
Abstract
Interferon-gamma (IFNG) has long been implicated as a central orchestrator of antitumor immune responses in the elimination stage of the immunoediting paradigm. However, mounting evidence suggests that IFNG may also have important and significant protumor roles to play in the equilibrium and escape phases through its regulatory effects on immunoevasive functions that promote tumorigenesis. These seemingly contradictory effects of IFNG undoubtedly play profound roles in not only the activation of inflammatory response to cancer but also in the determination of its outcome. In the face of the recent explosion of anticancer immunotherapeutic strategies in the clinic, it is critical that a complete understanding is achieved of the underpinnings of the mechanisms that determine the two faces of IFNG signaling in cancer. Here, the current state of this dichotomy is reviewed.
Introduction
Interferon-gamma (IFNG) is the only type II IFN cytokine, and has been shown to play crucial roles in the innate and adaptive immune responses. It was originally discovered to be a critical orchestrator of immune responses against viral infections, but it was soon realized that its immune-modulating activities reached much farther and wider (Boehm and others 1997; Pestka and others 2004; Schroder and others 2004). The downstream target genes of IFNG signaling pathway perform a wide variety of biological functions, which are primarily associated with modulation of innate and adaptive immune responses in host defense against viral and bacterial infections; but are also related to regulation of cell cycle, apoptosis, and inflammation (Schroder and others 2004). An important antimicrobial function of IFNG is regulation of the expression of the major histocompatibility complex (MHC) class I and II molecules, which are critical components of the antigen-presenting machinery (Shirayoshi and others 1988; Steimle and others 1994). In adaptive immunity, IFNG directly regulates the differentiation, activation, and homeostasis of Th1 cells; inhibits the development of Th2 cells; promotes the development of regulatory T cells (Treg); promotes the activity of natural killer (NK) cells; and activates the classic M1 subtype of inflammatory macrophages (Higuchi and others 1987; Gajewski and Fitch 1988; Maraskovsky and others 1989; Oriss and others 1997; Wang and others 2006; Curtsinger and others 2012). By inducing several chemokines, IFNG also plays important roles in recruitment of specific effector cells to different inflammatory microenvironments (McLoughlin and others 2003; Pak-Wittel and others 2013).
IFNG Signaling: An Overview
IFNG is predominantly produced by the NK and natural killer T (NKT) cells in the innate arm of the immune system, as well as by effector T cells such as CD4+ T helper (Th1) cells and CD8+ cytotoxic T lymphocytes (CTLs) in the adaptive arm of the immune system (Schoenborn and Wilson 2007). Recently, the noncytotoxic innate lymphoid cells have been shown to produce IFNG, which plays a key role in mucosal immunity against pathogens (Artis and Spits 2015; Elemam and others 2017). Varying degrees of IFNG are also secreted by mucosal epithelial cells and macrophages (Rouabhia and others 2002; Darwich and others 2009). The expression of IFNG is induced by a number of mitogens and cytokines, eg, interleukins (IL-) 2, 12, 15, and 18, as well as type I IFNs (Chan and others 1991; Carson and others 1995; Okamura and others 1995; Ye and others 1995; Nguyen and others 2000).
The canonical IFNG signaling is activated when the biologically active form of IFNG binds to the alpha subunit of its exclusive receptor, which is a heterodimer consisting of alpha (IFNGR1) and beta (IFNGR2) subunits (Platanias 2005). IFNGR2 subunit is responsible for the intracellular relay of the signal through activation of the nonreceptor tyrosine kinases known as janus-activated kinases JAK1 and JAK2, which phosphorylate the intracellular domains of IFNGR1/2 to create binding sites for the signal transducer and activator of transcription (STAT) factors. STAT1 is the principal activated effector molecule responsible for the downstream signaling mediated by IFNG (Haan and others 2006). Phosphorylation of STAT1 leads to its translocation to the nucleus where it binds to the consensus gamma-activated sequence (GAS) sites on the promoters of numerous target genes and activates their expression (Wen and others 1995; Varinou and others 2003).
