
Abstract
Interferons (IFNs) suppress viral infection through the induction of >400 interferon-stimulated genes (ISGs). Among ISGs, IFN-induced protein with tetratricopeptide repeats (IFITs) is one of the most potent and well-characterized ISGs. IFIT family consists of 4 cluster genes. It has been suggested that the antiviral action of each IFIT employs distinct mechanisms. In addition, it has been shown that each IFIT exhibits its antiviral properties partially in a pathogen-specific manner. To date, the expression profile of IFITs in the liver, as well as the antiviral potency of the individual IFITs in the regulation of hepatitis C virus (HCV) infection, is not yet fully defined. Our previous study found that the expression of hepatic IFITs is well correlated with the outcome of IFN-based antiviral therapy. This study explored the significance of each IFIT in the suppression of HCV. Our in vitro and in vivo studies with humanized liver chimeric mouse system revealed that IFIT1, 2, and 3/4 play an important role in the suppression of HCV. In addition, our in vitro experiment found that all IFITs possess a comparable anti-HCV potency. Follow-up studies collectively indicated that IFITs suppress HCV likely through 2 distinct mechanisms: (1) inhibition of internal ribosome entry site-dependent viral protein translation initiation complex according to experiments with bicistronic reporter assay as well as confocal microscopic analyses and (2) sequestration of viral genome based on an experiment using replication defective viral genome. In conclusion, our study defined the importance of IFITs in the regulation of HCV and also suggested the multifaceted antiviral actions.
Introduction
Hepatitis C virus (
Chronic HCV infection is one of the leading causes of end-stage liver diseases such as decompensated cirrhosis and hepatocellular carcinoma. Moreover, HCV-mediated end-stage liver disease is the primary reason for liver transplantation. Owing to the insidious nature of the disease, most of the affected population is unaware of the infection early in the chronic infection and thus fail to seek medical attention until they develop end-stage liver disease (Wedemeyer and others 2015). Thus, it is conceivable that HCV infection remains a significant threat to our public health despite the recent introduction of highly potent direct-acting antivirals (DAAs). Therefore, it is required to establish a definitive prevention strategy to mitigate the disease burden of HCV infection, which necessitates the furthering of our understanding of the immune response against HCV infection (Baumert and others 2014).
Previous studies investigating the genetic variants between the elite and poor controllers of HCV infection revealed that single nucleotide polymorphisms located near IL28B (also known as IFN-λ3), referred to as IL28B genotype, serve as a critical determinant whether the host achieves the spontaneous resolution or transitions to the lifelong persistent infection (Thomas and others 2009; Tillmann and others 2010). Numerous follow-up studies demonstrated that the IL28B genotype is coupled with the expression and induction efficiency of interferon (IFN)-stimulated genes (ISGs) (Honda and others 2010). Taken together, these notions collectively suggest that the potent activation of the endogenous IFN system plays a central role in shaping the clinical outcomes.
Upon viral infection, the recognition of pathogen-associated molecular patterns (PAMPs) by host pattern recognition receptors serves as a frontline defense program (Saito and Gale 2008). In the case of HCV infection, a host cytoplasmic viral RNA-sensing protein, retinoic acid-inducible gene-I (RIG-I), senses the viral genome as a PAMP, which then induces the activation of interferon regulatory factor (IRF) and NF-κB to the expression of ISGs as well as types I and III IFN (Saito and others 2008). The secreted IFNs then trigger activation of Jak-STAT signaling, resulting in the second wave of induction of ISGs. Thus, the virally infected cells are equipped with dual intracellular antiviral defense programs. Both IFN induction and IFN response pathways promote the expression of ISGs to fight against viral pathogen efficiently. To date, however, it largely remains elusive how exactly each ISG inhibits the lifecycle of a variety of viral pathogens.
Among ISGs, IFN-induced protein with tetratricopeptide repeats (IFITs) is relatively well characterized (Fensterl and Sen 2011, 2015; Diamond and Farzan 2013). IFIT family comprises 4 members, IFIT1, 2, 3/4, and 5, and all IFIT genes cluster on human chromosome 10. All IFITs share a comparable degree of homology between each protein sequence, with the highest between IFIT1 and IFIT5 at 54.3% and the lowest between IFIT1 and IFIT3 at 39.6% (Supplementary Fig. S1A). According to previous studies, the degree and efficiency of IFIT induction are highly dependent on the organ/cell type or induction stimuli such as IFN induction pathways or IFN response pathways (Katze and others 2002; Schoggins and Rice 2011). With regard to the antiviral mechanism, it has been suggested that each IFIT potentially possesses 2 distinctive antiviral properties. For example, IFIT1 has dual antiviral mechanisms, by which the N-terminus and middle region bind to viral RNA to compete with eIF4E, and the C-terminus binds eIF3E (also known as p48) (Fensterl and Sen 2011, 2015; Diamond and Farzan 2013). In contrast, IFIT2 appears to selectively interact with AU-rich RNA, suggesting the possibility that IFIT2 inhibits the viral lifecycle by modulating the abundance of viral genome, given the importance of AU-rich region in determining the stability of RNA (Sen and Fensterl 2012). Also, IFIT5 has also been shown to interact with 5′ triphosphate (ppp) RNA (Katibah and others 2014). Furthermore, it has been shown that IFIT2 and IFIT3 enhance the RNA binding activity of IFIT1 (Johnson and others 2018). These mechanisms are thought to inhibit the translation of the viral genome cooperatively and, to a lesser extent, cellular mRNA.
To date, intrahepatic induction of IFITs and their significance in the regulation of HCV infection have not been sufficiently studied. Therefore, this study was undertaken to assess the role of IFITs as anti-HCV effectors, including the induction kinetics of IFITs in the hepatocyte, determination of antiviral mechanism of IFITs using in vitro and in vivo approaches. Our studies with clinical samples and humanized liver chimeric mouse (HLCM) model system found that the degree of induction of IFITs in the liver well correlated with the outcomes of IFN-based antiviral therapy. Furthermore, a series of in vitro experiments revealed that all IFITs possess a comparable level of antiviral activity against HCV. In summary, our study results indicate that IFITs are one of the critical antiviral effector genes.
