
Abstract
Anemia is a complication of interferon-containing hepatitis C treatments. We characterized effects of interferon-based therapy on hepcidin and erythropoietin (EPO) production, iron metabolism, hemolysis, and hematopoiesis. Standard hemopoiesis [reticulocyte hemoglobin (Hb), reticulocyte production index (RPI), free Hb, and haptoglobin], iron biochemistry, hepcidin, and EPO levels were measured in 10 subjects over 12 weeks. There was a rapid decline in Hb during treatment, from a mean pretreatment (t = 0 weeks) Hb of 158.6 to 125.2 g/L at week 4 (P = 0.003) and 122.8 g/L at week 12 (P = 0.005). Paradoxically, the RPI (a measure of bone marrow responsiveness to EPO) decreased on initiation of hepatitis C virus treatment from 0.78% to 0.53% (P = 0.04). Despite worsening anemia, there was no significant increase in EPO levels. Hepcidin levels increased to >20 nM in 3 subjects from 5.8 to 27.5 nM (P = 0.009) compared with 9.6 to 12.3 nM (P = 0.5) for the remainder of subjects. Hepcidin levels peaked at week 1 before returning to baseline levels at week 4. Subjects who responded with a rise in serum hepcidin levels to >20 nM had a significantly greater drop in Hb (27.2 g/L, P = 0.008) and reticulocyte Hb (−1.4 g/L, P = 0.013) compared with the subjects who did not exhibit any change in hepcidin production. In conclusion, 30% of subjects treated with interferon exhibited significant transient increase in serum hepcidin levels, which was associated with more extreme anemia and decreased iron availability as evidenced by decreased reticulocyte Hb. In addition, there was a failure to upregulate EPO production in response to anemia and hemolysis (
Introduction
A
In many chronic conditions, anemia is also contributed to through dysregulation of the key iron-regulatory hormone hepcidin, which is produced in the liver. Hepcidin regulates iron uptake from the gastrointestinal tract and release from the marrow. Increased hepcidin levels reduce the availability of iron for erythropoiesis by inhibiting the absorption of iron from the gut and release of iron from the bone marrow macrophages. Conversely, reduced hepcidin production results in increased availability of iron through increased iron absorption from the gut and increased release of iron from the marrow. Thus, any condition which increases production of hepcidin will predispose to the development of anemia. This is now known to be the key mechanism underlying the development of anemia in chronic inflammatory conditions (Chua and others 2007; Olynyk and others 2008; Cullis 2011).
Hepcidin production is stimulated by iron (Chua and others 2007; Olynyk and others 2008). In addition, pro-inflammatory cytokines such as interleukin-6 (IL-6) and IL-1 are known to also increase hepcidin production through a STAT3-mediated signaling pathway, underlying the causation of anemia of chronic disease (Cullis 2011). Recent in vitro data indicate that interferon-alpha increases hepcidin production in monocytes (Zhang and Rovin 2010). A short-term study of subjects during the first 24-h of interferon-based therapy for chronic hepatitis C demonstrated a marked increase in serum hepcidin levels, which was associated with reduced serum iron and transferrin saturation levels (Ryan and others 2012). Thus, it is possible that pegylated interferon alpha-based therapy increases hepcidin production in humans, resulting in impaired iron availability for erythropoiesis and anemia. In this study, we examined the long-term effects of interferon and ribavirin-containing antiviral therapy on hepcidin production and its hematological ramifications through (1) induction of functional iron deficiency, (2) inhibition of marrow erythropoiesis, and (3) induction of hemolysis.
Materials and Methods
This observational study did not alter treatment of subjects, which were given standard of care hepatitis C virus (HCV) treatment at the time. Patients treated with Boceprevir received a 4-week lead-in using Peg-IFN and ribavirin followed by Boceprevir 800 mg/8 h, Peg-IFN-2b 1.5 μg/kg/week, and weight adjusted ribavirin 800–1,400 mg/day. Patients treated with Telaprevir received Telaprevir 750 mg/8 h, Peg-IFN-2a 180 μg/week, and weight adjusted ribavirin 800–1,400 mg/day. Patients on dual treatment with Peg-IFN/ribavirin received Peg-IFN-2a 180 μg/week and weight adjusted ribavirin 800–1,400 mg/day. All patients had normal renal function.