There is evidence that IFNG can also activate alternative noncanonical signaling pathways in the presence or absence of STAT1 (Green and others 2017). Gil and others (2001) had reported that IFNG can elicit a response in mononuclear phagocytic cells obtained from Stat1-knockout (Stat1−/− ) mice. In the STAT1-independent noncanonical pathway, IFNG can activate GAS-regulated genes through activation of STAT3 (Qing and Stark 2004; van Boxel-Dezaire and Stark 2007). IFNG has also been shown to induce antiviral genes regulated by the IFN-stimulated response element (ISRE) through activation of IFN-stimulated gene factor 3 (ISGF3), which is conventionally associated with type I IFN response (Morrow and others 2011). Moreover, IFNG can activate signaling through MAPK molecules (Ramana and others 2001; Matsuzawa and others 2014). For example, O'Donnell and others (2015) have shown that IFNG activates ERK1/2-mediated signaling in neurons, which confers prosurvival effects. These results suggest that IFNG can induce noncanonical signaling in a cell type-specific and context-dependent manner.
Dual Roles of IFNG in Cancer
In the context of cancer, IFNG has historically been considered a central player in antitumor immunity. It performs its antitumor functions through influencing both the tumor cells and the immune effector cells. For example, IFNG upregulates the expression of the MHC class I molecules in tumor cells, and thus enhances their antigenicity due to increased antigen presentation (Martini and others 2010). Moreover, IFNG enhances the cytotoxic activities of NK cells and CTLs (Young and Hardy 1995; Street and others 1997). These dual effects of IFNG synergize to make the tumor cells more susceptible to recognition and destruction by IFNG-activated immune effector cells by virtue of enhanced presentation of tumor-associated antigens (Martini and others 2010; Vigneron 2015).
IFNG has direct tumor cell-specific antitumor effects such as inhibition of proliferation by cell cycle arrest through activation of p21 and p27 tumor suppressors (Chin and others 1996; Harvat and others 1997). IFNG is also known to play antitumor role by inducing apoptotic cell death of multiple cancer cell types (Chawla-Sarkar and others 2003). Furthermore, IFNG can induce a necrosis-like regulated cell death termed necroptosis through activation of RIP1 serine-threonine kinase (Thapa and others 2011). Inhibition of tumor angiogenesis is another known antitumor activity of IFNG (Coughlin and others 1998; Ruegg and others 1998; Qin and others 2003). Mesenchymal stromal cell-mediated secretion of IFNG in the tumor microenvironment has been shown to repolarize the tumor-associated macrophages to the M1 inflammatory phenotype, which reduced tumor burden in a murine neuroblastoma model (Relation and others 2018). Although IFNG is known to support development of Treg that are immunosuppressive, recently it has been shown that IFNG also boosts antitumor immunity and tumor clearance by driving Treg fragility (Overacre-Delgoffe and others 2017). Recent evidence also implicates loss of IFNG pathway genes in acquiring resistance to immunotherapy, which suggests a critical role of IFNG signaling in immune-mediated tumor killing (Gao and others 2016).
Although IFNG quickly developed a conventional, well-recognized, and well-characterized reputation as an antitumor immune factor, its potential as a protumor factor also has a long history (Zaidi and Merlino 2011). One common theme that seems to emerge from the studies that show protumorigenic effects of IFNG is that tumors that are exposed to IFNG show greater immunoevasive capabilities. For example, several IFNG pathway target genes are known to be involved in immunosuppressive and immunoevasive mechanisms that are geared toward suppression of CTL- and NK cell-mediated antitumor immune responses. This is seemingly counterintuitive because of the fact that IFNG is a potent activator of the genes involved in antigen presentation, which should be expected to increase immunogenicity and CTL- and NK cell-mediated elimination of tumor cells. However, it has been noted in some models that IFNG deficiency or antibody-mediated systemic blockade increases immunogenicity of tumor cells as compared with those inoculated in fully immunocompetent mice (Shankaran and others 2001; Koebel and others 2007). It is plausible that exposure to prolonged and/or elevated IFNG levels exerts selective immune pressure on the tumor cells, which leads to a decrease or loss of the MHC class I and other genes involved in the antigen presentation machinery (Maeurer and others 1996; Algarra and others 2004). In fact, such a mechanism has been shown in M14 melanoma and CT26 colon cancer cell lines in which exposure to IFNG resulted in downregulation of antigen presentation and led to immune evasion (Beatty and Paterson 2000; Le Poole and others 2002).
