Viperin, MAVS, and tumor necrosis factor receptor-associated factor 3 (TRAF3). In addition, several other genes, such as TNF receptor-associated factor 6 (TRAF6), IRF3, IRF7, RIG-I, ubiquitously expressed transcript V1 (Uxt-V1), tripartite motif-containing protein 21 (TRIM21), and major vault protein (MVP), were detected in HCQ-treated murine J774A.1 macrophages (Table 2). Furthermore, the mRNA expression of inflammatory cytokines such as IL-6, IL-23 p19, TNFα, and IL-12 p40 were induced by HCQ in J774A.1 macrophages (Table 2). Thus, HCQ-controlled DENV-2 infection may be associated with the activation of the host innate immune response. HCQ induced the antiviral genes IFN-β, IFIT3, Viperin, and TRAF3 in both A549 and J774A.1 cells, which suggests that these genes might be the major effectors induced by HCQ to attenuate DENV-2 replication.
Total RNA was harvested from A549 cells without or with hydroxychloroquine (HCQ) treatment (50 and 100 μM) for 24 h. RT-qPCR of mRNA levels normalized to that of the internal control hypoxanthine phosphoribosyltransferase (HPRT) and fold induction over the solvent control (mean±SD).
IFIT3, IFN-induced protein with tetratricopeptide repeats 3; IFN-β, interferon beta; CXCL-10, C-X-C motif chemokine 10; MDA-5, melanoma differentiation-associated protein 5, Viperin/cig5, cytomegalovirus-induced gene 5 protein; MAVS, mitochondrial antiviral signaling protein 5; TRAF3, TNF receptor-associated factor 3.
J774A.1 mouse macrophages were treated with 50 μM HCQ or solvent control for 24 h. RT-qPCR of mRNA levels normalized to that of the internal control HPRT and fold induction over the solvent control (mean±SD).
IFN-β, interferon beta; Viperin/cig5, cytomegalovirus-induced gene 5 protein; IFIT3, IFN-induced protein with tetratricopeptide repeats 3; IRF3 and IRF7, interferon regulatory factor 3 and 7; TRAF3 and 6, TNF receptor-associated factor 3 and 6; MAVS, mitochondrial antiviral signaling protein; RIG-I, retinoic acid-inducible gene I protein; MDA-5, melanoma differentiation-associated protein 5, MAVS, mitochondrial antiviral signaling protein 5; UXT-V1, ubiquitously expressed transcript-variant 1; TRIM21, tripartite motif-containing protein 21; MVP, major vault protein; IL-6, interleukin-6, IL-23 p19, interleukin-23 p19; TNFα, tumor necrosis factor alpha; IL-12 p40, interleukin-12 p40.
HCQ activates cellular signaling of innate immunity
To address how HCQ induces the expression of these host defense genes, we measured the ability of HCQ to turn on the luciferase reporters driven by the IFN-β promoter, NFκB p65, and c-Jun/AP-1 binding sites (Doly and others 1998). HCQ dose-dependently increased IFN-β, NFκB and AP-1 reporter activity (Fig. 3A–C). Furthermore, stimulation with HCQ induced the phosphorylation of IRF3, which indicated IRF3 activation. Phospho-c-Jun, an indicator of AP-1 activation, was also induced after HCQ treatment. HCQ time-dependently activated NFκB by increasing NFκB p65 expression and degrading IκBα (Fig. 3D). HCQ significantly induced NFκB p65 translocation into the nucleus; the pattern was similar with the positive control of polyI:C stimulation (Fig. 3E, F). Thus, our reporter assays, immunoblot results, and NFκB p65 nuclear translocation analysis consistently show that HCQ can activate host antiviral signal transduction pathways.
