
Abstract
In breast cancer, an uncontrolled cell proliferation leads to tumor formation and development of a multifactorial disease. Metastasis is a complex process that involves tumor spread to distant parts of the body from its original site. Metastatic dissemination represents the main physiopathology of cancer. Inter- and intracellular communication in all systems in vertebrates is mediated by cytokines, which are highly inducible, secretory proteins, produced not only by immune system cells, but also by endocrine and nervous system cells. It has become clear in recent years that cytokines, as well as their receptors are produced in the organisms under physiological and pathological conditions; recently, they have been closely related to breast cancer metastasis. The exact initiation process of breast cancer metastasis is unknown, although several hypotheses have emerged. In this study, we thoroughly reviewed the role of several cytokines in breast cancer metastasis. Data reviewed suggest that cytokines and growth factors are key players in the breast cancer metastasis induction. This knowledge must be considered with the aim to development of new therapeutic approaches to counter breast cancer metastasis.
Introduction
In breast cancer an uncontrolled cell proliferation leads to tumor formation and the development of a multifactorial disease. Breast cancer tumors may grow in different breast areas, such as lobules, ducts, and connective tissue, being ductal cancer the most common of all (Nava-Castro and others 2014). However, the hallmark of cancer pathology is not the tumor itself, but the migration of transformed cells to different tissues of the tumor origin. In breast cancer, a patient with a primary tumor has a 5-year survival rate of 99%, whereas when metastasic tumors are present, this rate decreases to up to 23% (Siegel 2012). In this process, named metastasis, immunological factors, particularly cytokines, have a fundamental role. There are numerous risk factors linked to breast cancer tumorigenesis. The principal is the gender. According to World Health Organization (WHO), breast cancer is the most frequent women malignancy in incidence and mortality (GLOBOCAN 2012). Other aspects such as genetic background or breast cancer familial history, age, ethnic group, and higher mass index and nulliparity are other risk factors (Nava-Castro and others 2014). Of note, pregnancy reduces breast cancer risk, through breast cell maturation and diminishing the estrogen exposure during gestation (Russo and others 2005). Additional factors that enhance the estrogen exposure are an early beginning of menstrual cycle, a delayed menopause, and estradiol ingestion as hormone replacement (Nava-Castro and others 2014).
Metastasis in breast cancer
Metastasis is a complex process that involves tumor spread to distant parts of the body from its original site. To successfully colonize a distant organ in the organism, a cancer cell must complete a series of steps before it becomes a clinically detectable lesion. Cancer cells use 2 main dissemination pathways: the lymphatic pathway, leading to the invasion of the lymph nodes draining the organs where the tumor evolves; and the blood pathway that leads to the invasion of distant organs. In breast cancer, metastasis preferentially is targeted to the bones, lung, liver, and brain. The molecular mechanisms that promote the preferential migration to these organs are described extensively.
Different signaling pathways are involved in metastasis, such as the integrin pathway, the transforming growth factor beta (TGF-β) pathway, and different cytokine pathways among others. These pathways allow the possibility of therapeutic targeting, thanks to monoclonal antibodies or small molecules inhibiting the kinases involved in these signaling pathways. It is important to mention that, until now there is not a single antimetastatic drug for breast cancer, or any other type of cancer metastasis therapy.
The metastatic process
Metastasis is a complex phenomenon that comprises several processes which cancer cells must undergo, to transport themselves from the primary tumor to a distant organ. Briefly, we have categorized these processes into 3 stages, namely local invasion, migration, and colonization.
Initially, cancer cells reside within a well-defined primary tumor delimited by the adjacent stromal extracellular matrix, which separates it from the surrounding tissues, the lymphatic, and blood vessels. During local invasion, cancer cells undergo phenotypic changes toward more mobile ones, through a process known as epithelial–mesenchymal transition (EMT). EMT was initially involved in embryonic development in different animal species, in which mesenchymal cells acquire characteristics that allow them to migrate to different areas in the organ formation process (Thiery 2002).
There are 3 EMT subtypes based on biological processes (Kalluri and Weinberg 2009). Type I EMT is related with embryogenesis, embryo implantation, and initial placenta development, also with mesoderm and endoderm formation (epiblast–mesoderm transition) and the generation of mobile neural crest cells (Duband and Thiery 1982; Hay 1995; Vicovac and Aplin, 1996). Meanwhile, Type II EMT is associated with organ fibrosis mediated through the release of inflammatory factors and collagen, laminin, elastin, and tenancin (EMC components) by inflammatory cells and fibroblasts, for tissue reparation (Kalluri and Weinberg 2009). Finally, Type III EMT is related with tumor progression and metastasis. During this Type III EMT, polarized epithelial cells attached to basement membrane are transformed into mobile cells with the capacity of secrete ECM components and migrate to other tissues (Kalluri and Weinberg 2009; Wendt and others 2009).
In this context, Type III EMT is closely related to the development of metastasis and the transformation of mesenchymal cells by 3 different mechanisms: (1) morphology changes; (2) deregulation of epithelial markers [E-cadherin and cytokeratins 8 and 18 (CK-8 and 18)] and upregulation of mesenchymal markers (N-cadherin, vimentin, fibronectin, and SMA), which in turn, reduces intercellular adhesion; and (3) secretion of protein-dissolving enzymes that degrade extracellular matrix facilitating invasion and metastasis (Xu and others 2015). Since it allows cancer cells to become motile, Type III EMT contributes to metastasis favoring the dissemination of cancer cells to distant locations (Chang 2016). This process is highly promoted by angiogenesis, since it increases the number of available vessels (Baluk and others 2003).
Those cells (or cell clusters) that successfully survive to this process can then extravasate to a new organ parenchyma, where they will need to survive, adapt, and proliferate to generate a new tumor. This last process, called colonization, is favored in cells with a stem-like phenotype, known as cancer stem cells (CSCs), particularly breast cancer stem cells (BCSC's) (Al-Hajj and others 2003). These cells express CD44 but not CD24, which is consistent with a stem-like phenotype with multipotent differentiation ability. Other markers include EpCAM, CD47, and MET, which are usually found in metastasic lung, liver, and bone marrow tumors (Bacelli 2013). This kind of cells posess the gene expression signature Ras or HER2 overexpression, claudin-low, which is related to an increased Type IIIEMT potential (Geng 2014). It has been described that BCSC expresses the chemokine receptor CXCR4; whereas its ligand, the chemokine CXCL-12 is predominantly expressed in lymph nodes, lung, liver, and skeletal muscle–the common sites of metastasic tumors-(Muller 2001).
Another important condition that favors the breast cancer metastasis is angiogenesis, by which tumor promotes the formation of abnormal vasculature to provide the tumor with oxygen and nutrients. Among these proangiogenic factors are genetic mutations, mechanical stress, inflammation, the expression of angiogenic factors by tumor cells and of importance, hypoxia. This a vicious cycle since, in response to low oxygen levels, there is an induction of the expression of a series of factors related to proliferation, metabolism, angiogenesis, pH regulation, invasion, and metastasis and maintenance of CSC phenotype. The family of hypoxia-inducible factors, such as TGF-α, vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF)-2, and IGF-Bp2 (Favaro and others 2011) are the cause of all these processes.
In the present review, we will thoroughly discuss the roles that known cytokines have during breast cancer metastasis. To do this in an organized manner, we have listed all cytokines and their role on metastasis by numerical order.