Materials and Methods
Nucleic acid
RNA extraction from the liver needle biopsy was carried out using ToTALLY RNA™ kit (Ambion). The liver of HLCM, human hepatocytes, Huh7 cells, and PH5CH8 cells were first homogenized in DNA/RNA Shield (Zymo Research) followed by total RNA extraction using Quick-RNA™ MiniPrep (Zymo Research). cDNA was synthesized with qScript cDNA SuperMix (Quanta) using 1 μg of extracted RNA followed by 1:20 dilution for quantitative polymerase chain reaction (qPCR) with ABI HT7900 system. The reverse transcription quantitative polymerase chain reaction (RT-qPCR) primer sequences used in this study are the following: IFIT1 F 5′-TTCTCAAAGTCAGCAGCCAGT-3′, IFIT1 R 5′- TCCTTGGGTTCGTCTACAAAT-3′, IFIT2 F 5′-GGCTGGCAAGAATGGAACA-3′, IFIT2 R 5′- GGCTGGCAAGAATGGAACA-3′, IFIT3/4 F 5′-TCACTACCATCCTCAAGCTCA-3′, IFIT3/4 R 5′-ACTGGGCCGCCTGCTAA-3′, IFIT5 F 5′-CATTGCTATACTGGCCTCCTT-3′, IFIT5 R TGGAACGAGACTCTATGTTTG-3′, HCV TaqMan probe 5′-CTGCGGAACCGGTGAGTACAC-3′, HCV F 5′-CGGGAGAGCCATAGTGG-3′, and HCV R 5′-AGTACCACAAGGCCTTTCG-3′. The relative expression of a gene of interest (GOI) was determined by the delta Ct method with the relative expression of a reference gene (human GAPDH). The relative fold index was determined by setting the expression abundance of a GOI at the resting condition as +1. Plasmids used in this study are as follows: pRL-HL, a bicistronic reporter encoding the Cap-dependent Renilla and HCV-IRES-dependent firefly luciferase (a gift from S. Lemon), pCMVRluc-EFluc (a gift from Michael Katze) is a bicistronic reporter construct encoding Cap-Renilla, and encephalomyocarditis virus (EMCV)-IRES firefly luciferase. cDNA encoding the complete ORF of IFITs was isolated by RT-PCR from total cellular RNA extracted from PH5CH8 cells treated with IFN-β (100 IU/mL for 8 h) and cloned into pEF Myc/His Version C (Life Technologies) and propagated in DH5α under Ampicillin selection and purified with ZymoPure Plasmid Midiprep kit (Zymo Research). The primer sequences used for the amplification of ORF of IFITs are the following: IFIT1 F 5′-GCGGCCGCTAGTACAAATGGTGAT-3′, R 5′-GGTAACCCTAAGGACCTTGTCTCA-3′, IFIT2 F 5′-GCGGCCGCTAGTGAGAACAATAAGAATTCCT-3′, R 5′-TTCGAATCAGCAGTAGCCTAGTGGGCACCA-3′, IFIT3/4 F 5′-GCGGCCGCTAGTGAGGTCACCAAGAA-3′, R 5′-TTCGAATCAGTTCAGTTGCTCTGAGTTA-3′, IFIT5 F 5′-GCGGCCGCTAGTGAAATTCGTAAGGACACCT-3′, and R 5′-TTCGAATTAAATGGAAAGTCGGAGCTCA-3′. Mutant IFIT1 encoding amino acids 1 to 339 was prepared as described previously (Wang and others 2003). Bromo-UTP-labeled HCV-IRES RNA was synthesized with MEGAscript® T7 Kit (ThermoFisher) in the presence of BrdUTP (5-bromo-2′-deoxyuridine 5′-triphosphate) using a linearized HCV subgenomic replicon (SGR) encoding internal ribosome entry site (IRES) sequence. HCV PAMP RNA was synthesized and transfected as described previously (Saito and others 2008). In vitro transcription of HCV JFH-1 SGR (a gift from Charles Rice) and HCV JFH-1 SGR Luc GND (a gift from Takaji Wakita) genome was carried out as described previously (Kato and others 2006). IL28B genotyping of Huh7 cell was carried out as described previously (Elmasry and others 2016).
Cell culture
Huh7 human hepatoma cells (IL28B GT: rs12979860 CC), Huh7.5, and PH5CH8 cells, mouse embryonic fibroblast (MEF), were propagated in Dulbecco's modified Eagle's medium (DMEM; Cellgro) supplemented with 10% fetal bovine serum (Hyclone), nonessential amino acids, 2 mM
Virus infection and antiviral treatment
HCV infectious clone (JFH-1) was prepared as previously described (Ooi and others 2014) and used for in vitro infection at the indicated MOI. Humanized liver chimeric mice (PXB-mice®: cDNA-uPA+/+/SCID) were infected with HCV genotype 1a at 1.0 × 104copies/animal for the indicated period as described previously (Inoue and others 2007). Antiviral treatment was carried out with IFN-β-1b (R&D Systems), mouse IFN-β (R&D Systems), Peginterferon Alfa-2a (Pegasys®), IFN-λ1 (R&D Systems), and IFN-γ (R&D Systems), and ITMN-C (a gift from Dr. S. Seiwert at Intermune, Brisbane, CA) at the indicated dose. Lentiviral particles containing shRNA against each IFIT (Sigma-Aldrich) were used to establish stable cell lines with the infection titer followed by puromycin selection according to the manufacture's instruction. The shRNA sequences used for the establishment of stable cell lines are the following: IFIT1 5′-CTTCGGAGAAAGGCATTAGAT-3′, IFIT2 5′-GCAACCTACTGGCCTATCTAA-3′, and IFIT3/4 5′-GCGATGTACCATCTGGATAAT-3′. Sendai virus (SeV) (Cantell Strain) was purchased from Charles River and used for an in vitro infection at 100 HAU/mL.
