
Abstract
To evaluate the efficacy and safety of pegylated interferon–lambda-1a (Lambda)/ribavirin (RBV)/daclatasvir (DCV) for treatment of patients coinfected with chronic hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Treatment-naive patients were assigned to cohort A [HCV genotype (GT)-2 or -3] or cohort B [HCV GT-1(a or b) or -4]. All patients received Lambda/RBV/DCV for the first 12 weeks; cohort A received Lambda/RBV for an additional 12 weeks, followed by 24 weeks of follow-up, and cohort B received response-guided therapy. The primary endpoint was the proportion of patients who achieved a sustained virologic response at post-treatment week 12 (SVR12). In cohort A (n = 104), 84.6% achieved SVR12 (95.0% in GT-2; 83.1% in GT-3). In cohort B (n = 196), 76.0% achieved SVR12 (71.7% in GT-1a; 86.0% in GT-1b; 70.7% in GT-4). Rates of discontinuation due to adverse events (AEs) (3.8% and 6.1%) and serious AEs (5.8% and 6.1%) were low in cohorts A and B, respectively. In addition, treatment with Lambda/RBV/DCV had little impact on CD4 counts. SVR12 rates with Lambda/RBV/DCV in an HCV/HIV-coinfected population ranged from 71.7% to 95.0%. Treatment was generally well tolerated, with a low proportion of patients discontinuing due to AEs. Clinical trial registration NCT01866930.
Introduction
S
Historically, HCV in HCV/HIV-coinfected patients was treated with interferon (IFN)–alfa/ribavirin (RBV), with poor tolerability and observed sustained virologic response (SVR) rates that were lower than those seen in monoinfected patients. In 2003, pegylated interferon–lambda-1a (Lambda) was developed as a potentially better tolerated alternative to IFN-alfa. In contrast to the ubiquitous expression of IFN-alfa receptors in most tissues, Lambda receptors are largely restricted to cells of epithelial origin; therefore, Lambda was expected to have comparatively lower rates of hematologic and systemic toxicity (Kotenko and others 2003; Sheppard and others 2003; Doyle and others 2006; Zhou and others 2007). As anticipated, in clinical trials in patients with HCV monoinfection, Lambda has demonstrated improved tolerability, with efficacy comparable with that of IFN-alfa (Muir and others 2014).
With the availability of multiple, all-oral, direct-acting antiviral (DAA) regimens, treatment options for all HCV-infected patients, including those coinfected with HIV, have expanded, with recent clinical trials reporting much improved response rates compared with IFN/RBV regimens. However, as mentioned above, for HCV/HIV-coinfected patients, there are added considerations, including potential drug–drug interactions between HAART components and certain DAAs (Bichoupan and others 2014). Daclatasvir (DCV), a pangenotypic HCV NS5A inhibitor with low potential for drug–drug interactions, is approved in 59 countries and has been studied in over 13,000 patients (Foster and others 2015a). DCV is indicated in the United States for use with sofosbuvir, with or without RBV, for the treatment of patients with chronic HCV genotype (GT)-1 or GT-3 infection (Bristol-Myers Squibb 2016), including the challenging-to-cure patients with advanced cirrhosis, post-transplant recurrence of HCV, or coinfection with HIV (Wyles and others 2015). In the European Union, DCV is approved in combination with other medicinal products for the treatment of chronic HCV infection in adults (
Due to the recent treatment evolution of chronic HCV infection from IFN to DAA-based therapies, the Lambda clinical development program has been discontinued; however, results of IFN plus DAA studies are still of interest, particularly for patients in whom IFN-based therapy continues to be a viable treatment option. Therefore, the objective of this study was to evaluate the efficacy and safety of Lambda/RBV plus the DAA DCV for treatment of chronic HCV infection in HIV-coinfected patients.