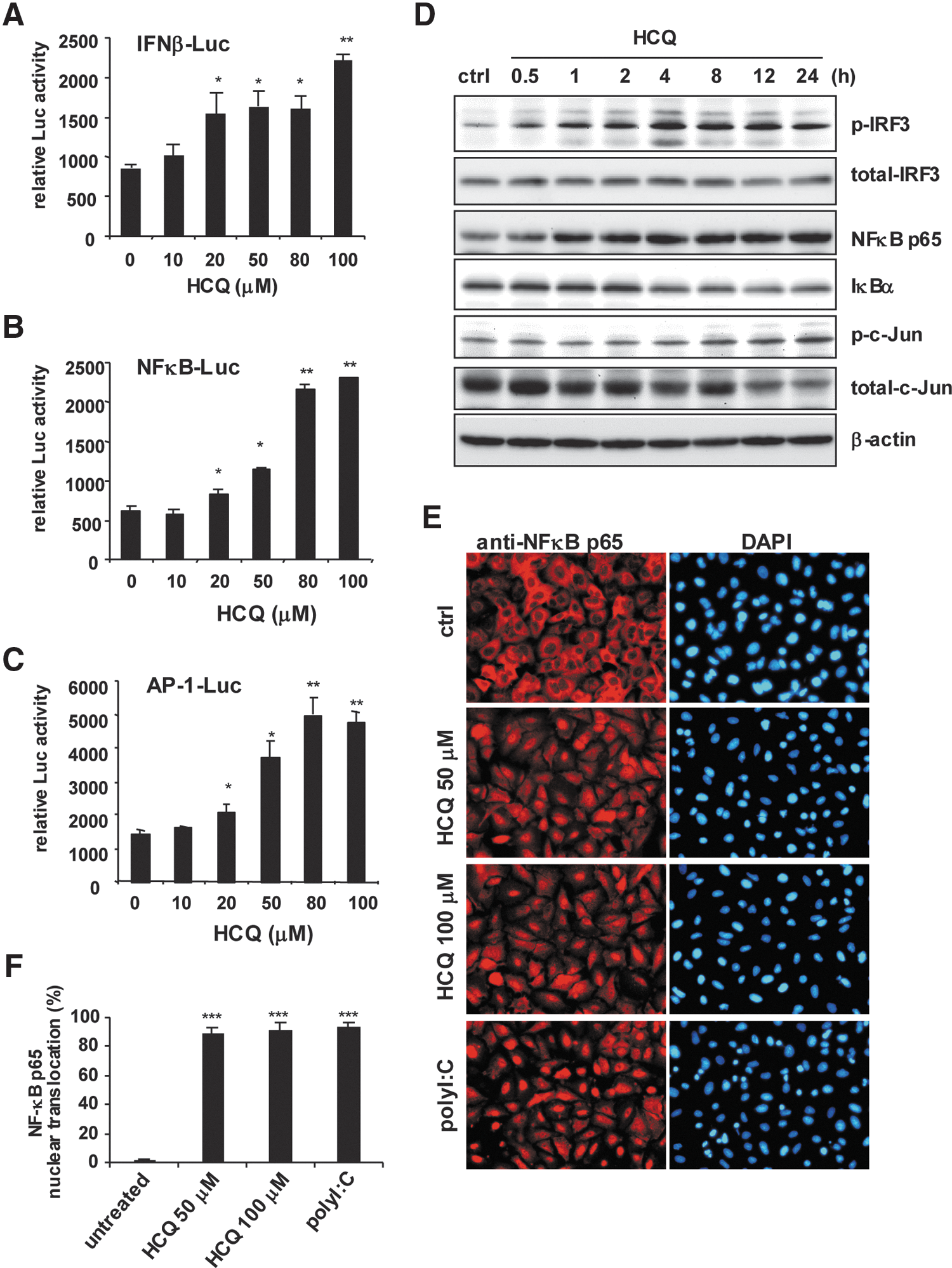
HCQ induces the activities of IFN regulatory factor 3 (IRF3), NFκB, and c-Jun.