Interleukin-1
Interleukin 1 (IL-1) is a pleiotropic cytokine involved in inflammatory processes. In different studies, elevated levels of this cytokine have been observed in breast cancer tumors and it has been proposed as a factor that promotes metastasis (Elaraj and others 2006). IL-1α and IL-1β are the main agonist proteins of the IL-1 family. In addition to being pleiotropic proteins, they bind to the same receptors and appear to have the same biological function. However, they are activated differently. Whereas IL-1β is only activated in its secreted form, IL-1α is activated intracellularly or bound to the plasma membrane and very little is secreted. Therefore, it has been proposed that IL-1β secreted into the tumor microenvironment activates inflammation and promotes invasion processes; whereas IL-1α is expressed inside tumor cells and it stimulates an antitumor immune response (Apte and others 2006). On the other hand, IL-1β has been associated with the induction of metalloproteinase expression (Fig. 1), such as the matrix metalloproteinase-1 (MMP-1) in BC-8701 breast cancer cells and fibroblasts (Rutter and others 1997) and the induction of the matrix metalloproteinase-9 (MMP-9) in the human breast cancer cell line MCF-7. Thereby, it has been proposed that the overexpression of these metalloproteinases mediated by IL-1β could favor the invasion ability of these tumor cells (Wang and others 2005). The group of Soria and col. suggests that the expression of IL-1β and tumor necrosis factor-α (TNF-α) has a role related to progression in breast cancer, since a greater amount of these proinflammatory cytokines were found in patients with invasive ductal carcinoma with relapse compared with samples of patients with ductal carcinoma in situ, and patients with ductal carcinoma without relapse. In addition, the continuous expression of these 2 cytokines led to Type III EMT in tumor cells, which could be favoring the observed relapse (Soria and others 2011).
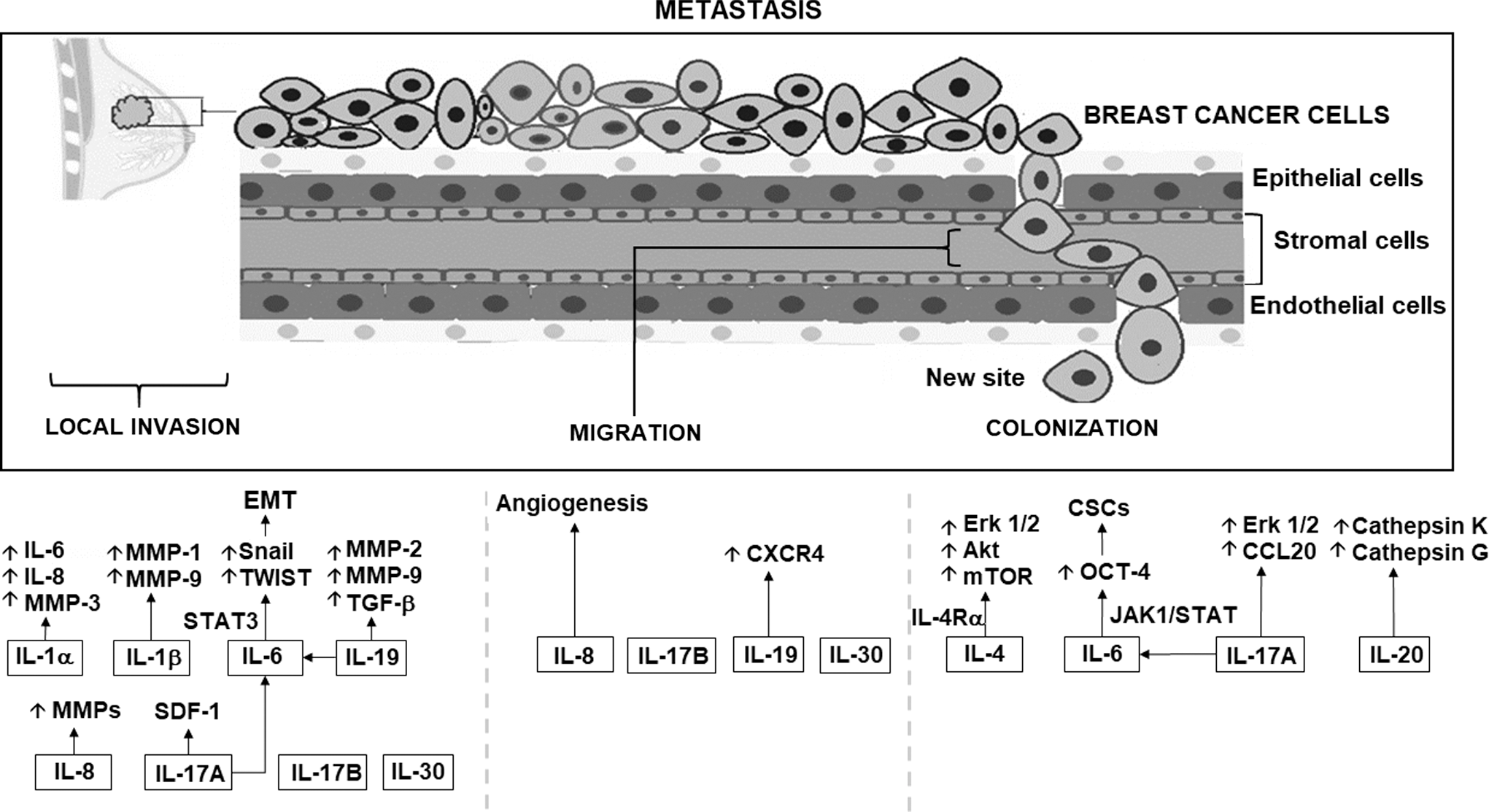
Regulation of cytokines that favor metastasis in breast cancer. The figure shows how interleukins 1, 4, 6, 8, 17, 19, 20, 25, and 30 regulate different signaling pathways that contribute to the process of metastasis in breast cancer cells, as well as the generalized pattern of migration of tumor cells to a secondary site. In this metastasis process, cytokines regulate the activation of metalloproteinases, kinases, chemokines, cellular processes, such as apoptosis, as well as the activation of other cytokines that are considered proinflammatory. MMP1,2,3,9,12,14, matrix metalloproteinase 1,2,3,9,12,14; IL-1Ra, interleukin 1 receptor antagonist; IL-4Rα, interleukin 4 receptor antagonist; Erk1/2, extracellular signal-regulated kinase; AKT, AKT serine/threonine kinase 1; mTOR, mechanistic target of rapamycin kinase; non-CSCs, nonstem cancer cells; CSCs, stem cancer cells; Jak1, janus kinase 1; Stat1,3, signal transducer and activator of transcription1,3; Oct-4, octamer-binding protein 4; Type III EMT, III epithelial–mesenchymal transition; HER2, human epidermal growth factor receptor 2; ER, estrogen receptor; SDF-1, stromal cell-derived factor 1; CXCR4, C-X-C motif chemokine receptor 4; TGF-β, transforming growth factor beta; KISS-1, KiSS-1 metastasis-suppressor; VEGF-A, vascular endothelial growth factor A; CXCL-10/IP10, C-X-C Motif Chemokine Ligand 10; CD11b, CD11 antigen-like family member B; Grl, glucocorticoid receptor.