Protein, immunoblotting, immunoprecipitation, and microscopic analysis
For immunoblotting analysis, cells were lysed in modified RIPA buffer [10 mMTris (pH 7.5), 150 mM NaCl, 0.5% sodium deoxycholate, and 1% Triton X-100] supplemented with protease inhibitor (PI) mixture (Roche) and phosphatase inhibitor mixture II (Calbiochem). Immunoblots were performed as described (Ooi and others 2014). For microscopic analysis, chamber slides were first fixed with 4% paraformaldehyde followed by permeabilization with 1% Triton X-100 solution. Then slides were blocked with 10% normal goat serum, sequentially stained with the appropriate dilutions of primary and secondary antibodies followed by application of Vectashield mounting medium (Vector Laboratories). Slides were then sealed under coverslips and examined using a Zeiss Laser Scanning Confocal Microscope. Antibodies used in this study are the following: HCV NS5A 9E10 (kindly provided by C. Rice), IFIT1 (a gift from G. Sen), eIF2α (Gale and others 1999), eIF3 (rabbit polyclonal, a gift from J. Hershey), GAPDH (Abcam; ab9484), Tubulin (Genetex; GTX101279), NS3 (U.S. Biologicals; H1920-22N1), Myc-epitope (Bethyl Laboratories; A190-205A), Flag-epitope (Sigma-Aldrich; F3165), and commercially available human serum obtained from HCV-infected individual (Access Biologicals). Antirabbit and antimouse horseradish peroxidase-conjugated secondary antibodies were obtained from PerkinElmer Life Sciences. Coimmunoprecipitation was performed using FLAG M2-agarose beads (Sigma-Aldrich) as described (Saito and others 2007). For [35S]methionine, metabolic labeling was carried out as described previously (Wang and others 2003). In brief, Huh7 cells were first infected with HCV-JFH-1 at MOI 0.5 FFU/mL for 24 h followed by transfection of each IFIT expression vector. After 24 h of an additional incubation in regular DMEM, the medium was replaced with methionine-free DMEM containing 10% dialyzed fetal bovine serum containing 200 μCi of [35S]methionine and labeled for 30 min. Cell extracts were prepared and subjected to immunoprecipitation followed by autoradiography exposure analysis.
Animal
PXB-mouse® (cDNA-uPA+/+/SCID mice harboring human hepatocytes) was established as described previously (Tateno and others 2015) and was used for the in vivo HCV infection followed by therapeutic IFN injection study.
Bioinformatics
Match function of TRANSFAC was used to scan for possible STAT1 binding sites in the [−5000, 1000] regions around the best supported transcription start sites of the each IFIT with minimized false positives (minFP) options (UCSC genome browser and TRANSFAC® release 2018.3) (Matys and others 2006). Preanalyzed ChipSeq data for the STAT1 occupancy within the [−5000, 1000] regions around the best supported TSS of each IFIT gene family in the cells treated with type II IFN were acquired from the publicly available database (Robertson and others 2007; Consortium 2012; Qiao and others 2013).
Results
Hepatic induction of IFITs in the liver of HCV-infected humanized chimeric mouse
According to previous studies by others, the rapid decline in serum HCV RNA titer upon therapeutic IFN injection, occurring within 48 h from the administration of IFN, has been thought to reflect the robust induction of ISGs in the liver (Neumann and others 1998). Our previous study also found that the therapeutic outcome of IFN-based antiviral therapy is well associated with the degree of intrahepatic induction of ISGs at 24 h after the administration of initial dose (Lau and others 2013). Accordingly, we found that the degree of IFIT1, 2, 3/4, but not IFIT5, induction well correlated with the therapeutic outcomes of IFN-based antiviral therapy such as rapid virological responder, the early virological responder, and nonresponders (Lau and others 2013). It is important to note that the gene expression analysis of the liver tissue reflects the abundance of gene transcripts in both the parenchymal cells of the liver (hepatocyte) and nonparenchymal cells such as Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and cholangiocytes.
Therefore, it is critical to assess the expression of each IFIT in hepatocytes, wherein HCV establishes its viral lifecycle, to better understand the significance of IFITs for the suppression of HCV infection. To this end, we have performed an in vivo HCV infection experiment using a HLCM model system. The in vivo injection of HCV genotype 1a successfully established a stable infection by day 7 postinfection (Fig. 1A). The serum HCV RNA titer showed a substantial decline in response to the single administration of pegylated (PEG)-IFN-α similarly to that of HCV-infected patients (Lau and others 2013) (Fig. 1A). The reduction in the abundance of HCV RNA is preceded in the serum followed by the delayed reduction in the liver tissue (Fig. 1B). We then assessed the expression profile of hepatic IFITs at 24 h after the first dose of IFN using specific primer sets to human IFITs (Fig. 1C). The result demonstrated the significant induction of IFIT1, 2, and 3/4, but not IFIT5 in parallel to the sharp decline of HCV RNA titer in the serum. Of note, the liver tissue of IFN-injected animal well maintained the gene expression abundance of IFITs, especially IFIT2 and 3/4, upon repetitive injection of PEG-IFN during which time the intrahepatic HCV genome abundance steeply declined (Fig. 1B). These results suggest the possibility that IFIT1, 2, and 3/4 indeed participate in the suppression of HCV in the infected hepatocytes. Moreover, our in vitro IFN treatment experiment with freshly isolated human hepatocytes from the liver of HLCM further confirmed the substantial induction of IFIT1, 2, and 3/4, but not IFIT5 in response to a therapeutic dose of IFN (Fig. 1D).

Induction of IFITs in the liver of humanized chimeric mice.
Taken together, these observations suggest that IFIT1, 2, and 3/4 are highly inducible in human hepatocytes and retain a high degree of expression during IFN treatment and may serve as one of the critical ISGs that limit HCV replication in human liver tissue.
Induction of IFITs in hepatocytes-derived cell lines
Our studies of the human liver tissue, as well as the HLCM model system, revealed that the expression of IFIT1, 2, and 3/4 well correlates with the degree of HCV suppression. To further understand the criticalness of each IFIT in the suppression of HCV, in vitro mechanistic investigations are required. As in vitro study of HCV lifecycle is restricted to the use of hepatocyte-derived cell lines, we, therefore, evaluated the expression profiles of each IFIT in 2 distinct human hepatocyte-derived cell lines, Huh7 cell and PH5CH8 cell (Ikeda and others 1998), a type of liver cancer cell line and an immortalized human primary hepatocyte, respectively. Our result demonstrated that IFIT1, 2, and 3/4 are highly inducible in both Huh7 and PH5CH8 cells in response to IFN-α and IFN-β at a comparable degree (Fig. 2A, B). Similar to our results with primary human hepatocytes, type I IFN failed to induce the expression of IFIT5 (Fig. 1C, D).