Subjects were recruited at Fremantle Hospital in Western Australia between 2013 and 2014. Ethics approval was granted by Human Research Ethics Committee South Metropolitan Health Service (reference 12/204). This study was prospectively registered as NCT01726400 on
Study subjects had blood samples obtained just before initiation of treatment (t = 0) and, thereafter, at t = 1, t = 2, t = 3, t = 4, t = 8, and t = 12 weeks of treatment. At each time point, and in addition to standard of care testing during treatment of hepatitis C, reticulocyte hemoglobin (Hb) concentration, serum iron levels, transferrin saturation, ferritin levels, IL-6 levels, erythropoietin (EPO) levels, serum haptoglobin levels, and free Hb levels were determined by a single NATA accredited laboratory (PathWest, Western Australia) using standard methods. Blood samples were collected before 10 a.m., 1 day before interferon injection.
Hepcidin-25 was isolated from patient serum by solid phase extraction and measured by liquid chromatography quadrupole time-of-flight mass spectrometry (LC-qTOF-MS) as previously described. Quantitation was by reference to an isotopically labelled hepcidin peptide internal standard (Peptides International, Inc.) (Gummer and others 2016). Reticulocyte Hb concentration was used as an indicator of functional iron availability for erythropoiesis (Mast and others 2002). Central marrow inhibition was quantified using the Reticulocyte Production Index (RPI = reticulocyte percentage/(subject hematocrit/0.45) × 0.5) (Poorana and Subhashree 2014). Hemolysis was assessed using serum haptoglobin and free Hb concentration.
Based on literature reports, serum hepcidin levels of 15 ± 5(SD) ng/ml in pretreatment subjects were anticipated. A sample size of 10 would have 90% power to detect a change of 13 ng/ml or greater with therapy. As there are no published data of hepcidin levels following therapy, we assume the same standard deviation of 5 ng/ml during therapy. Statistical analyses were performed using IBM SPSS (IBM). Contingency tables were analyzed using the chi-test or Fisher's exact test when expected frequencies were lower than 5. Quantitative parametric data were analyzed using the independent t-test for comparing different subjects and paired t-test for comparing different time points for the same subjects. All P-values were derived from 2-tailed statistical tests and deemed significant if <0.05. All authors had access to all materials and approved the final article.
Results
A total of 15 subjects were screened of which 5 subjects declined interferon-based treatment. The characteristics of the enrolled subjects are provided in Table 1. Of those, 80% were male and the median age was 51 years (range 46–57 years). Seventy percent had HCV genotype 1, 30% had HCV genotype 3, and 50% had cirrhosis. All subjects with genotype 3 were treated with interferon/ribavirin, whereas of the genotype 1 subjects, 3 received interferon/ribavirin/Telaprevir and 4 received interferon/ribavirin/Boceprevir. Dose reductions were required for 2 subjects receiving Telaprevir and 2 subjects on Boceprevir; no reductions were required for subjects on interferon/ribavirin only. One subject did not tolerate Boceprevir treatment and withdrew after 4 weeks and 2 other subjects completed treatment, but did not attain a SVR, both of whom had HVC genotype 1.
F, female; M, male.
There was a steady reduction in all hematological parameters throughout treatment. At 4 weeks, there were mean reductions in Hb of 33.4 ± 9.3 g/L (P = 0.003, confidence interval [CI] = 13.4–53.4), white cell count (WCC) of 4.9 ± 0.81 × 109/L (P < 0.001, CI = 3.2–6.6), platelets of 81.8 ± 21.0 × 109/L (P = 0.002, CI = 36.5–127.1), and hematocrit of 0.10 ± 0.03 (P = 0.003, CI = 0.023–0.19) compared to pretreatment. At week 12, reductions were Hb of 35.8 ± 7.5 g/L (P = 0.001, CI = 19.6–52.0), WCC of 5.0 ± 0.72 × 109/L (P < 0.001, CI = 3.4–6.5), platelets of 79.5 ± 30.5 × 109/L (P = 0.023, CI = 9.7–149.3), and hematocrit of 0.11 ± 0.025 (P = 0.001, CI = 0.046–0.17).