Different studies have shown that autocrine or paracrine activation of the cytokine family of IL-1 and its receptors in human breast cancer could favor mechanisms that lead to the expression of pro-tumorogenic cytokines such as IL-8, and thus initiate a process of angiogenesis, tumor proliferation and invasion (Pantschenko and others 2003). Although IL-1β has been associated with the promotion of tumor cell invasion in breast cancer, IL-1α has been shown to play an important role in the expression of pro-metastatic genes in breast cancer cells and in stromal cells. Studies where an antibody against IL-1α has been used, showed a reduction of pro-inflammatory cytokines, such as IL-6 and IL-8 in breast cancer cells and metalloproteinase-3 protein (MMP-3) in fibroblasts, which could inhibit the invasion and metastasis of these tumor cells (Nozaki and others 2000) (Fig. 1). Although the main source of IL-1 is activated macrophages, it has been observed that a subpopulation of CD44+/CD24- breast cancer cells express invasion-related genes such as IL-1α, IL6, and IL8 (Sheridan and others 2006). Studies in animals have shown that the inhibition of IL-1β by the IL-1 receptor antagonist (IL-1Ra) reduces both metastasis and tumor size, therefore, the use of monoclonal antibodies neutralizing the IL-1 pathway has been proposed as a complementary therapy to block both angiogenesis and metastasis (Dinarello 2010).
Interleukin-4
IL-4 is a cytokine produced mainly by innate immune system cells and Th2 cells; it plays an important role in the humoral immune response against parasites and allergic antigens. However, it has also been shown to be involved in tumor growth mediating increased proliferation and survival. On the other hand, this cytokine promotes differentiation of tumor-associated macrophages (TAM) toward a phenotype M2-like, thus increasing metastasis by activating epidermal growth factor receptor (EGFR) expressed in epithelial mammary cancer cells, in the presence of colony-stimulating factor-1 (CSF-1) (DeNardo and others 2009).
IL-4 binding to IL-4Rα (its surface cell receptor) promotes breast cancer colonization to metastasis sites such as lungs. This phenomenon occurs through the activation of extracellular signal-regulated kinases (Erk) 1/2, Akt, and mammalian target of rapamycin (mTOR) pathway (Fig. 1). Conversely, IL-4Rα knockout experimental models have shown a decrease in lung and liver metastasis (Venmar and others 2014). In the same way, neutralized IL-4 and IL-4Rα, in breast cancer experimental models, reduce metastatic foci in lungs (DeNardo and others 2009). Moreover, STAT6 activation by IL-4 is associated with proliferation (Hallett and others 2012). Alternatively, there are other ways in which IL-4 promotes tumor growth; one of them is related to glucose and glutamine metabolism (Venmar and others 2015).
Thus, interaction of IL4–IL-4Rα, promotes metastasis and tumor growth, its blockade could be a factor that decreases proliferation, metastasis, colonization, and survival in breast cancer cells.
Interleukin-6
IL-6 is a pleiotropic cytokine involved in inflammatory responses and hematopoiesis (Tanaka and others 2014). Also, IL-6 has an important role on many other physiological processes, such as steroidogenesis and neurotransmission. This cytokine signals through a receptor comprised by 2 chains, the IL-6-binding chain and a signal-transducing glycoprotein gp130 (Tanaka and Kishimoto 2014). The binding of IL-6 to its receptor induces the activation of the Janus kinases (JAK), JAK1, JAK2, and Tyk2. JAK-phosphorylation of gp130 allows further downstream signaling through the signal transducer and activator of transcription 1 and 3 (STAT1 and STAT3), SHP2, and phosphatidylinositol 3-kinase (PI3K) pathways (Fisher and others 2014).
One of the first described roles for IL-6 was the differentiation of activated B cells into plasma cells, but it also induces differentiation of CD4+ T lymphocytes into some effector T helper subsets. Together with TGF-β, IL-6 promotes differentiation into Th17 phenotype, while inhibiting the regulatory (Treg) phenotype usually endorsed by TGF-β (Tanaka and Kishimoto 2014). In addition, IL-6 induces the differentiation of CD8+ T lymphocytes into cytotoxic lymphocytes (Tanaka and others 2014). Furthermore, IL-6 also stimulates the production of acute-phase proteins (C-reactive protein, serum amyloid A, fibrinogen, hepcidin, and α1-antichymotrypsin) in the liver (Tanaka and Kishimoto 2014).
In breast cancer, several studies have shown a positive relationship between serum IL-6 levels and disease progression, suggesting a role as a negative prognostic marker (Knüpfer and Preiss 2007; Sullivan and others 2009). Particularly, IL-6 can promote metastasis by aberrantly activating STAT3 pathway, promoting Type III EMT and supporting CSCs (Fig. 1).
The activation of the IL-6/JAK/STAT3 pathway has been implicated in breast cancer progression. In the tumor milieu immune cells, adipocytes and fibroblasts secrete IL-6 as part of the inflammatory and wound-healing processes, but cancer cells have been shown not only to take advantage of it, but also to secrete and use this cytokine in an autocrine fashion to the point that they experience a constitutive activation of the STAT3 pathway (Dethlefsen and others 2013). The former gains relevance when looking at some of the STAT3 target genes, which control proliferation (Bcl-2, Bcl-xL, BIRC5, CCND1, c-Myc, Mcl-1), angiogenesis (HIF-1α, VEGF), and Type III EMT (VIM, Snail, TWIST, MMP-7, MMP-2, MMP-9) (Castellana and others 2015; Banerjee and Resat 2016). MMPs play a key role in the metastatic process; these enzymes facilitate the degradation of extracellular matrix, allowing cells to detach from the primary tumor. Furthermore, IL-6 activates STAT3 pathway leading to the transcription of Snail and TWIST, which are both repressors of E-cadherin. In this way, IL-6 has been shown to induce Type III EMT of cancer cells (Sullivan and others 2009; Huang and others 2011).
Although CSCs comprise a very small population within tumors, it has been proposed that nonstem cancer cells (non-CSCs) can become CSCs to maintain equilibrium. In that regard, it has been demonstrated that IL-6 is able to convert non-CSCs into CSCs in several breast cancer lines (Iliopoulos and others 2011). This is possible since secreted IL-6 from non-CSCs activates JAK1/STAT pathway, upregulating OCT-4 gene expression, which is one of the key genes involved in pluripotency (Kim and others 2013).
As it has been exposed, IL-6 is a critical factor in the development of breast cancer metastasis; in this sense, this cytokine could be considered as a prognostic marker as well as a therapeutic target for this disease.
Interleukin-8
Another interleukin that has an important role in tumor proliferation, angiogenesis, and metastasis is IL-8, also known as CXCL8. This interleukin is expressed in humans, but not in mice; its receptors are CXCR1 (IL-8RA) and CXCR2 (IL-8RB), and is secreted by fibroblasts, tumor, stromal, and endothelial cells (Zlotnik and others 2006; Todorovic-Rakovic and Milovanovic 2013).
Overexpression of this cytokine has been recently considered a cancer prognostic marker, especially in breast cancer, because of the coexpression of IL-8, human epidermal growth factor receptor 2 (HER2), and the lack of expression of estrogen receptor (ER), are correlated with a worse prognosis (Fig. 1) (Todorovic-Rakovic and Milovanovic 2013). It has been found that both ER-negative human breast cancer tumors and breast cancer cell lines express higher levels of IL-8 compared with breast cancer cell lines positive for the hormone receptor. In this regard, higher levels of IL-8 were found in sera of patients with lymph node metastasis and/or ER− and HER2+ breast tumors (Ma and others 2017).
Originally, IL-8 was described as a chemoattractant for neutrophils that produced proangiogenic factors that enhanced tumor vascular neoformation and growth. Nowadays, it is known that IL-8 is per se, a proangiogenic factor with autocrine and paracrine functions. For instance, the stimulation with IL-8 of endothelial proliferation and capillary development in vitro model enhanced production of MMP-2 (Li and others 2005). Also, the stimulation with IL-8 in another in vitro cancer model leads to the increase of VEGF and VEGF receptor expression (Martin and others 2009), suggesting that if this protein is present in breast cancer microenvirontment, it can contribute in the metastatic process of this cancer type.