These and other results clearly suggested a counterdogmatic and paradoxical protumorigenic role of IFNG within the context of the tumor microenvironment and the host's immunological response to tumor development. A flurry of recent reports have underscored the protumorigenic role of IFNG in cancer, bringing the IFNG paradox to the fore (Zaidi and Merlino 2011; Kursunel and Esendagli 2016; Lin and others 2017; Mojic and others 2017; Aqbi and others 2018; Castro and others 2018; Kosmidis and others 2018).
IFNG in Immunosurveillance and Immunoediting
Cancer immunosurveillance is the ability of the host immune system to recognize and destroy transformed cells (Kroemer and others 2015). Early indications of the role of IFNG in cancer immunosurveillance were provided by in vivo studies that showed that mice lacking Ifng (Ifng−/− ), as well as those lacking its receptor (Ifngr1−/− ) or downstream effector Stat1 (Stat1−/− ), were susceptible to chemical carcinogen-induced tumorigenesis (Shankaran and others 2001). Based on these results, Schreiber and others (2011) proposed an important role for IFNG in their cancer immunoediting paradigm. Immunoediting refers to the reprogramming of cancer cells to thwart the host immune system. This reprogramming enables cancer cells to not only survive the immune onslaught in a manner akin to Darwinian survival of the fittest (Schreiber and others 2011) but it also supports cancer growth.
Cancer immunoediting leads to modulations of the tumor immunogenicity through a 3-phase continuum; elimination, equilibrium, and escape (Teng and others 2015). The elimination phase is essentially comprised of immunosurveillance in which the innate and adaptive arms of the immune system cooperate to recognize and destroy any transformed cells and developing tumors before clinical manifestation. A small number of transformed cells may survive the elimination phase due to acquired capabilities but nonetheless are prevented from outgrowth due to immunological checks. This phase is termed equilibrium, as rare cancer cells lay dormant under the influence of adaptive immunity. IFNG has been shown to play important roles in the regulation of both elimination and equilibrium phases of immunoediting.
The Schreiber group extended their initial findings to show that in a genetic model of cancer that lacks the p53 tumor suppressor, mice that are unresponsive to IFNG signaling due to lack of Stat1 (p53−/− , Stat1−/− ) are significantly more susceptible to tumorigenesis with a much wider spectrum of tumor types as compared with those lacking p53 alone (p53−/− ) (Kaplan and others 1998). It was shown that the enhancement of tumorigenesis in the Ifng-deficient context was dependent on the unresponsiveness of the tumor cells themselves to IFNG signaling. While chemically induced tumors from Ifngr1−/− mice grew aggressively and progressively upon transplantation in either naïve Ifngr1−/− or wild-type hosts, this tumorigenic behavior was abolished when these cells were reconstituted with ectopic Ifngr1 expression. However, when the Ifngr1-reconstituted Ifngr1−/− tumor cells were inoculated into lymphocyte-deficient Rag2−/− mice, the tumors grew aggressively. These results suggested that insensitivity to IFNG signaling by the tumor cells was directly responsible for the enhanced chemically induced carcinogenesis in Ifngr1−/− mice, and that this phenomenon was dependent on the lack of tumor rejection mediated by an intact adaptive immune system (Kaplan and others 1998; Shankaran and others 2001). IFNG signaling-mediated tumor rejection was postulated to be effected in part by induction of expression of the MHC class I antigen processing and presentation machinery, which enhanced tumor immunogenicity and facilitated tumor recognition and elimination by the immune effector cells. It is clear from these studies that endogenous IFNG and lymphocytes cooperate to protect the host against tumorigenesis through an intricate array of cancer immunosurveillance mechanisms, which encompass the elimination and equilibrium phases of the immunoediting paradigm (Dunn and others 2005).