Hemolysis peaked at week 4 with haptoglobins decreasing from 1.44 to 0.79 mg/dL (P = 0.021, CI = 0.11–4.18) and free Hb nonsignificantly increasing from 98.70 to 185.00 mg/dL (P = 0.3, CI = −260.7–88.2). EPO levels exhibited a nonsignificant decrease of 14.0 ± 8.1 ng/mL (P = 0.18, CI = −3.5–31.5) despite worsening anemia.
The normal physiological response to decreasing Hb is increased renal EPO production leading to increased reticulocyte production [Reticulocyte production index (RPI)]. The RPI can be used to quantify the bone marrow response with a RPI >3%, indicating an adequate response (Mast and others 2002). Paradoxically, the RPI decreased on initiation of HCV treatment from 0.78% to 0.53% (P = 0.04, CI = 0.11–0.47), before peaking during week 4 at 1.5% (P = 0.009, CI = 0.21–1.27), still well below the expected adequate response threshold of 3%.
With regards to liver biochemistry, bilirubin increased from a baseline mean value of 12.1 to 28.2 mg/dL in week 1 (P = 0.002, CI = 7.1–25.1) before returning to baseline at week 12. ALT steadily declined throughout treatment from a baseline 167.2 to 34.0 U/L (P = 0.03, CI = 15.6–250.8) at week 12. No significant changes were observed in GGT, alkaline phosphatase, or albumin levels.
Serum iron parameters peaked at week 2 with mean serum ferritin levels increased from 532.4 to 929.3 ng/mL (P = 0.005), serum iron levels increased from 21.3 to 35.0 μg/dL (P = 0.004), and transferrin saturation increased from 32.1% to 57.1% (P = 0.002). All iron parameters returned to baseline by week 12, excepting serum ferritin, which remained significantly elevated from baseline (P = 0.012). Overall, serum hepcidin levels rose significantly following initiation of treatment, peaking at 1 week (P = 0.013) and returning to baseline by week 4. However, hepcidin levels increased to >20 nM for 3 subjects (from a mean value of 5.8 to 27.5 nM, P = 0.009) compared to nonsignificant changes of 9.6 to 12.3 nM (P = 0.5) for the remainder of subjects (Fig. 1).

Serum hepcidin levels in study subjects with a hepcidin response greater than 20 nM at week 1 following initiation of interferon-based therapy compared with subjects who did not exhibit any change in hepcidin production.
There were no significant pretreatment differences between subjects who either did or did not increase hepcidin levels during treatment (Table 2) with regards to the presence or absence of cirrhosis or exposure to protease inhibitor treatment during the first 4 weeks (data not shown). However, subjects who exhibited a hepcidin response of >20 nM exhibited a significantly larger drop in Hb (27.2 g/L, P = 0.008), hematocrit (0.1, P = 0.005), and reticulocyte Hb (−1.4 g/L, P = 0.013) compared with the subjects who did not have a significant change in serum hepcidin production at the same time point (Table 2; Figs. 2 and 3). There was also a trend toward a lower platelet count (P = 0.09) and higher ferritin levels in subjects who did not exhibit a hepcidin response (P = 0.057).