MAVS is required for HCQ against DENV-2
MAVS plays a pivotal role in the type I IFN activation pathway (Kumar and others 2009). Because MAVS is one of the antiviral genes induced by HCQ in A549 cells (Table 1), we aimed to confirm whether MAVS is involved in HCQ antiviral activity. Immunoblot results showed increased MAVS protein expression with HCQ (Fig. 4A), which is consistent with the mRNA induction of MAVS by HCQ. Reporter assay revealed that HCQ enhanced the MAVS-transactivated IFN-β-, ISRE-, NFκB-, and AP-1-driven luciferase activities (Fig. 4B). Exogenous MAVS expression was higher in HCQ-treated than untreated control A549 cells (Fig. 4C). These data implied that MAVS protein was stabilized or decreased by HCQ. We used siRNA transfection to knock down endogenous MAVS expression to further validate the role of MAVS in HCQ-restricted DENV-2. Immunoblot confirmed the efficiency of MAVS knockdown (Fig. 4D). MAVS knocked-down or negative control A549 cells were then infected with DENV-2. As compared with negative control siRNA-transfected cells, in MAVS knocked-down cells, HCQ failed to inhibit DENV-2 infection (Fig. 4E); the estimated data for DENV-2 infectivity on IFA and viral titers showed consistent results (Fig. 4E–G). Moreover, we examined the function of MAVS in the HCQ antiviral effect in our previously established A549 cells carrying shMAVS for MAVS knockdown or shLacZ for negative control (Yu and others 2010). RT-qPCR confirmed the MAVS knockdown efficiency by shRNA that HCQ-mediated MAVS mRNA expression was inhibited in shMAVS-A549 cells (Fig. 4H). HCQ-mediated nuclear translocation of IRF3 and NFκB p65 in control cells was downregulated in MAVS knocked-down cells (Fig. 4I, J). Therefore, MAVS is essential for HCQ-stimulated antiviral activity and activation of the innate immune pathway.

HCQ enhances mitochondrial antiviral signaling protein (MAVS) protein expression and its transactivity.
Blocking type I IFN and TBK1/IKKɛ signaling reduces the anti-DENV-2 activity of HCQ
To clarify that the type I IFN antiviral system plays a critical role in DENV inhibition of HCQ, we blocked type I IFN signaling with an IFN-α/β receptor antagonist in A549 cells; cells were then treated with HCQ and infected with DENV-2. DENV-2 infectivity and titration data indicated reduced efficiency of HCQ against DENV-2 in cells with blocked type I IFN signaling (Fig. 5A). HCQ also inhibited ADE-mediated DENV-2 infection in J774A.1 macrophages; again, blocking type I IFN signaling reduced HCQ inhibition efficiency (Fig. 5B). MAVS downstream IκB kinase (IKK)-related kinase IKKɛ and TBK1 phosphorylate IRF3 and IRF7 in type I IFN induction pathway (Sharma and others 2003). Treatment with TBK1 and IKKɛ inhibitors, BX795 (2 μM; Clark and others 2009) and Amlexanox (25 μM; Reilly and others 2013) attenuated HCQ anti-DENV-2 activity (Fig. 5C, D). Again, these data support that HCQ activated the host type I IFN antiviral machinery against DENV infection.
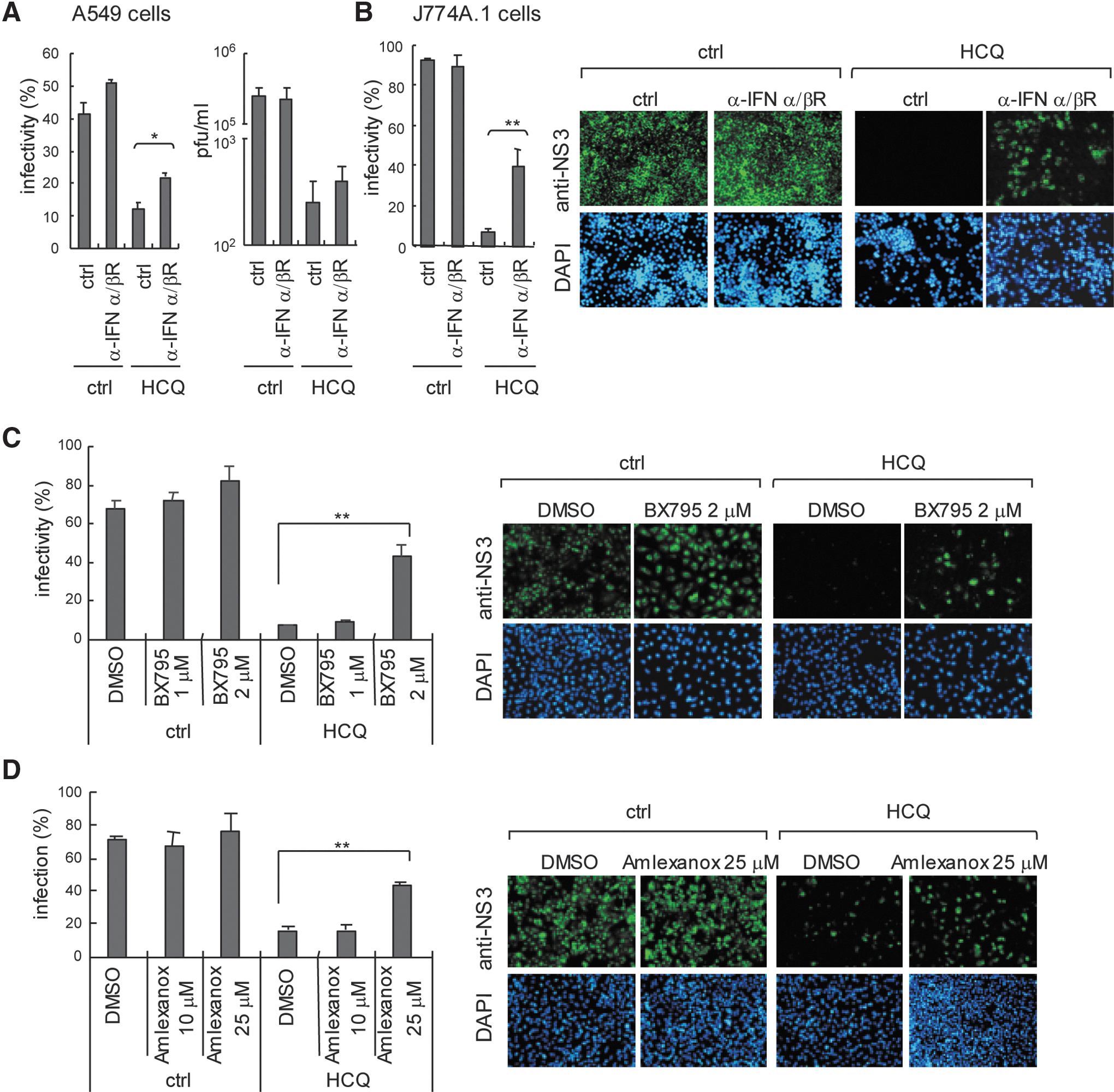
Blocking type I IFN signaling reduces HCQ anti-DENV-2 activity.
HCQ-induced ROS contributes to innate immune activation
ROS play an important role in normal cell signaling and homeostasis; ROS can also activate the host defence system, for example, promoting a TLR4 signaling pathway to drive cJun/AP-1 and NFκB activation (Kohchi and others 2009; Morgan and Liu 2011) and proinflammatory cytokine production (Bulua and others 2011). In addition, drug-induced oxidative stress in cells has been described (Deavall and others 2012). Therefore, we investigated whether ROS contributes to HCQ-stimulated innate immunity against DENV-2 infection. DCFH-DA assay was used for assessment of cells (Aranda and others 2013). We found the ROS signal of dichlorofluorescein (DCF) in HCQ-treated cells (Fig. 6A) and confirmed that HCQ induced significant ROS production (Fig. 6B). H2O2 stimulation was a positive control (Fig. 6A, B, right panels). Similar to the immunoblot results of HCQ stimulation (Fig. 3D), H2O2 stimulation increased the cellular MAVS and phosphorylated c-Jun level, with IκB degradation (Fig. 6C); so, ROS was involved in the innate immune signaling pathway. To substantiate the role of ROS in the HCQ antiviral effect, we used a nonspecific ROS scavenger, PBN (Fidanboylu and others 2011) to globally decrease cellular ROS level. PBN significantly reduced the level of HCQ-induced ROS generation (Fig. 6D). HCQ-restricted DENV-2 infectivity was attenuated with PBN (Fig. 6E). Along with the PBN-reduced HCQ-mediated ROS generation, HCQ-enhanced expression of MAVS, phospho-IRF3, and phospho-c-Jun was downregulated in PBN-treated cells, and HCQ-mediated IκBα degradation was also inhibited by PBN. (Fig. 6F). Importantly, PBN reduced ROS-inhibited HCQ and HCQ+MAVS-stimulated luciferase reporter activity of IFN-β, ISRE, and NFκB (Fig. 6G). Our data provide novel insights into the antiviral activity of HCQ mediated by ROS and MAVS involved in the host defense machinery, which indicates a key role of ROS in drug-activated host antiviral innate immunity.