Materials and Methods
Study design
This open-label, multinational phase 3 study enrolled HCV treatment-naive patients with chronic HCV GT-1, -2, -3, or -4 infection who were coinfected with HIV. Patients were assigned to cohort A (GT-2 or -3) or cohort B (GT-1 or -4) on the basis of their HCV genotype. All patients received Lambda (180 μg subcutaneous injection once weekly) plus RBV (cohort A: 800 mg per day orally; cohort B: weight-based RBV, 1,000 mg per day orally for patients weighing <75 kg and 1,200 mg per day orally for patients weighing ≥75 kg) plus DCV (90, 60, or 30 mg per day based on the concomitant HIV drug regimen) for the first 12 weeks. After the first 12 weeks, cohort A received Lambda/RBV for an additional 12 weeks, followed by 24 weeks of follow-up. Cohort B received response-guided therapy (RGT): if patients achieved an extended rapid virologic response (eRVR; defined as undetectable HCV RNA at weeks 4 and 12), the dosing regimen was 12 weeks of therapy; if patients did not achieve an eRVR, they received Lambda/RBV for an additional 24 weeks (for a total of 36 weeks), followed by 24 weeks of follow-up. Due to discontinuation of the Lambda clinical development program, the protocol was truncated at the SVR12 assessment.
Patients
Enrolled patients included adults (≥18 years of age) who were treatment naive (no prior HCV treatment with IFN-alfa or a DAA) with a screening HCV RNA >10,000 IU/mL and evidence of HIV-1 infection. Patients were excluded from the study if they were coinfected with hepatitis B virus or HIV-2; had evidence of AIDS-defining opportunistic infections within 12 weeks before study entry; or had other medical conditions contributing to chronic liver disease, pancreatitis, renal disease requiring dialysis, or cancer. Permitted HAART therapy included atazanavir plus ritonavir, efavirenz, nevirapine, rilpivirine, abacavir, tenofovir, lamivudine, emtricitabine, raltegravir, enfuvirtide, maraviroc, and fixed-dose combinations of these regimens, such as Atripla®, Epzicom®, Truvada®, and Complera®.
The study was conducted in accordance with good clinical practice, as defined by the International Conference on Harmonization, and with the ethical principles underlying the European Union Directive 2001/20/EC and the US Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study protocol with amendments and the patient informed consent received institutional review board/independent ethics committee approval/favorable opinion before initiation of the study.
Endpoints and assessments
The primary endpoint was the proportion of patients who achieved SVR at post-treatment week 12 (SVR12). Secondary efficacy endpoints included the proportion of patients with rapid virologic response (RVR; defined as undetectable HCV RNA at week 4 of treatment) and eRVR. Secondary safety endpoints included the proportion of patients with treatment-emergent cytopenic abnormalities and on-treatment IFN-associated symptoms (flu-like or musculoskeletal symptoms); the frequency of deaths; the rate of serious adverse events (AEs) and grade 3–4 laboratory abnormalities; and dose reductions and discontinuations due to AEs.
Plasma HCV RNA was assessed using the COBAS® TaqMan® HCV Test v2.0 (Roche Molecular Systems, Inc., Pleasanton, CA) with a lower limit of quantification (LLOQ) of 25 IU/mL. HCV genotype/subtype assessments were performed using the Versant® HCV genotype 2.0 assay (LiPA 2.0; Innogenetics, Ghent, Belgium). HCV RNA was assessed at baseline, at weeks 1, 2, 4, 6, 8, 12, 16, and 20 (and weeks 24, 28, 32, 36, 40, and 44, as appropriate), at end of treatment (EOT) weeks 24 or 48, and at post-treatment follow-up weeks 4, 12, and 24 (and weeks 36 and 48, as appropriate). HIV-1 RNA quantification was determined using the Abbott Real-Time HIV-1 assay and detection m2000 system platform (Abbott Molecular, Inc., Des Plaines, IL) with a linear range of 40–10,000,000 copies/mL [measured on day 1 and at weeks 4, 8, and 12 (and weeks 24, 36, and 48, as appropriate)].
In addition to discontinuation due to AEs, treatment was discontinued if any of the following futility criteria were met: HCV RNA ≥1,000 IU/mL at week 12; confirmed HCV RNA detected at week 24; or virologic breakthrough, defined as a confirmed increase in HCV RNA >1 × log10 above nadir or HCV RNA ≥LLOQ after a previous assessment of HCV RNA <LLOQ while on treatment.