IL-8 enhanced invasiveness of ER- breast cancer cells, but not their proliferation (Freund and others 2003). This IL-8 feature can be related to late stages, in which the expression of ER is suppressed and the cancer invasiveness is increased. According to this observation, in an in vitro study, IL-8 did not increase cancer cell proliferation; however, the IL-8 treatment enhanced endothelial cell proliferation and survival (Li and others 2003) indicating that it can modulate the tumor microenvironment.
As mentioned before, IL-8 receptors are expressed in epithelial and tumor cells (Miller and others 1998; Murdoch and others 1999). In a report, all breast cancer samples expressed CXCR1 and CXCR2, but only a half of normal breast tissues expressed CXCR1 or CXCR2 (Miller and others 1998). Also, CXCR1 expression is higher in breast CSCs (Sheridan and others 2006), and in vitro assays using antibodies anti-CXCR1, induced apoptosis (Ginestier and others 2010). Thus, the 3 most important features related to cancer metastasis, namely tumor vascularization, migration, and invasion are characteristics promoted directly by IL-8. As we have reviewed in this section, vascularization, migration, invasion, and metastasis are promoted by IL-8. In fact, its expression is closely related to metastasis in breast cancer patients, whereby, its molecular mechanisms should be deeply studied in the near future.
Interleukin-10
IL-10 is an immunoregulatory cytokine that inhibits the activity of proinflammatory helper T subsets, cytotoxic T cells, macrophages, and NK cells. This immune regulation can influx immune response toward a more permissive microenvironment but can also reduce the tissue damage associated to inflammation (Couper and others 2018).
This cytokine plays a fundamental role within the tumor microenvironment, but its role regarding the metastatic process particularly in breast cancer remains unclear. In the 90's, Kundu and collaborators transfected murine mammary tumor cell lines to stably express IL-10 and found that not only the transfected cells grew more slowly than the parental ones, but also the metastasis was almost abolished (Kundu and others 1996). Later, another group reported similar results in a melanoma model, finding that IL-10 diminished neovascularization of the tumors and decreased MMP-9 expression, thus inhibiting the metastatic dissemination (Huang and others 1999).
More recently, human cohort studies have reported dissimilar results regarding IL-10 role in metastasis. One group, focusing in invasive ductal carcinoma, found that higher IL-10 expression within the tumor was associated with lower risk of distant metastasis (Li and others 2014), whereas another group, focused in inflammatory breast cancer, reported that IL-10 stimulates carboxypeptidase B2 expression, which in turn is positively correlated with lymphovascular invasion (Mohamed and others 2018). The later result seems to be in agreement with Woo's report of higher IL-10 expression in sentinel lymph nodes of breast cancer patients (Woo and others 2007), but still, it is not clear whether IL-10 directly plays a role in cancer cell's lymphatic dissemination.
Interleukin-17
The cytokine IL-17 is a proinflammatory cytokine that is secreted primarily by Th17 cells, but it can also be produced by CD8+ T cells, γδ T cells, natural killer T (NKT), and lymph tissue inducer cells (LTi) (Ma and others 2014). IL-17A binds to and signals through IL-17 receptor A (IL-17RA), which is ubiquitously expressed in hematopoietic tissues, various myeloid cells, epithelial cells, fibroblasts, and endothelial cells. The linkage of IL-17A to its receptor induces the release of proinflammatory cytokines, chemokines, and MMPs that positively feedback the inflammatory cascade.
IL-17A has been found increased in many types of human cancers and murine models of tumors; however, its role in tumor development is controversial. Some studies have found antitumoral effects, whereas some others associate this cytokine to protumoral functions. Given the controversial results, several researchers agree that the effects of IL-17A depend on the context and the anatomical site where the tumor develops.
Some research groups have studied the role of IL-17A in invasive breast tumor pathogenesis. In this sense, it has been demonstrated mainly that IL-17A comes from different cell lines and exerts protumoral actions. For example, in a model of mammary tumors in mice, it was observed that IL-17A expression from γδ T cells, results in G-CSF-dependent expansion and polarization of neutrophils, suppressing cytotoxic T lymphocytes, which limit the establishment of metastasis (Coffelt and others 2015). Macrophages have also been identified as a major cellular source of IL-17 in breast tumors and have been shown to directly promote breast cancer cell invasion in vitro (Zhu and others 2008).
The ability of IL-17A to favor breast cancer metastasis may be related to the fact that this cytokine can also induce IL-6 and the chemokine (C-C motif) ligand 20 (CCL20) production in metastatic tumor cells, favoring the recruitment and differentiation of Th17 cells (Benevides and others 2015). On the other hand, IL-17A has the ability to modify the gene-expression profile and the behavior of nonmetastatic tumor cells, causing tumor growth in vivo. The molecular mechanisms by which IL-17A induces the secretion of prometastatic factors have not been fully described. However, it is probable that they occur in a mitogen-activated protein kinase (MAPK) pathway-dependent manner, since it has been observed that recombinant IL-17A upregulates ERK1/2 in human breast cancer cell lines (Fig. 1). Thereby, IL-17A promotes proliferation and resistance to conventional chemotherapeutic agents such as docetaxel (Cochaud and others 2013). Other possible mechanism might be related to the apoptotic pathways, as it has been observed that IL-17A suppresses apoptosis of several tumor cell lines in vitro (Nam and others 2008). Consistent with this result, a knockdown of the IL-17 receptor in 4T1 mouse mammary cancer cells enhanced apoptosis and decreased tumor growth in vivo.
Several studies have confirmed that the neutralization of IL-17A in breast cancer reduces the development of metastasis. Coffelt and others (2015) observed that the neutralization of IL-17A in the absence of γδ T cells prevents neutrophil accumulation and downregulates the T cell-suppressive phenotype of neutrophils. On the other hand, Roy and cols. demonstrated that breast cancer-associated secondary metastasis is significantly increased in proarthritic and arthritic conditions, and that blocking the IL-17A and cyclooxygenase-2 (COX-2) pathways may significantly reduce the rate of metastasis (Roy and others 2009). Finally, in 2 immune competent arthritic mouse models, in which the animals develop spontaneous breast cancer-associated bone and lung metastasis, it was observed that the systemic neutralization of IL-17A can block the C-X-C chemokine receptor type 4 (CXCR4)/stromal cell-derived factor (SDF)-1 signaling pathway. This phenomenon occurs as a result of the reduction of the expression of SDF-1 in the metastatic niches (Roy and others 2014).
More research is necessary to delve into the molecular mechanisms that allow IL-17A to be a factor that favors metastasis, since with the evidences exposed in the previous paragraphs suggest that the IL-17A axis represents an attractive target of potential prognostic or have a therapeutic value.
Interleukin-19, Interleukin-20, Interleukin-22, Interleukin-25, Interleukin-30, and Interleukin 17B
There are other cytokines that have been related to the process of metastasis in breast cancer as shown in Fig 1.
IL-19 is part of the IL-10 family, expressed in different types of tumor cells. In breast cancer, it has been expressed in advanced stages of the tumor, and it has also been observed to be involved in an increase in mitosis and in a significant increase in metastasis, which leads to a worse prognosis (Hsing and others 2012; Chen and others 2013). IL-19 is able to upregulate CXCR4, MMP-2, MMP-9, TGF-β, IL-1β, and IL-6, all factors that induce the migration of breast cancer cells (e.g., Hs578T and 4T1), and it has been involved in breast tumor metastastasis. Hsing and others (2012), have shown that overexpression of IL-19 in 67NR cells (which usually have low endogenous IL-19 levels), and also in MCF-7 cells, stimulates their proliferation and migration, enabling them to form larger tumors and metastastic micronodules in the lung.