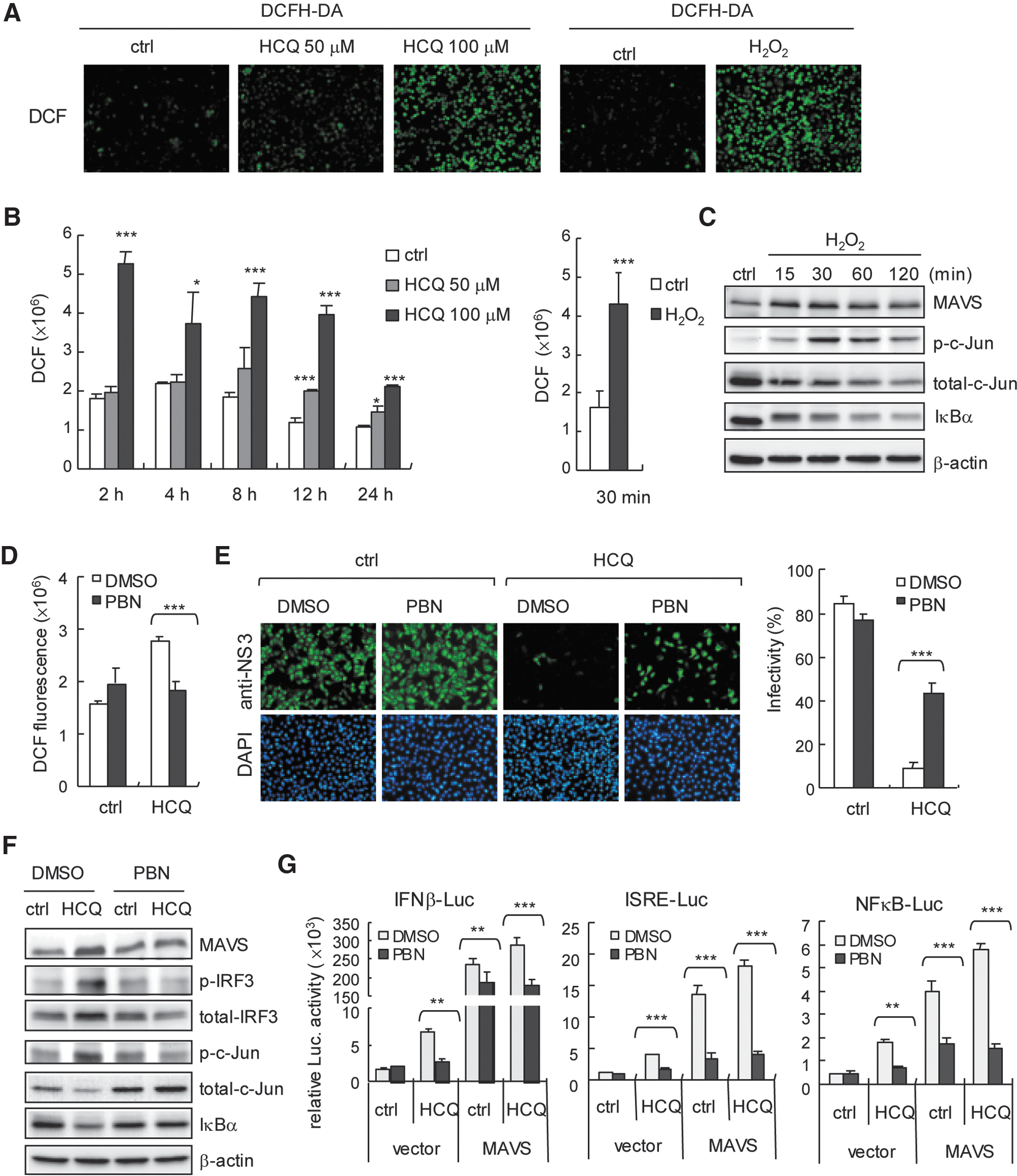
HCQ induces reactive oxygen species (ROS) generation.