Statistical analyses
It was determined that a target sample size of 200 patients for cohort B would provide a maximum width of the 95% confidence interval (CI) of ∼14% (±7%). While this study was not powered for comparison of SVR rates, this sample size would provide sufficient precision such that if the true SVR12 rate was 44% or higher, the lower bound of the 95% CI would exceed 36% [the upper confidence bound of the historical control rate (based on the APRICOT study)] (Torriani and others 2004). Similarly, for cohort A, a target sample size of 100 patients would provide a maximum width of the 95% CI of ∼20% (±10%).
The primary and secondary analyses were conducted using the modified intent-to-treat approach, where the numerator is based on the nonmissing data from patients meeting the response criteria, and the denominator is based on all treated patients for each population. Missing data were computed as failures (non-SVR).
Results
Patient disposition, baseline demographics, and disease characteristics
A total of 453 patients were screened, of which 153 were excluded and 300 enrolled, 104 in cohort A and 196 in cohort B (Fig. 1). Baseline patient demographics and disease characteristics are summarized in Table 1. Of those enrolled, 75.0% were from Europe, 19.0% from North America, and 6.0% from South America. The majority were males (77.3%) and white (88.0%) and had an HCV viral load ≥800,000 IU/mL (78.7%). Cohort A consisted of 19.2% GT-2 and 79.8% GT-3; cohort B consisted of 76.0% GT-1 (50.5% GT-1a and 25.5% GT-1b) and 20.9% GT-4. Most (54.8%) had no or mild fibrosis (whereas 6.7% had confirmed cirrhosis) and most (64.0%) had a non-CC interleukin 28B GT. In terms of HIV disease status, median HIV viral load was 1.59 log10 copies/mL, and median absolute CD4 count was 538.5 cells/mm3. Of those enrolled, 85.3% completed the treatment period (91.3% in cohort A and 82.1% in cohort B). The rates of and reasons for discontinuation in cohorts A and B are shown in Fig. 1. Of those patients who failed to complete treatment, 16 of 44 (5.3% of the total) did so due to AEs: 4 in cohort A and 12 in cohort B.
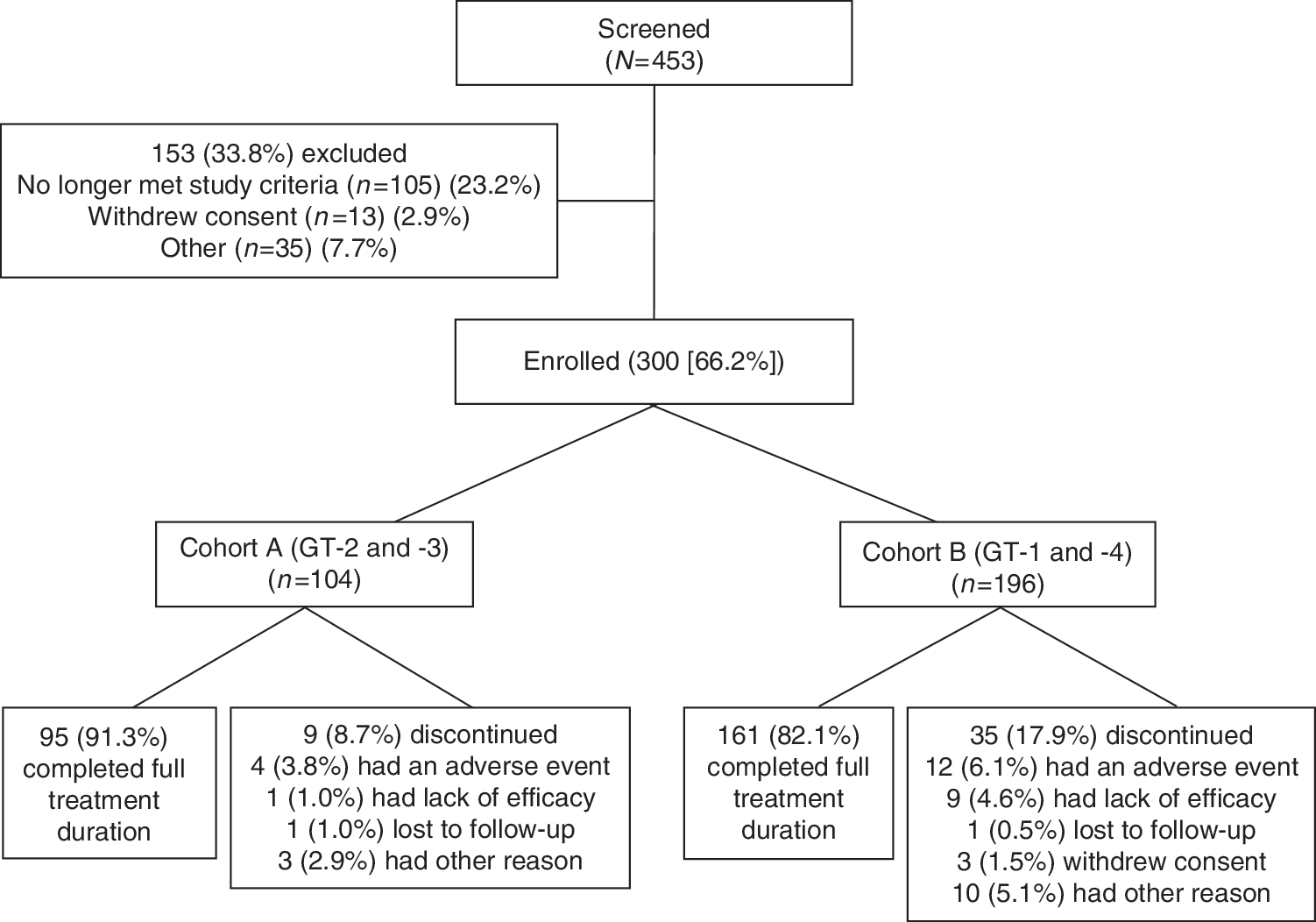
Patient disposition and reasons for discontinuation.