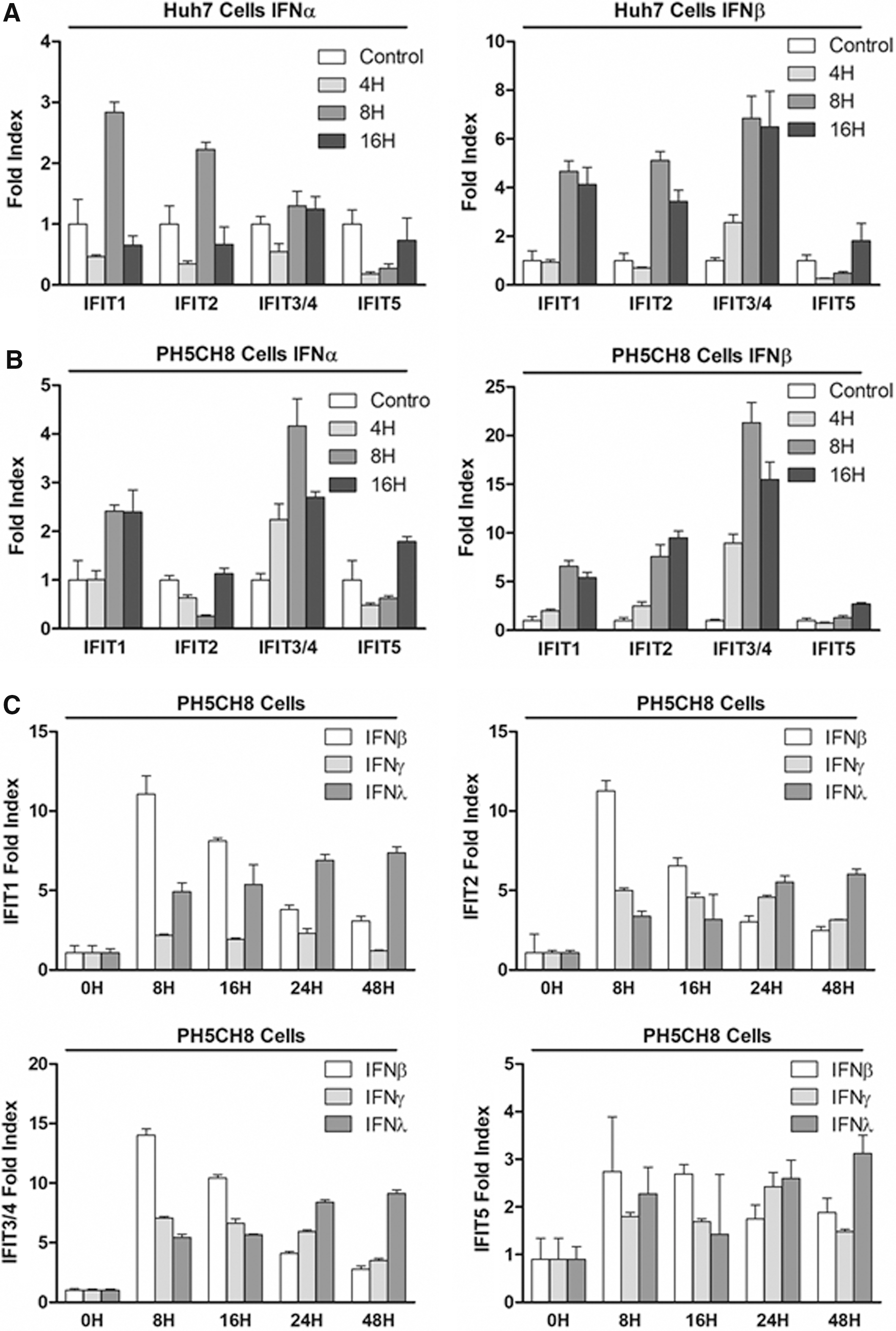
Induction of IFITs in hepatocytes-derived cell lines. Huh7 cells
Of important note, recent studies suggested that type III IFN, also referred to as IFN-λ, exerts a critical role in both the spontaneous clearance during acute infection and determining the therapeutic outcomes of type I IFN treatment (Ge and others 2009; Thomas and others 2009; Prokunina-Olsson and others 2013; McFarland and others 2014; O'Brien and others 2014). Moreover, studies by others also implicated that the contribution of type III IFN in the suppression of HCV is attributed to its regulatory effect on the expression of ISGs (Honda and others 2010). Lastly, the importance of type II, also referred to as IFN-γ, in the suppression of HCV as well as the induction of ISGs, has been implicated (Levy and others 1990; Improta and others 1992; Kokordelis and others 2014). Thus, we extended our investigation to determine the regulatory effect of type II and type III IFN in the induction of each IFIT (Fig. 2C). We found that type III IFNs also induced the expression of IFIT1, 2, and 3/4, but not IFIT5, similarly to that of type I IFN. We also found that types I and III IFN induced IFITs at distinct kinetics. The induction of IFITs by type III IFNs showed a steady increase throughout treatment and peaked at a much later time point than that of type I.
Moreover, we discovered that IFIT2 and 3, but not IFIT1 and 5, are significantly induced upon type II IFN treatment. In summary, we found that each class of IFN exhibits a distinct pattern and potency in the induction of each IFIT in hepatocytes. In addition, our result suggests a possibility that each class of IFN participates in the suppression of HCV partially through the induction of IFITs.
Induction of IFITs in response to DAA therapy
Since 2013, therapeutic IFN has been retired from the mainstay of antiviral therapy against HCV due to the introduction of highly potent DAAs. DAAs are anticipated to suppress HCV lifecycle by the specific inhibition of viral proteins, such as nonstructural (NS) 3/4A, 5A, and 5B (Aghemo and De Francesco 2013). Based on this notion, it is plausible that the antiviral potency of IFN-free DAAs combination therapy is independent of the activation of innate antiviral immunity. However, it is important to note that HCV-NS proteins have been shown to impair the ISGs-mediated antiviral innate immunity (Saito and Gale 2008; Bang and others 2016). Especially, the innate immune evasion by HCV-NS3/4A proteins indeed serves as one of the mechanisms for the establishment of persistent infection (Foy and others 2003, 2005; Saito and Gale 2008; Bang and others 2016). The serine protease activity of NS3/4A abolishes the RIG-I signaling-mediated induction of ISGs by cleaving mitochondrial antiviral signaling protein (MAVS) (Horner and others 2012). Thus, we hypothesized that the DAA-mediated inhibition of NS3/4A might result in the induction of ISGs by restoring the RIG-I-MAVS signaling cascade.