Another cytokine that has been related to metastasis in breast cancer is IL-20. In vitro, treatment with IL-20 upregulates MMP-9, MMP-12, cathepsin K, and cathepsin G, thus enhancing proliferation and migration of breast cancer cells. Interestingly, IL-20 is highly expressed in breast cancer bone metastasis (Hsu and others 2012).
IL-22, which belongs to the family of IL-10, has also been shown to be overexpressed in breast cancer. This cytokine is secreted by CD4+ effector T cells, such as TH1, TH17, TH22, and innate lymphoid cells (Lanfranca and others 2016). Both pro- and antitumor roles have been reported for this cytokine (Kim and others 2014). However, it has recently been shown that overregulation of IL-22 is associated with a worse prognosis in patients with breast cancer since it correlates with the expression of HER-2 in invasive ductal carcinomas (Zhang and Liu 2017). Additionally, the upregulation of this cytokine in parallel with the overexpression of a long noncoding RNA seems to favor growth, migration, and invasion through the PI3K/AKT signaling pathway in breast cancer patients (Riu and others 2017). More studies are needed on the role of IL-22 in the tumor microenvironment, however, it is proposed as a cytokine whose deregulation can function as cancer immunotherapy (Markota and others 2018).
Regarding IL-25 (also called IL-17E), the inhibition of its expression in an aggressive spontaneous breast tumor model (MMTV-PyMT), reduced the metastasis to the lung (Jiang and others 2017). However, other studies, in tumor-associated fibroblasts in a breast cancer model induced by transformed 4T1 cell injection, authors showed that the activity of IL-25 induced by a phytochemical (Q2-3) can provide antitumorigenic activities (Yin and others 2016).
Interleukin-30 (IL-30) is commonly expressed in breast cancer specimens; it has been associated with recurrence and with advanced stages of the disease. In vitro studies showed that IL-30 contributes to the overexpression of a pro-oncogenic program and in triple negative breast cancer cells, triggers a program of proliferation, invasive migration, and inflammation (Airoldi and others 2016).
Finally, another cytokine, named IL-17B promotes breast cancer cell migration (Molloy and others 2009; Goldstein and others 2010; De Luca and others 2012). Mesenchymal stem cells (MSCs) produce factors such as VEGF and IL-6 that, in addition to promoting angiogenesis, induce breast cancer cell migration and invasion, (Beckermann and others 2008; De Luca and others 2011, 2012). VEGF stimulates the invasion of breast cancer cells by activating MAPK and PI3K/AKT signaling (Price and others 2001).
Tumor Necrosis Factor Alpha
The TNF-α is a multifunctional proinflammatory cytokine that regulates different processes, such as inflammation, cellular apoptosis, coagulation, metabolism, tumor growth, and invasion, and also has vascular functions (Ji and others 2014). TNF-α plays an important role on the innate inflammatory responses by promoting the expression of cytokines, chemokines, adhesion, extravasation, attraction, and activation of the leukocytes at the site of the infection (Varfolomeev and Ashkenazi 2004). TNF-α induces its cellular effects by signaling through 2 receptors: TNFR1 (p55 or CD120a) that binds the soluble ligand and TNFR2 (p75 or CD120b), which binds mainly to the transmembrane form [5]. Both receptors have been found in hematopoietic cells, but TNFR1 has a wider distribution than TNFR2. In general, TNF-α may activate intracellular pathways leading to 2 different responses: cell survival, proliferation, and gene transcription or cell death. TNF-α is a two-edged sword molecule to tumors. It can induce tumor apoptosis (Donato 2004), but it can also induce tumor development (Balkwill 2006). As for cancer, TNF-α promotes breast tumor cell invasion as evidenced by in vitro experiments, upregulating several genes that are associated with proliferation, invasion, and metastasis. In this work, coculture of MCF-7 breast cancer cells with macrophages increases MCF-7 invasiveness. Microarray analysis of these cells demonstrated the enhanced expression of 39 genes, including MMP-9 and 13–related to Type III EMT; CD44–a marker of BCSC's; and CXCR-4 and its ligand CXCL-12–involved in tumor migration. Interestingly, KiSS1–a protein identified as a tumor suppressor was downregulated after TNF-α treatment (Yin and others 2009).
In addition, it has been shown that exposure of tumor cells to TGF-β and TNF-α induce Type III EMT, generating a BCSC phenotype characterized by the downregulation of claudins 3, 4, and 7 and cytokeratin 18 (Asiedu 2011). TNF and TNFR1 may be involved in type III EMT and metastasis, by the activation of the PI3K/AKT signaling pathway by 2 signaling cascades: (1) the activation of the Ras/Raf/MEK1/ERK pathway; in which Ras may also directly activate PI3K, allowing the activation of AKT and increased expression of the NF-kB p65 subunit (Downward 2003); and (2) by inducing GSK-3β ubiquitination and degradation, which in turn, impairs Snail degradation, inducing the phosphorylation of AKT (Wang and others 2013). In both cases, the final target is the stabilization of the Type III EMT-related transcription factor, Snail.
Finally, TNF-α-TNFR1-activated inflammatory macrophages produce high levels of VEGF-C to coordinately activate VEGFR3. TNF-α-stimulated lymphangiogenesis was completely abrogated in Tnfr1 −/− mice (Ji and others 2014). Even when there is no described evidence for breast cancer metastasis; it may be an important factor during the inital steps of tumor cell migration.
Growth Factors Involved in Metastasis
Colony-stimulating factor 1
It has been shown that colony-stimulating factor 1 (CSF-1) in breast tumors could mediate the recruitment of macrophages (Lin and others 2001). Interestingly, the proto-oncogene c-fos is the only gene that encodes to the only known CSF-1 receptor, (CSF-1R) (Sherr and others 1985; Dai and others 2002). Moreover, in neoplastic epithelial breast cancer cells, the expression of CSF-1 and its receptor correlated well with a poor prognosis, and it is predictive of ipsilateral recurrence (Scholl and others 1994; Maher and others 1998; Kluger and others 2004). CSF-1 promotes metastasis, stimulates angiogenesis, and participates in a paracrine loop with EGF to spur tumor cell invasion in mouse models (Lin and others 2001; Aharinejad and others 2002, 2004; Wyckoff and others 2004). Furthermore, it has been consistenly found that breast cancer cell lines express CSF-1 and CSF-1R, which sustains the proliferation of breast cancer cells (SK-BR-3 and MDA-MB-468) through ERK1/2 activation, stimulating c-Jun and upregulating c-myc and cyclin D1.
With the given background, we conclude that the role of the CSF-1R/CSF-1 system in breast cancer metastais is complex and requires further investigation as a therapeutic target.
Tumor Growth Factor Beta
The human TGF-β is a highly conserved superfamily of cytokines that are involved in controling the mammal development, a fact that may explain why their dysregulation has a close relation with tumorigenesis (Massague 2008).
TGF-β regulates innate and adaptative immune cell functions, and it is known that, depending on the tumor microenvironment, and thereby tumor stage, it performs suppressor or protumoral functions (Chen and Ten Dijke 2016). On the one hand, in early tumor development, TGF-β suppresses cell proliferation even when is mediated by ERα (Ewan and others 2005). Meanwhile, in late stages owing to abnormalities on TGF-β signaling, the cells lose their sensibility to antiproliferative signals. This event, along with the TGF-β ability to induce Type III EMT, leads to progression and dissemination of breast cancer (Wendt and others 2009; Esquivel-Velázquez and others 2015).