HCV GT subtype determined by local sites but not confirmed by central laboratory.
HCV, hepatitis C virus; HIV, human immunodeficiency virus.
Key efficacy endpoints
Key efficacy endpoint results are shown in Table 2. In cohort A (GT-2 and -3), an overall SVR12 rate of 84.6% (95% CI 76.2%–90.9%) was achieved, with SVR12 of 95.0% (95% CI 75.1%–99.9%) for GT-2 and 83.1% (95% CI 73.3%–90.5%) for GT-3. The RVR and eRVR rates in cohort A were 78.8% and 76.9%, respectively. In cohort B (GT-1 and -4), the overall SVR12 rate was 76.0% (95% CI, 69.4%–81.8%). For the individual GTs, SVR12 rates (95% CI) were 71.7% (61.8%–80.3%) in GT-1a, 86.0% (73.3%–94.2%) in GT-1b, and 70.7% (54.5%–83.9%) in GT-4. Rates of RVR and eRVR in cohort B were 76.0% and 70.4%, respectively. Of those patients in cohort B achieving an eRVR and thus receiving 12 weeks total of treatment, 87.0% achieved SVR12, whereas in patients not achieving an eRVR and thus receiving 24 weeks total of treatment, 50.0% experienced SVR12.
CI, confidence interval; eRVR, extended rapid virologic response; RVR, rapid virologic response; SVR, sustained virologic response.
In cohort A, 16 patients (15.4%) failed to achieve SVR12 (Table 3); among these, 9 patients (8.7%) completed treatment, 6 (5.8%) of whom had a confirmed relapse after achieving undetectable HCV RNA at EOT. Of the 7 patients (6.7%) who did not achieve SVR12 and also did not complete treatment, 2 patients (1.9%) experienced a confirmed relapse (Table 3). None of the patients who failed to respond to treatment in cohort A experienced virologic breakthrough or other on-treatment failure (Table 3). Data on resistance-associated variants were not available for patients with treatment failure.
Denominator is number of patients with EOT response, i.e., undetectable HCV RNA at EOT.
In cohort B, 47 patients (24.0%) failed to achieve SVR12 (Table 3); among these, 22 patients (11.2%) completed treatment, 17 (8.7%) of whom had a confirmed relapse after achieving undetectable HCV RNA at EOT. Among the 25 patients (12.8%) in cohort B who failed to achieve SVR12 and did not complete treatment, 14 (7.1%) experienced a confirmed relapse. Of those in cohort B who failed to respond to treatment, none experienced virologic breakthrough or other on-treatment failure (Table 3).