To test this hypothesis, we treated Huh7 cells and Huh7.5 cells harboring HCV JFH-1 SGR with ITMN-C, an HCV NS3/4A PI. The PI efficiently promotes the loss of HCV protein abundance in Huh7 cells and, to a much lesser extent, in Huh7.5 cells (Fig. 3A). Huh7.5 cells, a cell clone derived from Huh7 cells, are known to be defective in the RIG-I pathway as it endogenously expresses the dominant negative mutant of RIG-I. Therefore, Huh7.5 cell is unable to induce the expression of ISGs regardless of the presence of the RIG-I ligand such as HCV genome (Sumpter and others 2004; Saito and others 2007, 2008). Accordingly, our result revealed that the PI treatment only induced the expression of IFIT1 protein in Huh7 cells, but not in Huh7.5 cells (Fig. 3A). Similarly, a previous observation by others also reported that NS3 PI, but not NS5B polymerase inhibitor treatment, results in the induction of ISGs through the restoration of the RIG-I signaling (Liang and others 2008). This observation prompted us to further examine the significance of innate immune signaling restoration by DAA. To this end, we performed an RT-qPCR assay of PI-treated Huh7 SGR cells to evaluate the degree of each IFIT induction (Fig. 3B). The PI inhibitor efficiently induced the expression of all IFITs, especially, IFIT1, 2, and 3/4. More importantly, the degree of each IFIT induction by PI is comparable with that by types I, II, and III IFNs (Fig. 2).
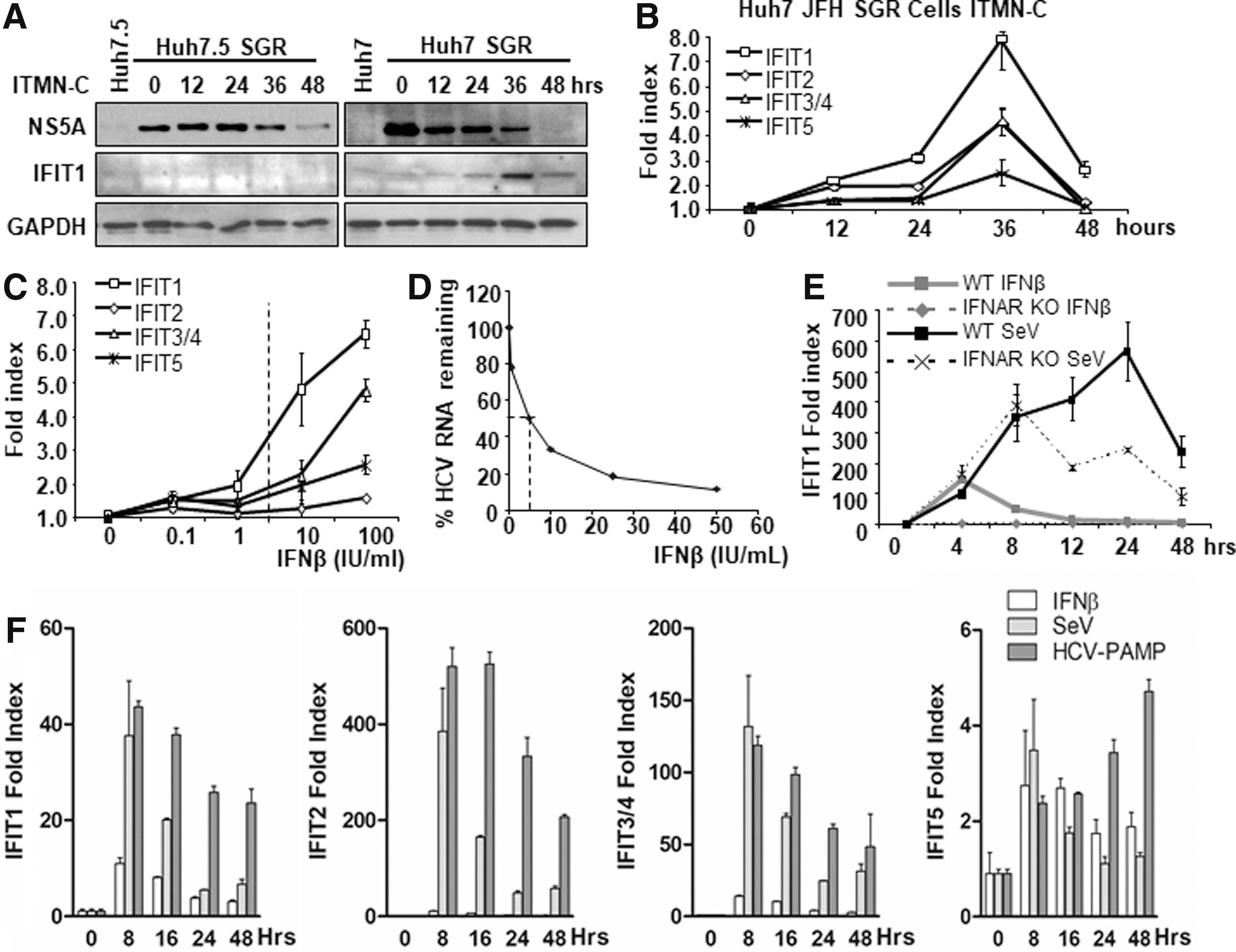
Induction of IFITs in response to RIG-I signaling activation.
Furthermore, the fold induction of each IFIT by the PI treatment appears to be equivalent to that of type I IFN treatment (100 IU/mL), which is much greater than the IC50 dose (Fig. 3C, D). These results indicate the significance of induction of IFITs by the PI-mediated restoration of RIG-I signaling. Regarding the mechanism by which the restored RIG-I signaling induces the expression of IFITs, it is plausible that 2 distinct pathways are involved: (1) direct induction by the activated IRF3/7(Grandvaux and others 2002) and (2) indirect induction by the Jak-STAT pathway activation through secreted IFNs.
To specifically assess the potency of RIG-I signaling in the induction of each IFIT, we employed MEF extracted from wild-type and type I IFN receptor-deficient mice, IFNAR1(−/−). This experiment allows us to exclude the contribution of secreted IFN as a result of RIG-I signaling activation that facilitates the second round of induction of ISGs. The challenge with SeV, which is known to specifically induce RIG-I signaling (Loo and others 2008), induced IFIT1 much greater than that induced by the therapeutic dose of type I IFN treatment (Fig. 3E). We also observed the significant difference in the inducibility of IFIT1 between WT and IFNAR KO cells upon SeV infection. This result indicates that the secreted IFNs play an important role in the expression of IFITs especially at the later time point. This observation suggests that the PI-treated HCV-infected hepatocytes may induce IFITs through the activation of both RIG-I and IFN-Jak-STAT pathways. Thus, we assessed the degree of type I IFN induction in the PI-treated Huh7 SGR cells (Supplementary Fig. S1B). The result demonstrates that the restoration of RIG-I signaling is capable to induce the expression of type I IFN in both IFN-β-promoter luciferase reporter assay and RT-qPCR assessment of IFN-β abundance. Next, we tested the significance of IFNs secreted as a result of RIG-I signaling restoration using naive Huh7 cells incubated with the conditioned medium of PI-treated Huh7 SGR cells. The result demonstrated neither the signatures of type I/III IFN-Jack STAT signaling activation nor the induction of IFITs through an assay with luciferase reporter regulated by IFIT1 promoter or RT-qPCR of IFITs (data not shown). These data suggest that the PI-treated Huh7 SGR cells induce IFITs predominantly in an RIG-I signaling-dependent manner. Of important note, our Huh-7 cells-based observations do not preclude the possibility that endogenous IFN plays a role in the secondary induction of IFITs as it has been shown that primary human hepatocytes are capable of producing a significant quantity of types I and III IFNs in response to a variety of stimulus (Thomas and others 2012).