Discussion
In this study, we validated HCQ as a potential DENV infection inhibitor that activates the innate immune signaling pathways of IRF3/ISRE, NFκB, and c-Jun/AP-1 and their downstream genes. HCQ induced the expression of antiviral factors such as IFN-β, IFIT3, CXCL-10, Viperin, MDA-5, MAVS, TRAF3, and TRIM21 and the cytokines IL-6, TNFα, IL-12 p40, and IL-23 p19. With MAVS knockdown, we showed the essential role of MAVS in HCQ-inhibited DENV-2 infection. Blocking the type I IFN pathway with an anti-IFN-α/β receptor antibody and TBK1/IKKɛ inhibitors reduced HCQ anti-DENV activity. In addition, HCQ-stimulated ROS generation was a key factor triggering host innate immune activation to restrict DENV-2. Our data reveal an emerging role of HCQ activating the host defence machinery against viral infection.
The role of ROS in inflammation and host defense has been established; mice lacking ROS activity have high susceptibility to bacterial infection (Shiloh and others 1999; Kohchi and others 2009; Bulua and others 2011). Similar to our finding, ROS-promoted host antiviral machinery was demonstrated in study of herpes simplex virus (HSV) infection in that natural-compound stilbenoids showed an anti-HSV effect by inducing ROS expression (Chen and others 2012). ROS might be a signaling messenger during HSV infection for activating NFκB and IRF3 pathways and producing type I IFN and inflammatory cytokines (Gonzalez-Dosal and others 2011; Hu and others 2011). A more sophisticated regulation mechanism was revealed in that ROS directly inducing the posttranslational modification of TRAF3 and TRAF6 with S-glutathionylation is important in activating the innate immune pathway (Gonzalez-Dosal and others 2011). Thus, elucidating whether HCQ promotes post-translational modification of signaling proteins to regulate innate immune responses would be of interest.
We revealed that ROS are critical in HCQ-activated innate immune signaling and the antiviral response and that ER stress was affected by HCQ treatment. HCQ enhanced the expression of IRE1α of the ER stress pathway but not immunoglobulin heavy chain-BiP/GRP78 or PERK. The level of chaperone proteins of ER, such as calnexin, Ero1α, CHOP, and PDI, in the ER was not changed after HCQ treatment. Along with the HCQ, dithiothreitol treatment was a positive control that could activate IRE1α, PERK, BiP, and Ero1α (Supplementary Fig. S2). (Nadanaka and others 2007). Thus, the IRE1α pathway of ER stress might be involved in HCQ-induced innate immunity activation. Because IRE1α has a role in the innate immune response, IRE1α is able to activate signaling of c-Jun/AP-1 and NFκB via TRAF2-JNK, -p38 MAPK and TRAF2-IKK pathways, for the expression of genes associated with host defense (Xu and others 2005). Also, the activation of IRE1α by ER stress is synergistic with TLR activation for cytokine production in macrophages against bacterial infection (Martinon and others 2010), so understanding the role of IRE1α in HCQ against DENV-2 infection would be important. Moreover, ROS production and ER stress activation show a crosstalk feature, which also supports our findings of HCQ (Malhotra and Kaufman 2007).