Protumor Roles of IFNG
The selection pressure exerted on the tumor cells by the immune system during the elimination and equilibrium phases of immunoediting results in removal of highly immunogenic tumor cells and favors generation of genetically variant tumor cells that exhibit reduced immunogenicity, which would render them “invisible” to the immune system (Schreiber and others 2011; Malladi and others 2016; Saranchova and others 2016). Such evolutionarily selected tumor cells would be capable of evading the host immune system and forming gross tumor outgrowth, entailing the escape phase of immunoediting. Recent evidence shows that IFNG also plays an important role in the escape phase, which characterizes the protumorigenic face of IFNG and epitomizes the paradoxical nature of IFNG signaling pathway in cancer.
It was shown as early as 1987 that IFNG enhanced lung colonization of intravenously inoculated B16 mouse melanoma cells (Taniguchi and others 1987). Activation of expression of the MHC class I molecules H-2Kb and H-2Db by IFNG was shown to render B16 cells resistant to NK cell-mediated cytolysis. IFNG also activates expression of MHC class II molecules, which is associated with an aggressive phenotype in human melanoma (Brocker and others 1988). In fact, human melanoma cells respond to IFNG treatment in culture by exhibiting an aggressive phenotype characterized by inhibition of dendrite formation and increased expression of ICAM1 and other melanoma progression markers (Garbe and others 1990). Treatment of NIH3T3 cells with IFNG accelerates their proliferation by activating expression of the guanylate binding protein 2 (GBP2) (Gorbacheva and others 2002). Ectopic expression and autocrine IFNG signaling in TS/A mammary adenocarcinoma cells augmented metastatic capabilities by enhancing resistance to NK cell-mediated immunity (Lollini and others 1993).
IFNG signaling is regulated through diverse mechanisms, with many mechanisms regulating receptor expression, lipid raft association, and endocytosis/recycling. Another critical regulator of IFNG is the protein SOCS1. SOCS1 is a negative regulator of the IFNG-mediated JAK/STAT signaling. It has been reported that Socs+/− mice exhibit enhanced carcinogen-induced hepatocellular carcinoma (Yoshida and others 2004). To rule out the effect of reduction/lack of SOCS1 on factors other than IFNG signaling as the cause of such tumorigenic effect, Hanada and others (2006) restored SOCS1 expression specifically in T and B cells in a Socs−/− genetic background, and showed that the carcinogen-induced colon cancer was dependent on constitutive IFNG signaling. Despite the conventionally accepted antitumor role of IFNG produced by the T cell compartment, chronic IFNG production by the CD4+ T cells that harbor intrinsic deficiency of the deubiquitinase USP15 has been shown to enhance carcinogen-induced fibrosarcomas (Zou and others 2015). Moreover, IFNG produced by adoptively transferred cytotoxic T cells was shown to promote growth of leukemia stem cells (Schurch and others 2013). Chronic exposure to IFNG has been shown to enhance tumor growth in transplantable models of H22 hepatoma, MA782/5S mammary adenocarcinoma, and B16 melanoma (He and others 2005). These results clearly suggest that exposure to IFNG in the tumor microenvironment has the potential to promote cancer growth. Moreover, recent evidence suggests that IFNG also promotes tumor metastasis. Xu and others (2018) have shown that IFNG induces gastric cell proliferation and metastasis at least partially through upregulating integrin β3-mediated NF-κB signaling. Ni and others (2017) have reported that while IFNG induces angiostasis, IFNG-mediated signaling also dissociates perivascular cells from blood vessels, which contributes to the acceleration of tumor metastasis.