All members of TGF-β superfamily share structurally related membrane receptors type I (TRβI), type II (TRβII), and type III (TRβIII). These receptors transduce principally through Smad intracellular proteins, and by noncanonical signaling pathways (Massague 2008; Imamura and others 2012; Zarzynska 2014). In breast tissue, TRβI and TRβII are normally expressed, but this expression is decreased during carcinoma in situ, and increased in invasive carcinoma. This may be due to the initial losing of TGF-β sensibility signals in early tumor stages, and the turnover in advanced stages, associated with Type III EMT induction and metastasis (Hachim and others 2016).
With regard to the TGF-β role in metastasis, this cytokine supports tumor cell release and organ colonization. The releasing of tumor cells is mainly promoted by Type III EMT induction and its effects of adherent and tight junction gene downregulation such as (E-Cadherine, CDH1), the upregulation of motility genes' expression and the loss polarity in epithelial cells, thus promoting a mesenchymal invasive phenotype (Wendt and others 2009). Besides, TGF-β and the invasive potential of breast cancer cell lines, correlates with junctional adhesion molecule-A (JAM-A) decreased expression (Naik and others 2008), associated with TGF-β/Smad and TGF-β/p54 JNK pathways, promoting breast cancer cell release and further invasion (Wang and Lui 2012).
TGF-β has a main role in tumor progression when cells arrive to a metastatic niche. Despite the original conception that colonizing cancer cells retain their mesenchymal state in metastasis sites after Type III EMT, clinical evidence shows that cells undergo a mesenchymal-to-epithelial (MET)-type regression, revealed by the presence of tumor cells with epithelial morphology in metastatic sites (Thiery 2002; Tarin 2005). This EMT–MET change may be helpful in the colonization process, when cells proliferate in the metastatic tumor site (Stankic and others 2013). TGF-β induced MET mediated by Id1 gene in lung colonization breast cancer cells, but only in the ones that first underwent Type III EMT, a feature that might enhance local settlement (Yang and others 2008).
In a low-oxygen-level condition, MSCs produce cytokines and growth factors, such as TGF-β1, TGF-β2, and TGF-β3, which enhance proliferation and invasiveness of breast cancer cells (Hung and others 2012a, 2012b). TGF-β in a hypoxic microenvironment (through HIF-1α) regulates different gene expressions (CTGF, OPN, MMP-1, IL-6, and IL-8) to promote bone metastasis and increases VEGF and CXCR4 expression (Dunn and others 2009). Also, in spheroid assays, TGF-β promotes the invasiveness in MCF-10A1 (M1) (noncarcinogenic epithelial cells) and M1-derived MCF-10AneoT (M2) (RAS-transformed cells) (Naber and others 2011).
Meanwhile, in bone metastasis, TGF-β is released from bone matrix by osteoclastic activity, promoted by cancer cells, and in turn, this TGF-β stimulates the release of osteoclastic cytokines by tumor cells, forming a loop of bone matrix degradation (Kingsley and others 2007).
Therefore, either by eliciting Type III EMT–MET of cancer cells, by enhancing angiogenesis, or by its immunosuppressive effects, TGF-β is a crucial cytokine involved not only in tumor development and proliferation, but also in cell release, migration, and survival in metastasic sites.
Vascular Endothelial Growth Factor
In breast cancer, a key regulator in the metastasis is VEGF, which has been related to the neovascularization process, one of the first steps for metastasis. VEGF is a glycoprotein, member of the platelet-derived growth factor family (Ferrara and Davis-Smyth 1997). There are several subtypes of VEGF (VEGF A-F), all of them related to angiogenesis (Shibuya 2011). VEGFs exert their angiogenic functions throughout the binding of their specific tyrosine kinase receptors VEGFR-1 (Flt-1), VEGFR-2 (Flk-1/KDR), and VEGFR-3 (Flt-4), which are mainly expressed in endothelial cells (Ferrara and Davis-Smyth 1997). It has been described that the angiogenesis, lymphangiogenesis, migration, and invasion processes in breast cancer are mainly driven by VEGF-A and VEGFR-C (Mandriota and others 2001; Alitalo and Carmeliet 2002). In this sense, it was observed that VEGFR-3 and their ligands, VEGF-C, are overexpressed in human aggressive breast cancer tumor tissues, and their coexpression correlates with poor prognosis (Su and others 2006). Furthermore, the expression of VEGF-C in plasma has been correlated with the expression of HER2-positive breast cancer patients (Yang and others 2002; Hoar and others 2003). In addition, elevated plasma concentrations of VEGF-A have also been found in hormonal positive breast cancer patients that are in remission, and with metastatic disease instead of in patients with local tumors. On the other hand, an interesting interaction between VEGF and chemokine receptors has been described. VEGF throughout the binding of a nonclassical receptor that lacks canonical signaling sites, neuropilin-1 (NRP-1), activates invasion and migration actions in breast cancer. This ligand–receptor interaction has also been linked to the protein upregulation of chemokine receptor CXCR4 and its ligand, SDF-1, driving the migration of breast cancer cells to secondary sites (Bachelder and others 2002).
The coexpression of VEGF/NRP-1 was found in a large number of mammary tumors and they were associated positively with lymph node metastasis. This interaction was associated with the enhancement of the Type III EMT process and the modulation of the transcription factor NF-κB, β-catenin, and glycogen synthase kinase-3 (GSK-3β) signaling. It is worth mentioning that the knockdown of both proteins (VEGF/NRP-1) induced apoptosis and the reexpression of adhesion proteins such as E-cadherin in aggressive breast cancer cells. Additionally, this knockdown inhibited the development of liver metastatic nodules in a mice model (Luo and others 2016). Thus, VEGF/NRP-1 axis may be a valuable target for the design of antimetastatic therapies in breast cancer.
Another biointeraction that favors the metastasis in breast cancer is the interplay between tumor suppressor genes and VEGF modulation. Recently, loss of expression of p16 gene suppressor was reported in human primary breast carcinomas (Silva and others 2003). This protein is important because it can downregulate VEGF gene and protein expression, resulting in the suppression of angiogenesis and metastasis in breast cancer. The pathway by which this suppressor gene acts in breast cancer cells is through the change of localization from nuclear to cytoplasm of HIF-1α, preventing the transactivation of VEGF promoter (Zhang and others 2007). Thus, the reactivation of the p16 activity will be an innovative approach to counteract the metastasis in breast cancer.
HIF-1α is a transcription factor that controls the expression of VEGF (Krock and others 2011). In this sense, a specific drug known as 2-benzoyl-3-phenyl-6,7-dichloroquinoxaline 1,4-dioxide (DCQ) that reduces the low oxygen microenvironment was able to block the metastatic process in breast cancer cell lines through the inhibition of 2 target genes of HIF-1α implicated in angiogenesis and Type III EMT, VEGF, and TWIST, respectively (Ghattass and others 2014). These findings open the possibility for the search of new drugs that not only inhibit proteins involved in the metastasis process, but also drugs that counteract the tumor microenvironment.