HCQ and CQ are weak basic drugs that accumulate in the acidic environment of cellular organelles to inhibit the replication of different viruses by interfering with endosome/lysosome trafficking or viral protein maturation during virion maturation. For example, CQ can change the structure of newly produced HIV gp120 glycoprotein (Savarino and others 2001a) and block the proteolytic processing of DENV prM to M protein in acidic post-Golgi vesicles (Randolph and others 1990). We also demonstrated a similar pneumonia of HCQ interrupting the endosome/lysosome pathway by monitoring the cellular expression pattern of transferrin receptor (TfnR), a marker for the early/recycling endosome (Shin and others 2004). Our data showed that TfnR-associated endosome vesicles were enlarged or accumulated and formed punctate structures in HCQ-treated cells (Supplementary Fig. S3). Because CQ was found to suppress JEV internalization (Zhu and others 2012), we do not exclude the possibility of HCQ blocking DENV-2 entry; however, this requires further investigation.
HCQ and CQ are well-known autophagy pathway inhibitors (Rubinsztein and others 2012). Double-stranded RNA stimulation was found to switch cellular activity from autophagy to innate immunity response with TBK1-dependent IRF3 activation (Simicek and others 2013), which suggests a mutually antagonistic regulation between autophagy and innate immunity, and ROS activity might be involved. For example, autophagy can modualte NACHT, LRR, and PYD domain-containing protein 3 (NALP3) inflammasome and ROS-mediated inflammtory cytokine expression (Nakahira and others 2011); autophagy also inhibits ROS-mediated MAVS aggregation to downregulate type I IFN production (Zhao and others 2012). Because autophagy can enhance JEV and DENV replication (Lee and others 2008; Heaton and Randall 2010; Li and others 2012), which might reflect flaviviruses downregulating the innate antiviral response by activating the autophagy pathway for replication. These findings may support our results. Nevertheless, the detailed role of autophagy in virus replication and host defence remains for further validation (Jounai and others 2007; Ke and Chen 2011a; Dong and Levine 2013).
We found that HCQ pretreatment induced the expression of multiple antiviral-associated proteins such as type I IFNs, IFIT3, Viperin, RIG-I, MDA5, and MAVS, demonstrated to suppress flavivirus infection (Shresta and others 2004; Diamond and others 2000; Perry and others 2009; Jiang and others 2010; Nasirudeen and others 2011; Szretter and others 2011; Teng and others 2012). However, the anti-DENV activity of other signaling proteins such as TRAF3, TRAF6, UXT-V1, and MVP remains to be validated (Konno and others 2009; Perez de Diego and others 2010; Huang and others 2012; Liu and others 2012). Because UXT-V1 is an integral component of the MAVS signalosome on mitochondria, which is critical in host antiviral activity (Huang and others 2012), understanding whether HCQ regulates UXT-V1-MAVS signalosome activity is critical.
In our study, HCQ greatly inhibited viral replication in cells treated with HCQ before but not after DENV-2 infection. Thus, the cellular environment change with HCQ treatment may be crucial to restrict DENV-2 infection, whereas the late stage of interference in virion maturation might not be the major mechanism of HCQ anti-DENV-2 activity. Thus, HCQ preventive treatment in the area or season of a dengue pandemic might be a feasible strategy to reduce the severity and spread of DENV outbreak. Moreover, results from clinical trials showed that patients receiving HCQ combined with other antiviral drugs, hydroyurea and didanosine, showed significantly reduced HIV viral load and increased CD4 count (Paton and Aboulhab 2005). Therefore, combined therapy with HCQ and other antiviral drugs could be considered in DENV infection control. For example, an anti-DENV-2 iminosugar derivative (Wu and others 2002) may be used with HCQ to prevent DENV infection.
In conclusion, we demonstrated that HCQ could restrict DENV infection by activating ROS and a MAVS-mediated host IFN antiviral pathway. HCQ is a marketed drug that may be developed for clinical therapy of DENV infection.
Footnotes
Acknowledgments
This work was supported by grants from Kaohsiung Veterans General Hospital (VGHKS102-004 and VGHKS103-107) and in part by the Zuoying Branch of Kaohsiung Armed Forces General Hospital, Kaohsiung, Taiwan (ZAFGH 102-16). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.