Exposure to IFNG was also shown to cause loss of melanocytic antigens melan-A and gp100 in human melanoma cell lines, which led to immune evasion by loss of recognition by cytotoxic T cells (Morel and others 2000). IFNG has been shown to enhance development of Treg and suppress CTL activity by inducing the expression of indolamine 2,3-dioxygenase (IDO) in melanoma cells (Katz and others 2008; Prendergast 2008; Brody and others 2009). IFNG-induced IDO also cooperates with gangliosides shed by tumor cells to impede dendritic cell capacity to activate T cells in the tumor microenvironment, leading to immunosuppression (Dillinger and others 2018). It is noteworthy that in addition to the classical MHC class I genes, IFNG activates nonclassical MHC class Ia genes (eg, HLA-G, HLA-E, and HLA-F), which have been shown to be involved in immune escape in a number of cancers by inducing resistance to CTL and NK cell responses (Gobin and van den Elsen 2000; Wischhusen and others 2007). IFNG-mediated activation of the nonclassical MHC class Ia genes was implicated in evasion of melanoma cells from CTL-mediated cytolysis, which led to the clinical failure of melanoma peptide vaccines (Cho and Celis 2009). IFNG-induced expression of HLA-E and shedding by melanoma cells protected them against CTL-mediated cytolysis (Derre and others 2006). Suppression of anticancer T cell response is also attributed to the influx of monocytic and granulocytic myeloid-derived suppressor cells to the tumor microenvironment, which is dependent on IFNG signaling (Ostrand-Rosenberg and Sinha 2009).
There is steadily increasing evidence that IFNG signaling pathway regulates immunoevasion by activating immune checkpoint genes. One of the most important IFNG-regulated immune checkpoint mechanisms is PD-L1 and PD-L2 expression on the surface of tumor cells as well as immune infiltrating stromal cells. IFNG-induced PD-L1/2 ligands bind to their immune inhibitory receptor PD-1, and this interaction suppresses the immune effector activities of T cells and NK cells, which can promote cancer progression (Abiko and others 2015; Bellucci and others 2015; Garcia-Diaz and others 2017). Induction of PD-L1 and PD-L2 expression in cancer cells has been postulated to be an important determinant of adaptive immune resistance to immune checkpoint inhibitor immunotherapy (Sharma and others 2017). Benci and others (2016) have shown that prolonged exposure of cancer cells to IFNG signaling in the tumor microenvironment activates expression of a number of ligands for T cell inhibitory receptors, which also promotes resistance to immune checkpoint inhibitor immunotherapy in a PD-1/PD-L1-independent fashion. Moreover, PD-L1 has been found to be autonomously responsible for promoting tumor-initiating cells that exhibit drug-resistant behavior, and contributes to relapse of melanoma and ovarian cancers (Gupta and others 2016).
Recently, it has been shown that IFNG also regulates the expression of CTL antigen 4 (CTLA4) in human primary melanocytes, and human melanoma cell lines and tissues (Mo and others 2018). CTLA4 is the most potent inhibitor of CTL activation, and thus plays a key role in the negative regulation of antitumor immune response. Expression of CTLA4 on T cells contributes to T cell tolerance and anergy, which can lead to tumor immunoevasion. The finding that IFNG signaling highly activates expression of CTLA4, in addition to PD-1/PD-L1, on melanoma cells has important implications for the conventionally accepted role of IFNG in tumor immunosurveillance and immunoevasion. It is plausible to speculate that IFNG-induced CTLA4 expression on melanoma cells can directly inhibit CTL activity. Such melanoma cell-mediated inhibition of T cell cytotoxicity can have profound and yet unappreciated immunoevasive functions.
IFNG as a Therapeutic Agent
IFNG rapidly gained a reputation as an antitumor agent and prompted its advancement into the clinic as a therapeutic option against a variety of cancers. However, recombinant IFNG (rIFNG) showed only marginal and mixed results as an immunotherapy agent. The initial clinical trials suggested that maximum tolerated doses of IFNG were generally unsuccessful; rather, the effectiveness of IFNG in modulating immune responses followed a Gaussian dose–response curve; ie, low to moderate doses of IFNG exhibited greater biological effects than higher doses (Balachandran and Adams 2013). IFNG therapy was shown to prevent recurrence of bladder cancer after surgical resection (Giannopoulos and others 2003) and produced lasting remissions in T cell leukemia patients (Tamura and others 1987). Adenoviral vector-based delivery of IFNG-expressing construct exhibited positive clinical response in patients with T and B cell lymphomas (Accart and others 2013; Dreno and others 2014).