Notch pathway has been related to multiple processes such as cell–cell communication, cell differentiation, and angiogenesis through VEGF (Kofler and others 2011; Benedito and others 2012; Garcia and Kandel 2012; Hernandez and others 2013). Besides, aberrant Notch pathway activity is involved in breast cancer progression (Reedijk 2012). Interestingly, a feedback between VEGF and one ligand of Notch pathway, delta-like 4 ligand (ΔII4), has been described. In this sense, VEGF can increase the expression of ΔII4. This protein can also be stimulated by hypoxic conditions and in this way it can stimulate at the same time the VEGF expression (Patel and others 2005). However, little is known about this feedback regulation in breast cancer, leaving the possibility of new research to inhibit both pathways.
A close relationship between VEGF expression and MMP-9 was found in breast cancer tumors (Dore-Savard and others 2016), highlighting their interaction to promote tumor metastasis. Furthermore, this detailed work found the presence of 23 cytokines related with angiogenic/metastatic processes in the tumor interstitial fluid of breast cancer xenografts termed as “angiogenic secretome,” proposing the collection of this fluid instead of plasma from patients with aggressive cancers with the aim to design effective personalized treatments (Dore-Savard and others 2016).
Moreover, it has been described as a “metastatic signature” driven by VEGF in a human orthotopic model of ER-positive and -negative breast cancer cells (Pathak and others 2013). VEGF has a narrow interaction in the extracellular matrix remodeling, driving the metastasis toward the lung and lymph nodes mediated by its participation as activator of fibrin degradation and promoter of extracellular proteolysis (Pathak and others 2013). VEGF also was able to increase breast cancer cell invasion mediated by its association with MMPs, and by the activation of MAPK and PI3K pathways with the consequent activation of the proto-oncogene c-Fos (Miralem and others 2001; Chen and others 2017a). In relation to the works mentioned above, it is clear that not only VEGF is responsible for the “metastatic phenotype,” but there are also several targets that need to be taken into account to improve the therapies and even predict the ability of tumors to have metastasis.
Also, in ER- and HER2-negative breast cancer, it has been described that a group of cytokines, such as VEGF-A and VEGF-C, forms a paracrine loop to promote metastasis in breast cancer (Lee and others 2014a, 2014b). A more recent work complemented this information in different breast cancer tumor sample subtypes. This work described a blend of prometastatic factors, where VEGF ligands and its receptors are involved (VEGF-A, VEGF-C, VEGFR-2, VEGFR-3, NRP-1, and NRP-2) (Fertig and others 2015). The overall conclusion of this research is that VEGF-A and VEGF-C mRNA are overexpressed in both ER/HER2-negative and HER2-positive breast cancer more than ER-positive subtype (Fertig and others 2015). This is partly because there is a crosstalk between EGFR and angiogenesis signaling in breast cancer (Alameddine and others 2013).
Broadening the previous crosstalk, heregulin-1β (HRG-1β), a ligand of the EGFR family, upregulates the VEGF-C gene and protein expression throughout the p38 MAPK and NF-κB signaling pathways in human breast cancer cells (Xiong and others 2001; Tsai and others 2003). Additionally, HRG-1β has also been shown to upregulate the expression of VEGF-A in breast cancer cells, inducing the neovascularization process (Yen and others 2000). Overall, this protein modulation panel is involved in lymphangiogenesis, metastasis, and drug resistance, pointing out NF-κB as an important therapeutic target in this pathology (Godwin and others 2013). On the other hand, the alterations in HRG-1β expression, impact on the invasive and metastatic process in breast cancer, since this protein has shown to regulate the expression of proteins implicated in Type III EMT, such as MMP-9 (Yao and others 2001). In summary, the cooperation between EGFR family ligands and VEGF signaling confers an enhanced metastatic potential (Fig. 2), which eventually may result in a poor prognostic condition in breast cancer patients.
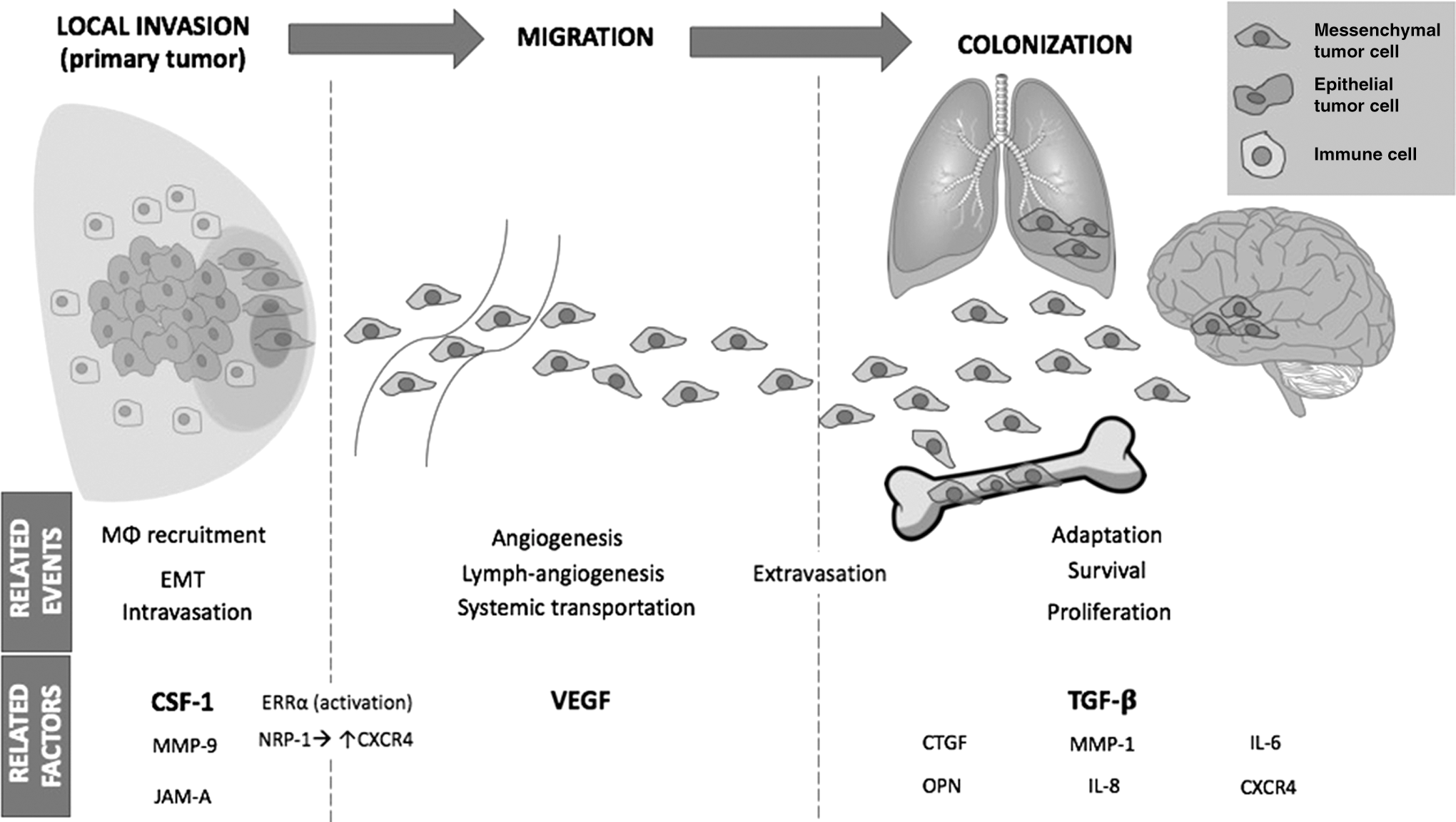
Growth factors involved in the development of breast cancer metastasis. The figure shows how VEGF, CSF-1, and TGF-β regulate different signaling pathways that contribute to the development of breast cancer cell metastasis. This process is evolving starting from epithelial tumor cells in the breast tumor (primary tumor), the Type III EMT and the intravastion, extravasation, and colonization of other organs as mesenchymal tumor cells. VEGF, vascular endothelial growth factor; CSF-1, colony-stimulating factor 1; TGF-βR, transforming growth factor beta receptor; MØ, macrophage; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; NRP-1, neuropilin 1; NFkB, nuclear factor kappa-light-chain-enhancer of activated B cells; HIF1α, hypoxia-inducible factor 1 alpha subunit; GSK-3β, glycogen synthase kinase 3 Beta; III EMT, type III epithelial–mesenchymal transition; CTGF, connective tissue growth factor; OPN, osteopontin; ERRα, estrogen-related receptor alpha; CXCR4, C-X-C Motif Chemokine Receptor 4; JAM-A, junctional adhesion molecule-A; JNK, c-Jun N-terminal kinase.