In summary, our result suggests that the activation of RIG-I-MAVS signaling serves as the predominant induction pathway of IFITs in hepatocytes, at least in Huh7 cells. Based on this notion, we assessed the significance of RIG-I signaling in the induction of IFITs in hepatocytes using Huh7 cells. We found that both SeV infection and HCV PAMP RNA transfection potently induced IFITs, predominantly IFIT2, and, to a much lesser extent, IFIT1 and 3/4 (Fig. 3F).
IFITs are the potent anti HCV effectors
Based on the correlation between the clinical outcome and the induction pattern of IFITs in the hepatocytes, we hypothesized that IFITs could be one of the critical antiviral effectors in the suppression of HCV. To examine this hypothesis, we overexpressed each IFIT or a defective mutant of IFIT1 lacking c-terminus end of the protein in Huh7.5 cells followed by HCV (JFH-1) infection (Fig. 4A). The result showed that the expression of IFITs and the presence of HCV are mutually exclusive, indicating that each IFIT indeed serves as potent antiviral host factors that suppress HCV replication in hepatocytes. In contrast, the cells overexpressing the mutant IFIT1, which abolishes the interaction with the p48 subunit of eIF3 (Wang and others 2003), failed to exclude HCV replication.

IFITs are the potent anti-HCV effectors:
Next, we experimented to ensure the antiviral potency of each IFIT with a loss of function approach. We established Huh7 cell lines that stably expressed shRNA against each IFIT (Supplementary Fig. S1C). Upon confirming the adequate silencing of each IFIT, these cell lines were infected with HCV followed by treatment with either IFN or PI (Fig. 4B, C). We found that the silencing of each IFIT appears to significantly increase the susceptibility to HCV infection compared with its control cells. Moreover, the cell lines lacking individual IFITs exhibited the enhanced resistance to the anti-HCV treatment, further suggesting that IFITs indeed function as antiviral effector molecules in the context of both IFN- and DAA-based antiviral therapies.
IFITs exert its antiviral properties through the inhibition of viral protein translation
Our results in Fig. 4A showed that the mutant IFIT1, which lacks the capacity to interact with eIF3, is not equipped with the antiviral properties against HCV. This notion is also supported by our previous study showing that IFIT1 inhibits HCV viral protein translation through the interaction with eIF3 p48 (also known as eIF3E) subunit (Wang and others 2003). This interaction disturbs the formation of 43 S preinitiation complex (PIC), which consists of eIF3, eIF2, GTP, and Met-Trna (Sokabe and Fraser 2014). Thus, it has been thought that the instability of the PIC caused by IFIT1 results in the inhibition of protein translation. To date, however, it has not been fully understood whether other IFITs also similarly exhibit their anti-HCV property.
First, we assessed how each IFIT impacts both Cap- and IRES-dependent protein translation using a bicistronic expression construct encoding the 5′ cap-dependent Renilla and IRES-dependent firefly luciferase expression reporter construct (Fig. 5A). The ectopic expression of each IFIT globally suppressed both Cap- and IRES-dependent protein translation for both EMCV and HCV IRES. Moreover, we noted that IFIT1 and 3/4 significantly suppressed HCV IRES-dependent translation over Cap-dependent translation.
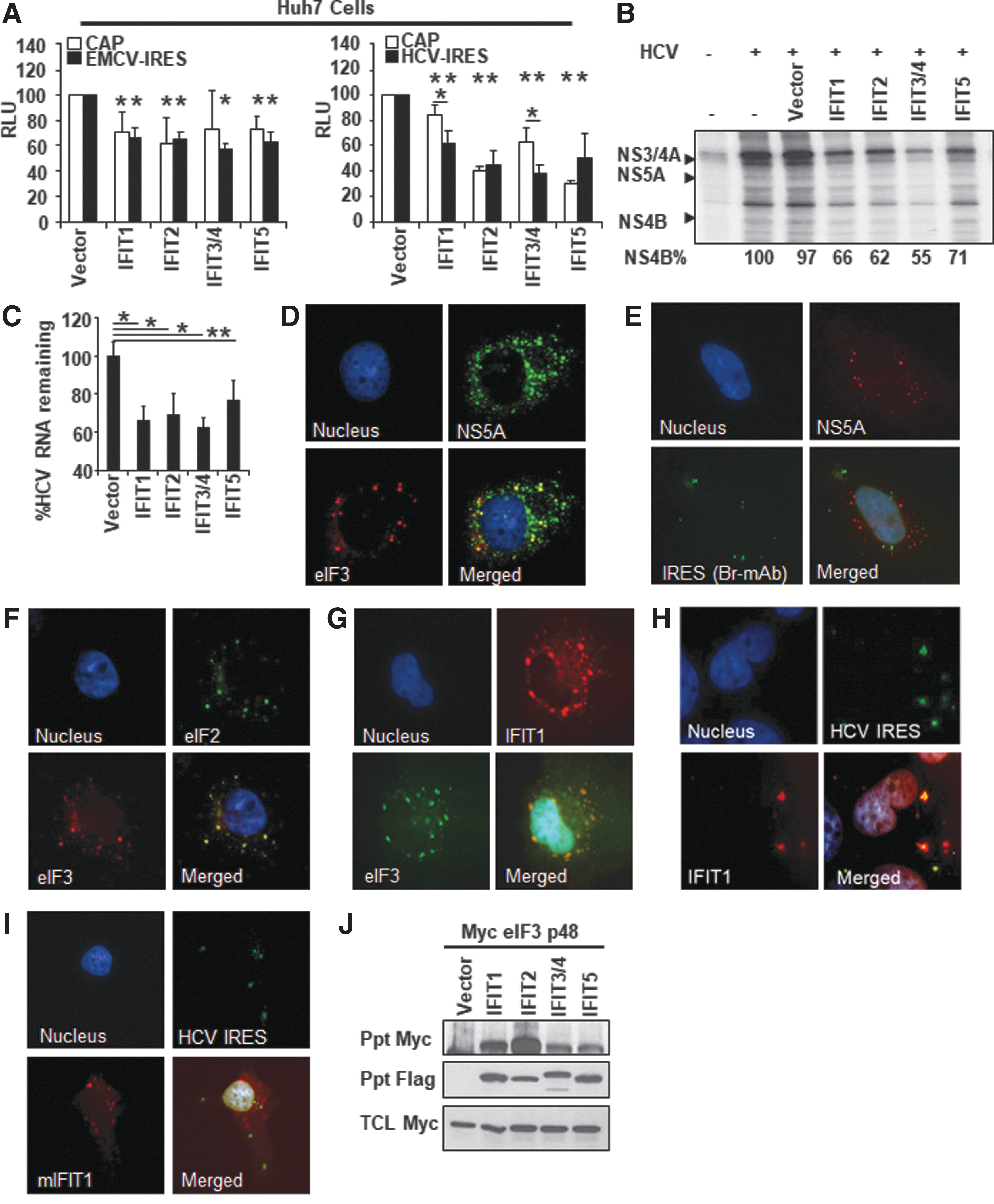
IFITs exert its antiviral properties through the inhibition of viral protein translation.