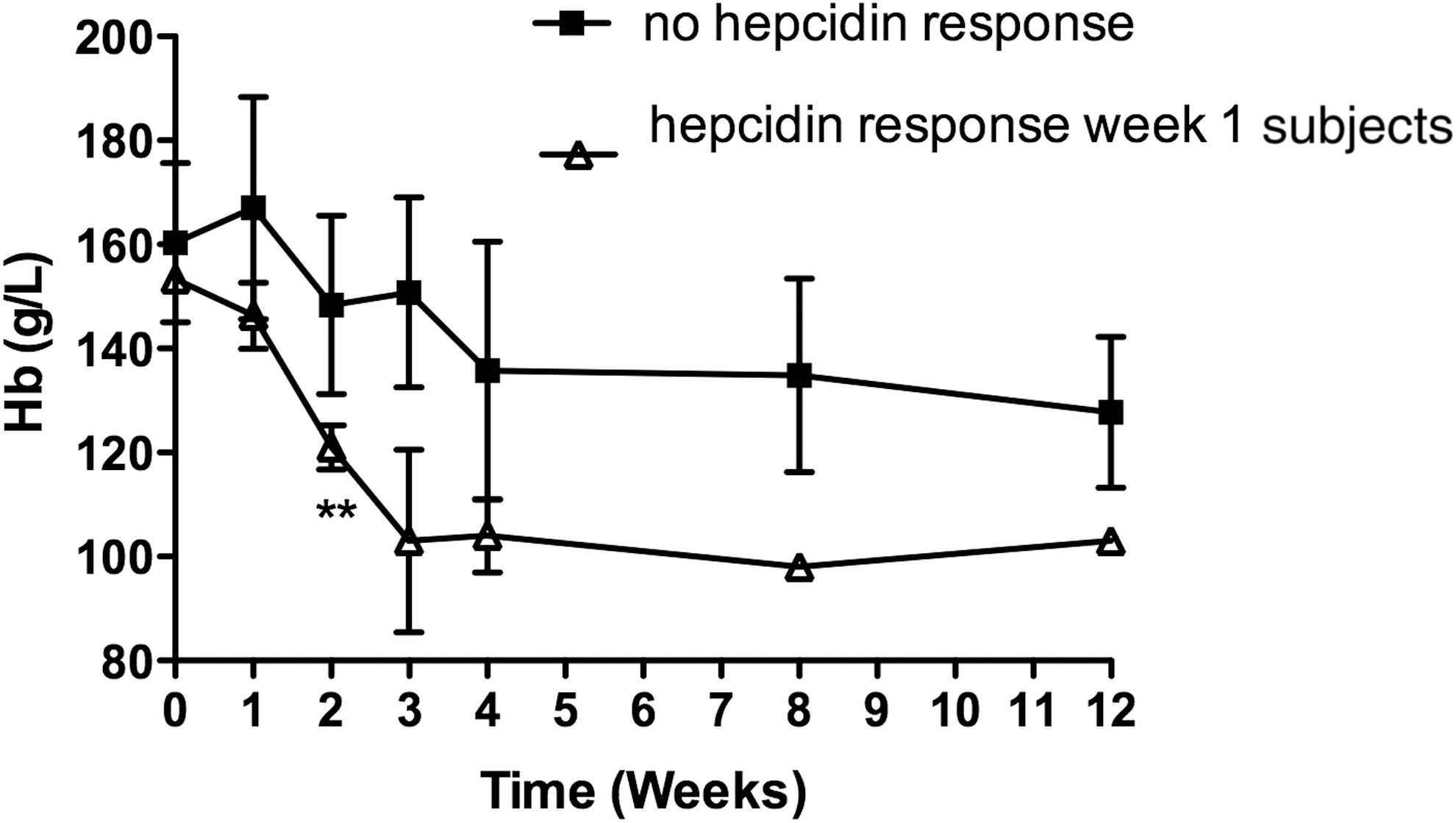
Hemoglobin concentrations in study subjects with a hepcidin response greater than 20 nM at week 1 following initiation of interferon-based therapy compared with subjects who did not exhibit any change in hepcidin production.