Safety analysis
Based on the type and frequency of AEs, the Lambda/RBV/DCV regimen was generally well tolerated in this HCV/HIV-coinfected patient population. AEs leading to discontinuation were uncommon in both cohorts: 4 (3.8%) in cohort A and 12 (6.1%) in cohort B.
As part of the secondary safety endpoint evaluation, the following AE rates were noted in cohort A: cytopenic abnormalities, 3.8%; flu-like symptoms, 5.8%; and musculoskeletal symptoms, 5.8%. Serious AEs occurred in 6 patients (5.8%) in cohort A and were predominantly hepatobiliary (n = 2) or psychiatric (n = 2; 1 dysthymic and 1 psychotic disorder). Grade 3–4 total bilirubin elevation occurred in 26/104 (25.0%) patients in cohort A; however, a direct bilirubin fraction >50% of total was seen in only 1 of 26 patients in cohort A, and no cases of concurrent alanine aminotransferase (ALT) and total bilirubin elevation were noted, indicating that the origin of bilirubin elevation was likely not hepatobiliary in the majority of patients. The most common AEs of any grade occurring in at least 15% of patients in cohort A were asthenia (21.2%) and insomnia (15.4%; Table 4). With regard to baseline values, on-treatment mean changes to absolute CD4, lymphocyte, and platelet counts in cohort A were minor and not clinically significant.
Three patients died during the treatment period (1 patient due to multiorgan failure and 2 patients due to sudden death). The patient with multiorgan failure discontinued due to an AE of anemia and then died 10 days later, and this death was considered by the investigator to be related to study drugs. The other 2 were considered not related to study drugs.
Serious AEs in 2 patients each per cohort: cohort A: jaundice; cohort B: jaundice, sudden death. All other SAEs were reported by 1 patient each.
AEs leading to discontinuation of study drug reported in 2 or more patients included: jaundice, hyperbilirubinemia, and AST increased in 2 patients each, and ALT increased in 3 patients.
Most common (>2.0%) grade 3–4 AEs: hypertransaminasemia and AST increased (2.9% each) in cohort A; hyperbilirubinemia (4.6%), and jaundice and fatigue (2.0%) in cohort B.
Hemoglobin <10 g/dL, and/or ANC <750 mm3 and/or platelets <50,000 mm3 during the treatment period.
AEs, adverse events; ALT, alanine aminotransferase; AST, aspartate transaminase.
In cohort B, safety endpoint results included the following: 7.7% of patients experienced cytopenic abnormalities, 9.7% flu-like symptoms, and 10.7% musculoskeletal symptoms (Table 4). Serious AEs occurred in 12/196 (6.1%) patients in cohort B and were predominantly due to hepatobiliary disorders, with 1 patient experiencing grade 3–4 liver function laboratory abnormalities. Grade 3–4 total bilirubin elevation occurred in 63 patients (32.1%) in cohort B, with a direct bilirubin fraction >50% of total seen in 9 of 63 patients (14.3%). However, only 2 cases of concurrent ALT and total bilirubin elevation were reported, again indicating that bilirubin elevation was likely not hepatobiliary in origin in the majority of patients. In both cases, the patients were also receiving atazanavir, which is associated with asymptomatic elevations in indirect bilirubin (Bristol-Myers Squibb 2015). As in cohort A, minor clinically insignificant changes in absolute CD4, lymphocyte, and platelet counts were observed during treatment among patients in cohort B (Table 4).
Three deaths occurred during the study, all in cohort B. One patient death was due to multiorgan failure and 2 patient deaths were due to sudden death. The patient with multiorgan failure discontinued due to an AE of anemia and then died 10 days later; this death was considered by the investigator to be related to study drug. The other 2 deaths occurred while on treatment (weeks 2 and 48) and were considered not related to study drug. One sudden death was attributed to cardiac failure following use of methylenedioxymethamphetamine, and the cause of the second sudden death was reported as unknown.
