
Abstract
Since its discovery in 2013, interferon lambda 4 (IFN-λ4) has received a reputation as a paradoxical type III IFN. Difficulties in detecting IFN-λ4, especially in secreted form even led to questions about its existence. However, the genetic ability to generate IFN-λ4, determined by the presence of the rs368234815-ΔG allele, is the strongest predictor of impaired clearance of hepatitis C virus (HCV) infection in humans. Significant modulation of IFN-λ4 activity by a genetic variant (P70S) supports IFN-λ4, and not other type III IFNs encoded in the same genomic locus, as the primary functional cause of the association with HCV clearance. Although the ability to produce IFN-λ4 is associated with decreased HCV clearance, the recombinant IFN-λ4 is active against HCV and other viruses. These observations present an apparent conundrum—when and how does a presumably good IFN, with anti-HCV activity, interfere with the ability to clear HCV? In this review, we discuss findings that suggest potential mechanisms for explaining this conundrum.
Introduction
The family of class-2

IFN-λ4 as a member of the class-2 cytokine family. Protein sequences were downloaded from the UniProt Knowledgebase and structures from the RSCB Protein Data Bank. Computational tools were used to predict structure of IFN-λ4, for which crystal structure is not yet available. Multiple sequence alignments were generated, and phylogenetic reconstruction was carried out using the Markov Chain Monte Carlo Bayesian evolutionary analysis by sampling trees (MCMC BEAST, v1.7.4). The phylogenetic tree was graphed with FigTree v1.3.1. Clustering is based on protein sequence similarity, with the length of connecting lines representing the relative evolutionary distance from a common ancestral gene. IFN-λ4, interferon lambda 4.
Since the discovery of the genetic association with spontaneous and treatment-induced clearance of hepatitis C virus (HCV) infection (Suppiah and others 2009; Tanaka and others 2009), and the eventual identification of IFN-λ4 as a functional cause of this genetic association (Prokunina-Olsson and others 2013), efforts of ours and many other groups have focused on understanding the function of IFN-λ4. We now know that IFN-λ4 is a poorly secreted IFN with strong antiviral activity, including against HCV, Sendai, dengue, coronavirus and West Nile virus (Hamming and others 2013; Hong and others 2016; Obajemu and others 2017). In addition to inducing a potent antiviral response, IFN-λ4 overexpressed in cell lines also shows faster antiviral kinetics and stronger induction of negative regulators of the IFN response than other type III IFNs (Fan and others 2016; Obajemu and others 2017).
Despite these advances, several questions remain to be answered, such as when and why a type III IFN, which is generally antiviral and, specifically, with anti-HCV activity, impairs HCV clearance. Also, why is this effect restricted to HCV (so far) despite type III IFNs being recognized as important players in many other viral infections. In addition, the antiviral potency of IFN-λ4 remains debated, given its transient expression, significant intracellular retention, and poor detection of the secreted protein (Hamming and others 2013; Prokunina-Olsson and others 2013; Hong and others 2016; Obajemu and others 2017). All these questions define the IFN-λ4 conundrum that is discussed in this review.
IFN-λ4 Is a Poorly Secreted but Highly Potent Type III IFN
Analysis of hepatic expression in HCV patients with IFNL4 genotype (carriers of the rs368234815-ΔG allele) shows higher expression of IFN-stimulated genes (ISGs) and lower HCV levels (Uccellini and others 2012; Lu and others 2015), suggesting some level of viral control provided by IFN-λ4, but insufficient for complete viral clearance. Using stable inducible hepatoma cell lines, we showed that the concentration of secreted IFN-λ4 is 20-fold lower than that of IFN-λ3, yet IFN-λ4 is twice as potent (Obajemu and others 2017). These results differ from what was reported by other studies that used transient overexpression (Hong and others 2016), or treatment with recombinant protein (Hamming and others 2013). The continuous release of endogenously produced and properly folded IFN-λ4 might be essential for the observed difference in its activity between studies. Furthermore, in our experimental system, IFN-λ4 acted faster, reaching peak activity within 8 h, whereas the activity of IFN-λ3 was slower and more sustained. Both results suggest that, compared with other IFNs, the effect of IFN-λ4 may be paracrine, with only cells immediately adjacent to virally infected cells being fully exposed to the effect of the secreted IFN-λ4.