The overexpression of SIX1, a transcription factor, has been associated with lymph node positivity and poor prognosis in breast cancer (Iwanaga and others 2012). Moreover, this protein is involved in the regulation of multiple protumorigenic genes (Yu and others 2006; McCoy and others 2009; Micalizzi and others 2009). In fact, its expression has been closely related to later stages of breast cancer metastasis through the upregulation of TGF-β signaling (Micalizzi and others 2010). In this sense, the promoting activity of SIX1 upon VEGF-C/VEGR3 lymphangiogenesis and lymphatic metastasis actions has been demonstrated (Wang and others 2012). SIX1 could become a diagnostic protein for breast cancer patients as a preventive metastatic process.
In addition, another transcription factor, STAT3, has been correlated with VEGF-C, VEGF-D, and VEGFR-3 expression in lymph node metastasis in breast cancer tissues. Thereby, this suggests that STAT3 may be an important molecular marker for predicting metastasis to axillary lymph nodes in breast cancer, which is associated also with cancer recurrence and poor prognosis (Chen and others 2017b). Moreover, a clear interaction among STAT-3/VEGF-C/VEGFR-3 expressions was established with lymphangiogenesis, whereas STAT-3/VEGF-D/VEGFR-2 expression was associated with carcinogenesis in breast cancer (Chen and others 2017b). The main cross of these pathways is that STAT3 can bind to one of the binding sites of VEGF, upregulating its expression (Niu and others 2002).
In addition, a coexpression of 2 growth factors, VEGF and angiopoietin-2, was related to breast cancer brain metastasis, development, and progression, in a mice model where hypoxic conditions are critical for the maintenance and stimulation of the secretion of angiogenic factors. Further works should be driven to determine if simultaneous blocking of these proteins can be effective to reduce breast cancer brain metastasis (Bohn and others 2017).
VEGF was also associated with breast cancer bone metastasis through the activation of the orphan estrogen-related receptor alpha (ERRα) (Fradet and others 2011). This protein is increased in breast cancer cells and it is associated with its recurrence (Ariazi and others 2002; Suzuki and others 2004). The mRNA expression of target genes of ERRα, such as VEGF, MMP-1, and MMP-13 was increased in breast cancer biopsies that contained elevated ERRα expression. Therefore, the mechanism by which ERRα favors bone metastasis processes is through the modulation of proteins involved in angiogenesis and invasion (Fradet and others 2011). This orphan receptor represents a novel guide for breast cancer treatment.
Regarding breast cancer liver metastasis, the main pathways suggested that govern this process are MAPK, NF-κB, and VEGF. This genetic network was clarified with different databases. Interestingly, these proteins are associated with distinct stages of breast cancer liver metastasis (Tsai and others 2003; Chen and others 2017a). The best molecular understanding of this condition is important because breast cancer liver metastasis is a disease that once detected, it only allows a short survival time (Adam and others 2006).
Recently, in a breast cancer lung metastasis mice model, an interesting relationship between VEGFR-1 and a population of metastasis-associated macrophages (MAMs) was identified (Qian and others 2015). These cells have shown to stimulate tumor extravasation and they also sustained distal metastasis (Qian and others 2009). Moreover, MAMs express VEGR-1 in their surface, and it acts as a chemotactic molecule and vascular permeability enhancer. It was also associated with the inflammatory signaling through stimulation of a key regulator of macrophage lineage-specific growth, the CSF-1 (Murakami and others 2008; Qian and others 2015), which in turn stimulates the VEGF expression in macrophages (Curry and others 2008). Of note, VEGFR-1 expression in MAMs was extremely remarkable, whereas in lung resident macrophages this expression was not found. Significantly, VEGFR-1 ablation resulted in a decrease of breast cancer lung metastasis (Qian and others 2015). This angiogenic/immunological modulation invites to extend the cancer therapy options not only to breast cancer cells and VEGF signaling but also to their immune cell counterpart.
In Table 1, we sumarize the available information regarding the role of different cytokines involved in breast cancer metastatic process.
Role of Cytokines in Breast Cancer Metastasis
BCBoM, breast cancer bone metastasis; BCBrM, breast cancer brain metastasis; BCLiM, breast cancer liver metastasis; BCLuM, breast lung metastasis; CAFs, cancer-associated fibroblasts; CSC, cancer stem cell phenotype; CXCR, chemokine receptors; CSF-1, colony-stimulating factor 1; MCP-1/CCL2, derived monocyte chemotactic protein-1; EMT, epithelial–mesenchymal transition; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; GSK-3β, glycogen synthase kinase-3; HER2, human epidermal growth factor receptor 2; IL, interleukin; JAM-A, junctional adhesion molecule-A; MMP, metalloproteinase; MAMs, metastasis-associated macrophages; MAPK, mitogen-activated protein kinase; miR, microRNA; MCP-1, monocyte chemotactic protein 1; NF-κB, nuclear Factor-Kappa B; ERRα, orphan estrogen-related receptor alpha; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; STAT, signal transducer and activator of transcription; SDF-1, stromal-derived factor-1; TAMS, tumor-associated macrophages; TNF-α, tumor necrosis factor alpha; TGF-β, transforming growth factor beta; VEGF, vascular endothelial growth factor.
Concluding Remarks
Metastasis is the major cancer pathophysiology and there are a lot of interactions of different signaling pathways that lead to the possibility of developing specific therapeutic target drugs. To counteract breast cancer metastasis, in this study, we described different molecular targets that should be blocked together, with the aim to offer a promisig antimetastatic drug regardless of the expression of hormonal receptors. We also want to highlight that not only cancer cells must be taken into account but also the modification of the tumor microenvironment and the surrounding cell metabolism play a key role in attacking the metastatic breast cancer process and it would result in a better patient clinical outcome. In addition, the immune cells and cytokines are key factors whose modulation would be important as an adjuvant drug option in breast cancer metastasis.
Footnotes
Acknowledgments
Financial support: Grant IN208715 from Programa de Apoyo a Proyectos de Innovación Tecnológica, Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México to J. Morales-Montor. Rocío Alejandra Ruìz-Manzano is a PhD student at Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México and received a fellowship from CONACyT. Margarita I Palacios-Arreola and Mariana Segovia-Mendoza, both are Postdoctoral fellows from Dirección General de Asuntos del Personal Académico (DGAPA), from Universidad Nacional Autónoma de México (UNAM), and receive a fellowship from DGAPA. Lucía Angélica Méndez-García is a CONACyT Postdoctoral fellow.
Author Disclosure Statement
Authors declare that no competing financial interests exist.