These observations suggest that IFITs may suppress HCV at the stage of viral protein translation. Next, we tested the potency of each IFIT in the suppression of viral protein translation with S35-methionine metabolic labeling assay. The result revealed that overexpression of each IFIT potently suppressed HCV protein translation (Fig. 5B). In addition to the inhibition of HCV polyprotein translation, the overexpression of IFITs efficiently inhibited the HCV genome replication (Fig. 5C). These results suggest that all IFITs inhibit the HCV lifecycle at the stage of viral protein translation followed by the suppression of genome replication given the reciprocal relationship between these 2 steps.
To further delineate the anti-HCV mechanism of IFITs, we conducted a series of immunofluorescence microscopic analyses using Huh7 HCV SGR cells. First, we found that eIF3, as well as RNA encoding HCV-IRES, does not largely colocalize with HCV NS5A protein, which serves as a marker for the HCV replication complex (Fig. 5D, E). Also, we found that eIF3 colocalizes with eIF2 as well as IFIT1 (Fig. 5F, G). Furthermore, we confirmed that wild-type, but not mutant IFIT1, colocalizes with RNA encoding HCV-IRES (Fig. 5H, I). These results indicate that IFITs, at least IFIT1, exhibit the inhibitory effect on HCV replication through disturbance of PIC. Lastly, we tested whether other IFITs also interact with eIF3 p48 through an immunoprecipitation assay. The result revealed that IFIT2, 3/4, and 5 also bound to the p48 subunit of eIF3 similarly to that of IFIT1 (Fig. 5J). We have also tested whether p110 subunit of eIF3 interacts with IFITs with an immunoprecipitation assay and found no interaction between these 2 molecules (data not shown). Taken together, these data collectively suggest that the antiviral effect of these IFITs may also require the interaction with the p48 subunit of eIF3.
Although these results suggest that IFITs inhibit HCV lifecycle at the stage of viral protein translation, our experiment results do not sufficiently exclude the possibilities that other mechanisms contribute to the suppression of HCV. In particular, emerging evidence indicated that IFITs sequestrate viral genome from the protein translation machinery and perhaps genome replication (Abbas and others 2013). Given the reciprocal relationship between the viral protein translation and genome replication, we further assessed whether IFITs specifically inhibit the viral protein translation. To this end, we analyzed the efficiency of HCV-IRES-dependent translation in Huh7 cells using JFH-1 SGR genome encoding a replication-defective mutant (GND) (Supplementary Fig. S1D, E). The result suggested that IFITs do not have a direct inhibitory effect on the HCV-IRES-dependent translation; however, expression of IFIT1 and 2 significantly reduced the abundance of HCV genome. In contrast, the expression of IFIT3/4 and IFIT5 had no effect on the HCV genome abundance. These results collectively suggest the potential possibility that the interaction between viral genome and IFITs, especially IFIT1 and 2, may promote the decay of viral RNA, resulting in the suppression of viral replication. Lastly, our results indicate that IFIT3/4 and 5 do not have a direct impact on the viral protein translation as well as viral genome stability.
Discussion
Recently, the standard antiviral therapy against HCV has transitioned from a combination of PEG-IFN-α and Ribavirin to IFN-free DAA combination regimens. The DAA combination therapy is highly effective, and nearly all treated patients attain a sustained virologic response. Thus, furthering our understanding of IFN biology, including the IFN-inducible antiviral effector genes, has lost its relevance in the context of antiviral therapy.
However, in contrast, the incidence of acute HCV infection, at least in the United States, has been exponentially growing due to the high opioid endemic and associated injection drug use (Liang and Ward 2018). It is well known that more than half of acute HCV infection transitions to the lifelong persistent infection. Owing to the insidious nature of the disease, the vast majority of the affected population are either unaware of the infection or do not seek medical attention until they develop end-stage liver diseases.
Therefore, an establishment of a definitive prevention strategy, such as a vaccine, plays a central role to mitigate the disease burden of HCV infection. To this end, it is extremely important to further our understanding of the immune event that determines the clinical outcomes, whether the patient achieves spontaneous resolution or transitions to the chronicity. A recent genome-wide association study comparing the elite and poor controller of acute HCV infection redirected our attention to IFN biology once again even in the era of DAA as a recent seminal study implicated that the efficient activation of endogenous IFN system plays a central role in the spontaneous clearance of HCV (Thomas and others 2009). Moreover, the findings of other studies collectively indicate that the efficient activation of the endogenous IFN system including the robust induction of ISGs serves as a prerequisite for the potent induction of adaptive immunity, which is required for the resolution of the infection (Lauer and Kim 2006; Honda and others 2010; Raghuraman and others 2012; Prokunina-Olsson and others 2013). Taken together, it remains extremely important to advance our understanding of the IFN biology and its antiviral effector molecules, ISGs for the better management of HCV infection.