Discussion
Before the use of DAAs, IFN-alfa/RBV was used for the treatment of HCV/HIV-coinfected patients with only modest results (SVR range of 27%–44%) (Carrat and others 2004; Chung and others 2004; Laguno and others 2004). As in HCV-monoinfected patients, the addition of a DAA to IFN/RBV regimens led to improved SVR rates in patients coinfected with HCV and HIV; however, these improvements have only been clearly demonstrated for coinfected patients with HCV GT-1 (Sulkowski and others 2013a, 2013b). In the present study, DCV combined with Lambda/RBV in HCV/HIV-coinfected patients resulted in an overall SVR12 rate of 84.6% in patients with HCV GT-2 and -3, with a particularly high on-treatment SVR rate of 95% in GT-2 patients. In the HCV GT-1 and -4 cohort, the overall SVR12 rate was 76.0%, with the highest rate, 86.0%, seen in the GT-1b subgroup. Notably, in the HCV GT-1 and -4–infected patients who achieved an eRVR, an 87.0% SVR12 rate was observed.
Based on the known GT-dependent variation in response rates to antiviral treatment regimens as well as the GT-specific composition of the cohorts in this study, historical comparisons of outcomes are more appropriate than comparisons between treatment arms.
Genotype 2
In a large, multicenter phase 3 study (Foster and others 2015a), HCV GT-2–monoinfected patients treated with Lambda/RBV/DCV for 12 weeks achieved an SVR12 rate (90%) similar to that demonstrated in this study (95%). In the Foster and others (2015b) study, in HCV GT-2–monoinfected patients receiving IFN-alfa/RBV/sofosbuvir, a similarly high rate of SVR12 (94%) was observed. These response rates are comparable with SVR rates associated with current IFN-free DAA therapies (
Genotype 3
In contrast to GT-2, compared with the 83.1% SVR12 rate observed in GT-3-coinfected individuals in this study, HCV GT-3–monoinfected patients treated with IFN-alfa/RBV/sofosbuvir achieved an SVR rate of 93% (Foster and others 2015b). In addition, the IFN-free all-oral combination of sofosbuvir plus RBV provided cure rates in coinfected individuals that were also substantially lower for GT-3 (67%–85%) compared with GT-2 (88%–93%) (Sulkowski and others 2014b; Zeuzem and others 2014). These results are consistent with the current understanding that HCV GT-3-infected patients have lower response rates compared with GT-2 patients, with variability in their responsiveness to different DAAs (Foster and others 2015b). Importantly, HCV GT-3 is more prevalent among intravenous drug users, a major risk factor for HCV/HIV coinfection (Ampuero and others 2014). As a result, with more substantial gains in treatment of HCV GT-1 with DAAs, HCV GT-3 has emerged as the most challenging-to-cure population.
Genotype 1
Compared with other HCV genotypes, there are more data available on response rates of patients coinfected with HCV GT-1 and HIV across the spectrum of HCV treatment regimens. It has been established that for HCV GT-1, overall response rates to IFN/RBV/DAA regimens are similar between mono- and HCV/HIV-coinfected patients, and the results of the present study confirm these findings. In 2014, Hézode and others (2015) reported that the treatment of HCV GT-1-monoinfected patients with IFN-alfa/RBV/DCV resulted in SVR12 rates of 60%–64%, while SVR rates reported for GT-1-coinfected patients treated with IFN-alfa/RBV/DAA (in other studies using either boceprevir or telaprevir) have ranged from 63% to 74% (Sulkowski and others 2013a, 2013b). In the present study, we observed a similar overall SVR12 rate of 77% for HCV GT-1-coinfected individuals. We also observed differences in response rates between GT-1a and -1b (72% GT-1a and 86% GT-1b) that are similar to differences previously observed for GT-1 subtypes in monoinfected patients (55%–57% GT-1a and 76%–77% GT-1b) (Hézode and others 2015). One study (Flisiak and others 2016) evaluated HCV cure rates in monoinfected GT-1b patients treated with Lambda/RBV/DCV and found an SVR12 rate (89%) that was comparable with the present study (86%). More recently, in the ALLY-2 study (Wyles and others 2015), a 96% response rate was achieved in HCV GT-1-coinfected patients (70% of whom were GT-1a) following 12 weeks of treatment with DCV plus sofosbuvir (vs. 77% in the present study). These data are consistent with the evidence that IFN-free DAA-containing regimens can provide higher response rates than those including IFN in HCV GT-1-infected patients.