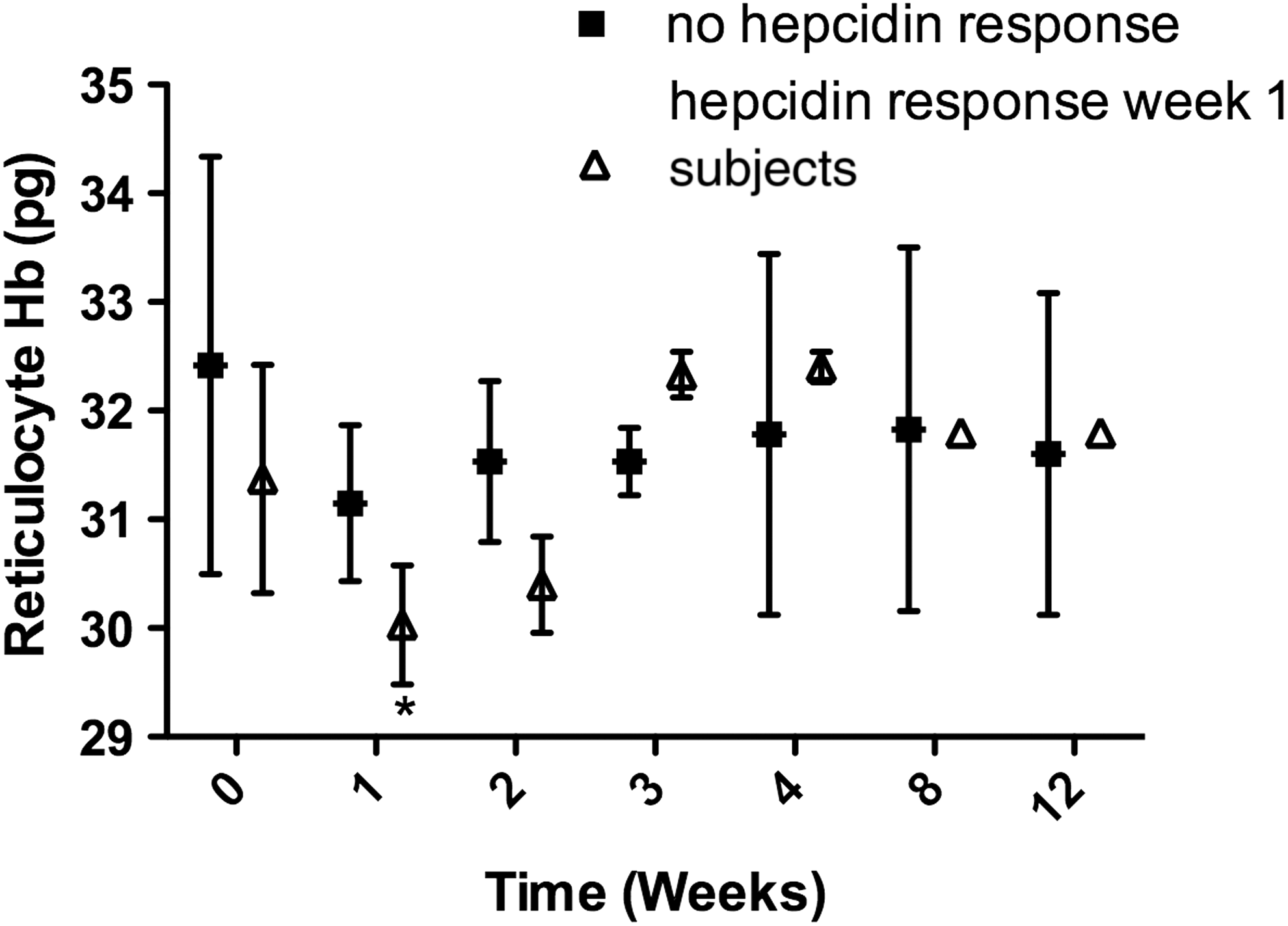
Reticulocyte hemoglobin levels in study subjects with a hepcidin response greater than 20 nM at week 1 following initiation of interferon-based therapy compared with subjects who did not exhibit any change in hepcidin production.
EPO, erythropoietin; MCH, mean cell hemoglobin; MCV, mean cell volume; WCC, white cell count.