In contrast to these minor successes, several other clinical trials showed none or negative responses to IFNG therapy. Adjuvant therapy with IFNG following surgical resection in colon cancer patients did not show any clinical benefit (Wiesenfeld and others 1995). IFNG therapy trials in melanoma patients as an adjuvant application either failed to show any efficacy (Schiller and others 1996), or were prematurely terminated because the IFNG-treated patient arm exhibited worse survival than the control arm (Meyskens and others 1990, 1995). Similar results were obtained in IFNG therapy applications in ovarian cancer patients. While initial trials showed improved survival rates in ovarian cancer patients treated with IFNG (Pujade-Lauraine and others 1996; Marth and others 2006), other studies showed adverse outcomes and shorter overall survival (Alberts and others 2008).
These inconsistent, and in many cases adverse outcomes of IFNG as a therapeutic agent has not deterred continued efforts to test its clinical applications. There are ongoing clinical trials for rIFNG therapy for HER2-positive breast cancer, ovarian cancer, fallopian tube cancer, primary peritoneal cancer, and soft tissue sarcoma (NCT03112590, NCT02948426, and NCT03056599). Moreover, the recent successes of the immune checkpoint inhibitor immunotherapies, eg, antiprogrammed death (anti-PD-1) (nivolumab) and anti-CTLA4 (ipilimumab) monoclonal antibodies, have revived interest in rIFNG as a combination therapeutic that might provide clinical benefit. One such clinical trial is testing nivolumab and rIFNG combination in patients with advanced solid tumors (NCT02614456). Interestingly, rIFNG has shown promise in determining efficacy of adoptive T cell therapy against large tumors (Spear and others 2012; Textor and others 2014).
Conclusions and Future Directions
The counterdogmatic protumorigenic role of IFNG may originate from its homeostatic functions (Nirschl and others 2017). While IFNG induces an inflammatory cascade in response to microbial infections as well as tumorigenesis to eliminate damaged cells, it might also simultaneously function to protect normal cells from collateral damage caused by the inflammatory tissue remodeling. For example, we have previously shown that IFNG induces an immunoevasive/survival gene expression signature, including CTLA4, in skin melanocytes in the context of exposure to genotoxic ultraviolet radiation (UVR), which may play an important role in protecting melanocytes from eradication by the UVR-induced inflammatory response (Zaidi and others 2011, 2012). Such protective functions of IFNG signaling can be exploited by cancer cells to evade immune-mediated destruction and to survive long term in a state of equilibrium until they accumulate enough mutations to get fully transformed. In effect, IFNG signaling may simultaneously induce tissue inflammation and immunosuppressive/tolerogenic tissue microenvironment. Such a dual function would aptly explain the paradoxical and seemingly contradictory results that show antitumor as well as protumor faces of IFNG signaling.
It is not very clear exactly what downstream effectors determine which face of IFNG signaling would prevail in a particular tumor microenvironment. It is likely that a combination of a variety of factors is involved, including tumor-type specificity, presence or absence of specific microenvironmental cues, and the spatiotemporal dynamics of IFNG signaling (Fig. 1). A concerted and continued effort is needed to delineate the upstream and downstream determinants of the eventually winning face of IFNG signaling pathway in the tumor microenvironment. The potential implications of identifying such molecular determinants are profound, as these may serve as useful biomarkers for distinguishing between cancer patients who might benefit from recombinant IFNG therapy and those who are likely to be unresponsive or even exhibit adverse outcomes. Moreover, a provocative possibility exists that a subset of cancer patients may benefit from inhibition of IFNG signaling as a viable therapeutic strategy.