Genotype 4
For HIV- and HCV GT-4-coinfected patients, there is a gap in available data regarding the response to IFN/RBV/DAA treatment regimens. In monoinfected patients, treatment with the IFN-alfa/RBV/DCV combination resulted in SVR12 rates that were higher for GT-4 (75%–100%) compared with what was observed in the present study (71%) (Hézode and others 2015).
In terms of safety, overall, Lambda/RBV/DCV was generally well tolerated, with discontinuation rates for any reason of 8.7% in cohort A and 17.9% in cohort B. Of the patients who did not complete treatment, most discontinued due to an AE. These discontinuations occurred more often in cohort B (GT-1 and -4), which included weight-based, and thus higher, doses of RBV, as well as longer treatment durations as part of RGT. These relatively low rates of discontinuation, which likely contributed to the high observed SVR rates, may have been influenced by inclusion/exclusion criteria that resulted in enrollment of a relatively healthy coinfected population: absolute CD4 counts were high, with a median of ∼540 cells/mm3. In addition, as anticipated from differences in the cell populations expressing receptors for IFN-alfa and Lambda, as well as previous studies comparing treatment with IFN-alfa and Lambda in HCV-monoinfected patients (Muir and others 2014), lower rates of systemic AEs were observed in this study with Lambda (17%) versus a similar regimen that included IFN-alfa (56%) (Ampuero and others 2014). Three deaths occurred in cohort B, 1 of which was considered to be related to study treatment, and 2 of which occurred while on treatment, but were not considered by investigators to be related to study drug.
Due to the approval of all-oral IFN-free regimens for the treatment of HCV, the Lambda clinical development program has been discontinued. Nevertheless, results of IFN/RBV/DAA combination therapies continue to be relevant, particularly for coinfected patients who develop HCV drug resistance after treatment failure of a non-NS5a-based regimen, as well as in certain parts of the world where the use of all-oral DAA regimens is cost prohibitive and/or access is limited. In addition, this study supports the safety and tolerability of DCV in HCV/HIV-coinfected patients (no previously unreported AEs were noted) and highlights the importance of anticipating and proactively managing treatment-related toxicity with IFN-containing HCV treatment regimens.
Conclusions
Treatment of HCV/HIV-coinfected individuals with Lambda/RBV/DCV resulted in genotype-dependent SVR12 rates ranging from 72% for GT-1a to 95% for GT-2. Overall, treatment was generally well tolerated, with a low proportion of patients discontinuing due to AEs. The IFN/RBV/DCV combination represents a viable option in coinfected patients requiring an IFN-based therapy. The safety and tolerability of DCV demonstrated in this study, as well as currently available clinical trial and real-world data, support the use of DCV in HCV/HIV-coinfected patients.
Footnotes
Acknowledgments
The authors would like to acknowledge Rosemary Scricca for her significant contributions to the execution of this study as well as Cindy Schultz, PhD, for assistance with medical writing and Joshua Safran for editorial support. The analysis, writing, and editorial support for this work were fully funded by Bristol-Myers Squibb.
Author Disclosure Statement
M.N.: Honorarium: AbbVie, Bristol-Meyers Squibb, Gilead, MSD; Travel funding: AbbVie, Bristol-Meyers Squibb, Gilead, MSD. R.R.: Grant: AbbVie, Janssen; Honorarium: AbbVie, Bristol-Meyers Squibb, Gilead, Janssen, MSD, Viiv. A.L.: Declared no interests that might be perceived as posing a conflict or bias. S.R.: Declared no interests that might be perceived as posing a conflict or bias. A.L.: Grant during conduct of study: Bristol-Meyers Squibb; Grant outside submitted work: AbbVie, Gilead, Merck, Pfizer. B.C.: Research: AbbVie, Bristol-Meyers Squibb, Gilead, Merck; Travel support: AbbVie, Bristol-Meyers Squibb, Gilead, Merck. J.M.M.: Grant: Merck, Gilead; Advisory board: Bristol-Meyers Squibb, Gilead, Merck, Viiv. D.X., S.S., and S.P.: Employees of Bristol-Myers Squibb.