Discussion
In this study, we aimed to elucidate the factors contributing to anemia during treatment of chronic HCV with specific emphasis on hepcidin and its relationship to erythropoiesis over a 12-week period. To our knowledge, this study is the first to report changes in hepcidin production in relation to hematological, iron, and erythropoietic changes during long-term interferon-based treatment of chronic hepatitis C. We found that hepcidin levels increased significantly following initiation of treatment, peaking after 1 week of therapy, and then returning to baseline; this is primarily due to a subpopulation of 30% of subjects who exhibit a rise in serum hepcidin levels to greater than 20 nM 1 week after starting interferon-based treatment. Interestingly, subjects who exhibited the higher hepcidin levels at 1 week also had lower levels of Hb, Hct, and reticulocyte Hb at 2 weeks following initiation of treatment. Reticulocyte Hb has the highest sensitivity and specificity for decreased marrow iron availability (Mast and others 2002). These observations suggest that elevated hepcidin production in this subgroup of subjects may have reduced marrow iron bioavailability for Hb production, contributing to anemia, which can complicate interferon treatment.
Our results extend the previous observations of Ryan and others to indicate that hepcidin induction as a response to interferon therapy is neither universal nor maintained over long time periods (Ryan and others 2012). The earlier study of these investigators was limited to the first 24-h of therapy and short-term in vitro studies. Our study demonstrates that extrapolation of the physiological relevance to human long-term therapy requires specific assessment in that setting as evident by the changing pattern of hepcidin production with continued therapy. Furthermore, the failed response of anemia to generate an appropriate increase in EPO production in the setting of interferon-based treatment suggests that interferon, either directly or indirectly, may be capable in inhibiting renal production of EPO. While interferons alpha and beta are known to inhibit human erythroid colony forming units through direct and indirect mechanisms (Means and Krantz 1996), there are no published data regarding the direct effects of such agents being mediated by impaired renal EPO production. Alternatively, it is known that EPO responses may be attenuated by circulating cytokines or other factors in chronic disease and it is possible that this may also contribute to the development of anemia (Cullis 2011). Further work examining the effects of interferon-based therapies on EPO production is required to elucidate this further.
Limitations of the current study are a relatively small sample size, the concurrent use of drugs other than interferon, and the multiple pathways contributing to anemia. To extrapolate the potential causes of anemia, a large number of hematological and biochemistry parameters were investigated. An increased hepcidin response was found in patients who were not receiving protease inhibitors at the time, indicating that hepcidin production was not an effect of protease inhibitor treatment. Hemolysis related to ribavirin took a number of weeks to reach maximal effect (4 weeks). This contrasts with the increased hepcidin production, which developed within 1 week, indicating that it is unlikely that ribavirin was the causative factor for increased hepcidin production. In the current study, we show that hepcidin levels are elevated early during treatment before returning to baseline. It remains unclear as to the exact mechanism of adaptation or negative feedback leading to hepcidin level normalization after a number of weeks or why not all patients exhibited a large increase in hepcidin levels. Importantly, this study shows that the increase in hepcidin is clinically significant as it leads to reduced iron availability as evidenced by a lower reticulocyte Hb and a larger drop in Hb levels.
With the advent of Direct Antiviral Agents (DAA) for treatment of chronic HCV, there has been a dramatic reduction in usage of interferon. However, the elucidation of new mechanisms for development of anemia as a result of interferon therapy or in other chronic diseases raises the potential for discovery of new therapies. For example, we have recently shown that anemia related to increased IL-6, and hepcidin production in chronic kidney disease can be treated with pentoxifylline, which successfully reduced both IL-6 and hepcidin production, significantly increasing Hb levels (Gummer and others 2016; Ferrari and others 2010).
In conclusion, during interferon-based treatment of chronic hepatitis C, 30% of subjects exhibit transient but significantly increased levels of serum hepcidin, which are associated with more extreme anemia and depression of erythropoiesis. There is a failure to upregulate EPO production in response to anemia and hemolysis in subjects undergoing this type of treatment, indicating that the currently known marrow suppressive and hemolytic effects of interferon and ribavirin, respectively, are compounded by a state of EPO failure.
Footnotes
Acknowledgments
Financial Support—this work was supported by unrestricted grants from MSD Australia, The Fremantle Hospital Medical Research Foundation, and the National Health and Medical Research Council of Australia. J.K.O. is a National Health and Medical Research Council of Australia Clinical Practitioner Fellow. Trial registration number—NCT01726400 on
Author Disclosure Statement
No competing financial interests exist.