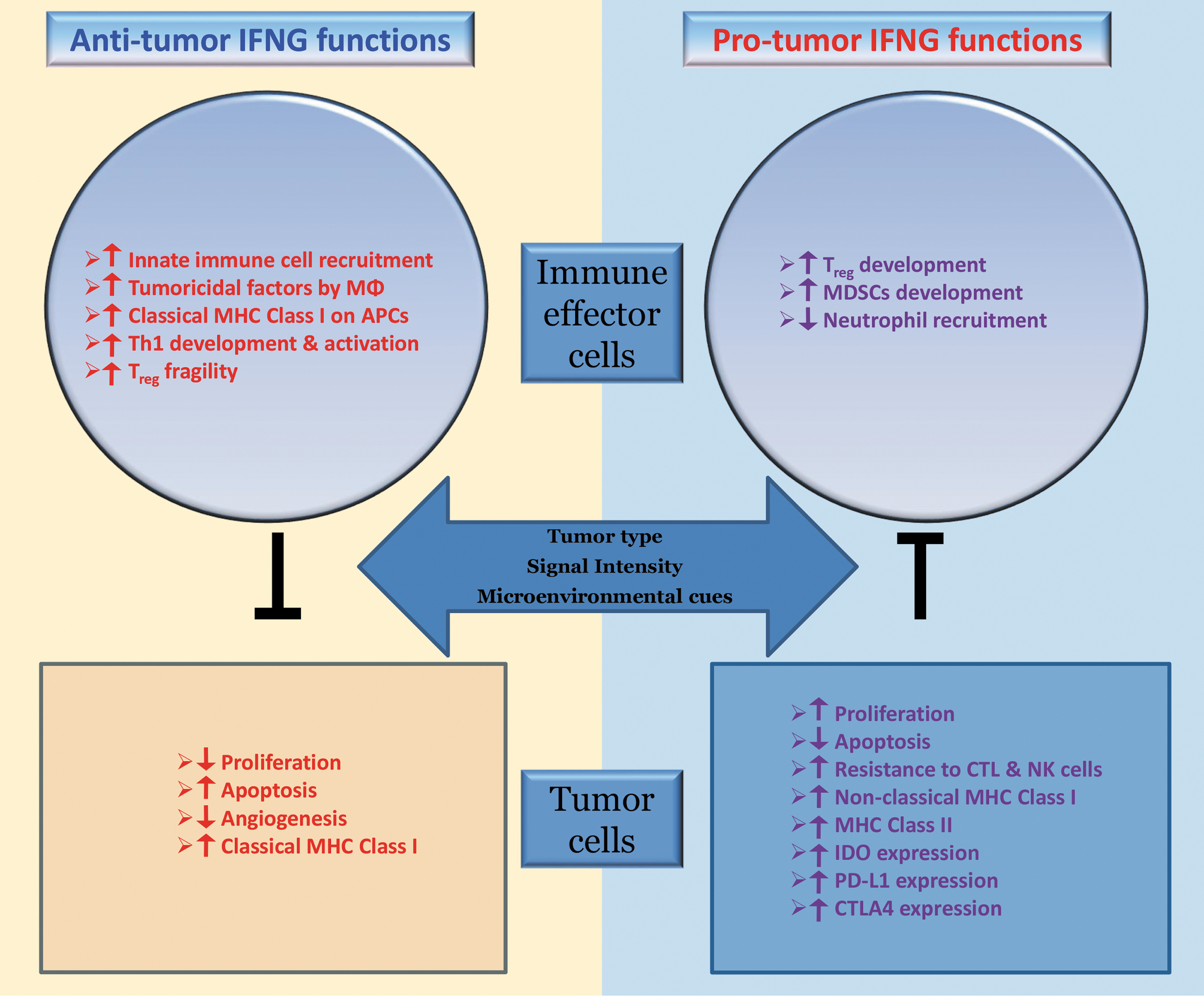
Putative mechanisms of the antitumor and protumor roles of IFNG in the tumor microenvironment. IFNG signaling affects both the immune cells and tumor cells. Which face of IFNG dominates in a particular tumor microenvironment is likely dependent on the tumor-type specificity, local signal intensity, and/or other microenvironmental cues. IFNG, interferon-gamma.
Footnotes
Acknowledgment
This work was supported by National Cancer Institute, National Institutes of Health grant number R01CA193711 to M.R.Z.
Author Disclosure Statement
No competing financial interests exist.