IFN exhibits its antiviral properties through the induction of ISGs. However, it remains still unclear how exactly >300 ISGs corporately suppress the viral lifecycle in infected cells. In addition, only a little is known about the expression profiles of each ISG in individual cell types, their antiviral potencies, and the mechanism of action to individual viral pathogens. A recent work demonstrated that the antiviral potency of each ISG is highly variable depending on the viral pathogen (Schoggins and others 2011). Thus, the antiviral potency of each ISG needs to be carefully characterized in a pathogen-specific manner using the relevant cell types. With regard to the mechanistic aspect of each ISG, to date, only a few ISGs have been well characterized. These include RNA-dependent protein kinase, adenosine deaminase acting on RNA 1, Viperin, OAS/RNase L, ISG20, and IFITs. With regard to the anti-HCV effect of IFITs, only IFIT1 has been studied in the past (Wang and others 2003; Sumpter and others 2004; Raychoudhuri and others 2011).
It has been demonstrated that the IFN-mediated decline of HCV titers occurs in a biphasic manner, which divides into 2 phases: 0–48 h and after 48 h from the administration of IFN (phases 1 and 2, respectively) (Neumann and others 1998; Lanford and others 2007). It has been proposed that phase 1 serves to exert its antiviral effects by blocking the production of de novo viral particle and phase 2 to potentiate the death of infected cells. Of note, the rapid decline in the viral load during phase 1 is thought to be mediated through the induction of ISGs. Moreover, the degree of viral load decline during the first phase tightly correlates with clinical outcome, suggesting that the ISGs highly expressed during the phase I IFN response likely to be the critical effectors that define the therapeutic outcome. Our previous study with the human liver tissue of a HCV-infected patient harvested at 24 h after the first dose of IFN injection indicated that the induction of not only IFIT1 but also IFIT2 and 3/4 well correlates with the therapeutic outcomes (Lau and others 2013).
In this study, our study with HCV-infected HLCM and human primary hepatocytes discovered that IFIT1, 2, and 3/4, but not IFIT5, are highly induced upon IFN treatment. A series of in vitro experiments with hepatoma cell line also confirmed that IFIT5 less likely plays a role in anti-HCV defense program of the liver. With regard to the type of stimulus for the induction of each IFIT, we found that the pattern of induction is highly distinctive among different types of IFNs such as delayed induction kinetics by type III IFN as well as the selective induction of IFIT2 and 3/4 by type II IFN. Moreover, we also found the robust induction of IFIT1, 2, and 3/4 by the activation of RIG-I signaling as compared with that of IFN-Jak-STAT signaling activation. With regard to the anti-HCV potency of each IFIT, both our gain and loss of function approaches collectively indicated that IFIT2 and 3/4 possess the most inhibitory effect on HCV replication and all the rest showed a comparable degree of HCV suppression. These observations together indicate that all IFITs except IFIT5 certainly play an important role in the suppression of HCV in the infected hepatocytes. However, our study did not sufficiently delineate the mechanism by which type II IFN selectively induce IFIT2 and 3/4. In addition, the exact mechanism of how IFITs inhibit HCV replication remains unclear.
To seek the potential explanation for the selective induction of IFITs by type II IFN, we performed a series of bioinformatic searches using the match algorithm of TRANSFAC and also preanalyzed ChipSeq database. Type II IFN signaling triggers the phosphorylation of STAT1 followed by the formation of STAT1-homodimer, namely gamma interferon activation factor (GAF). Then, the GAF translocates into the nucleus and induces gene transcription through binding to a defined DNA sequence called gamma activated sequence. Thus, our bioinformatics search sought the occurrence of consensus STAT1 binding motif within the hypothetical promoter region of each member IFIT gene family. In addition, we also assessed the peak identified in the ChIPSeq data of type II IFN-treated cells within the same promoter region. Although the search result revealed a significantly high frequency of STAT1 binding site within the hypothetical promoter region of each IFIT as compared with that of randomly selected genes, there was no significant difference between each IFIT (Supplementary Table S1). Similarly, the analysis of ChipSeq data did not demonstrate significant association with the inducibility of each IFIT gene. Thus, our bioinformatics analysis did not uncover the explanation on how type II IFN exerts the selective induction of IFITs. This might be attributed to the cell-type-based differential response to type II IFN or the discrepancy between the bioinformatics algorithms and the actual STAT1 occupancy. Alternatively, the selective induction of IFITs by type II IFN might be mediated by the activation of noncanonical response pathways (Green and others 2017). Thus, further investigations are required to understand better the mechanism by which type II IFN exhibits selective induction of some IFITs.
Regarding the anti-HCV effect of IFITs, our study result suggests that IFITs–eIF3 p48 subunit interaction serves, at least in part, as the mechanism that inhibits the HCV lifecycle by inhibiting the translation of the viral protein. However, of great interest, we found that the degree of HCV suppression does not well correlate with the affinity between IFITs and p48 subunit, indicating that the destabilization of PIC may not be a sole mechanism for the antiviral activity of IFITs. In fact, recent studies by others showed that IFIT proteins recognize and bind to 5′ppp RNA that lacks 2′-O methylation such as HCV genome. This event prevents PIC to interact with viral RNA, thereby inhibiting the translation of viral protein (Daffis and others 2010; Zust and others 2011). Another potential antiviral mechanism of IFIT1 is to sequestrate the uncapped 5′ppp RNA such as HCV genome from the replicase complex (Pichlmair and others 2011). Indeed, our study with HCV-SGR genome containing replication-defective mutation (GND) indicated that the association between IFITs and viral genome might be a predominant mechanism for the suppression of HCV at least for IFIT1 and IFIT2. The mechanism of how IFIT3/4 inhibits HCV remains largely unclear based on our study result. One potential explanation is that IFIT3/4 inhibits viral infection through the augmentation of expression of ISGs (Liu and others 2011). Thus, further investigations are required to define the antiviral mechanism by which IFITs exert anti-HCV properties as a sole effector or in combination.
In summary, this study investigated the criticalness of IFITs in anti-HCV innate immune defense program of the liver. Our result collectively suggested that IFITs, except IFIT5, possess potent anti-HCV properties and thus are likely to be involved in determining the clinical outcomes in response to the antiviral therapy as well as during the acute infection.
Footnotes
Acknowledgments
We thank Dr. Chunfu Wang for the technical assistance. This work was supported by funds from NIH NIAAA (R21AA022751: T.S.), NIH NIAID (R21AI139954: T.S.), NIH NIDDK (RO1DK101773: T.S.), NIH NIAID (R01AI127463: M.G.), NIH NIAID (R01AI118916: M.G.), and research funding from PhoenixBio (T.S.).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1