Despite some technical issues with IFN-λ4 protein detection, it is now clear that IFN-λ4 is produced and secreted during viral infections, but at relatively low levels (Obajemu and others 2017; Minas and others 2018). So, how is it possible that the poorly secreted IFN-λ4 induces high ISG expression? With functional data demonstrating the enhanced antiviral activity of IFN-λ4 compared with IFN-λ1–3, one assumption would be that the distinct sequence of IFN-λ4 affects its interaction with the IFN-λ receptors (Mendoza and others 2017). However, owing to the poor expression and limited solubility of IFN-λ4, analysis of its interactions with receptors remains challenging. Another possibility is that the transient expression of IFN-λ4 somehow leads to epigenetic changes that enhance ISG induction. For example, changes in histone methylation patterns have been shown to modify ISG induction in response to IFN-α (Fang and others 2012).
The activity of secreted IFN-λ4 protein may also be enhanced if it is associated with the plasma membrane, continuously and locally activating neighboring cells, as has been described for IL-15 (Bergamaschi and others 2008). IFN-λ4 undergoes N-glycosylation that does not appear to affect its function but seems to be required for optimal secretion (Hamming and others 2013). However, for some unknown reason, a large proportion of IFN-λ4 remains intracellular. It is unclear whether intracellular IFN-λ4 plays any role in enhancing ISG induction, although our studies suggest that ISG activation requires IFN-λ4 secretion (Obajemu and others 2017).
IFNL4 genotype has been associated with increased ISG induction in peripheral blood mononuclear cells (PBMCs) of HCV-infected patients (Honegger and others 2016; Price and others 2016). It is unlikely that this effect is caused by IFN-λ4 secreted from HCV-infected hepatocytes, which opens a possibility that IFN-λ4 might be produced from PBMCs through the action of virally associated particles on antigen presenting cells or a direct effect of HCV replication in PBMCs (Gong and others 2003; Stone and others 2013).
A further potential consequence of the poor secretion and localized effect of IFN-λ4 is destabilization of the immune response by shifting it from a more adaptive to a more innate response, thus compromising viral clearance (Honegger and others 2016; Price and others 2016). It was reported that HCV-infected pregnant women with IFNL4 genotype had lower T cell responses to HCV but increased ISG signatures (Honegger and others 2016; Price and others 2016). Strong induction of localized innate immune responses by IFN-λ4 without having a broader effect on the adaptive immune system could compromise the development of effective adaptive immunity, possibly by dysregulating cytokine production. As an example, we observed high levels of IP-10 secretion after IFN-λ4 overexpression in primary human hepatocytes (Onabajo and others 2015), and high plasma levels of IP-10 have been associated with chronic infection and autoimmune disease (Antonelli and others 2004; Oliver and others 2010). It will be interesting to determine whether IFN-λ4-induced expression of IP-10 has similar consequences for the development of adaptive immunity.
Interestingly, in a Sendai virus infection model in primary human hepatocytes, we were unable to demonstrate increased ISG induction in individuals with versus without the IFNL4 genotype, in contrast with our observations in stable inducible cell lines engineered to express IFN-λ4 (Obajemu and others 2017). However, we observed a kinetic difference, with IFN-λ4–expressing hepatocytes showing faster ISG induction than hepatocytes from individuals genetically unable to produce IFN-λ4 (Obajemu and others 2017). However, the sample sizes were small, and further studies using larger number of donors are necessary, as well as using other viral infection models.
IFN-λ4 Expression Increases Negative Regulation of the IFN Response
It is well established that the IFN response, however beneficial for controlling viral infections, is also quite detrimental to tissues (Arimoto and others 2018). Indeed, most of the tissue damage from viral infections involves the effectors of the innate antiviral response in the host. As a result, the IFN response is subject to multiple layers of negative regulation that serve to limit tissue damage among other events (Arimoto and others 2018). In HCV, ubiquitin-specific peptidase (USP18) has emerged as a common negative regulator with possible consequences of its differential expression between individuals with versus without IFNL4 genotype (Randall and others 2006; Francois-Newton and others 2011; Fan and others 2016; Obajemu and others 2017). USP18 is a specific peptidase for ISG15 that cleaves ISG15 from target proteins, in the ISGylation process (Malakhova and others 2006). USP18 also binds the IFNAR2 subunit and blocks its interaction with JAK1, effectively dampening the activity of IFN-α (Malakhova and others 2006) (Fig. 2). Indeed, in HCV-infected cell lines, USP18 overexpression has been shown to blunt the antiviral effect of IFN-α, and to a lesser extent—type III IFNs (Randall and others 2006; Francois-Newton and others 2011; Fan and others 2016; Blumer and others 2017). Recently, we showed that IFN-λ4 treatment of primary human hepatocytes or stable expression of IFN-λ4 in HepG2 cells led to increased USP18 expression compared with cells similarly treated with IFN-λ3 (Obajemu and others 2017). Another report showed that treatment of HCV-infected cells with IFN-λ4 inhibited antiviral activity of IFN-α through increased induction of USP18 and ISG15, suggesting that IFN-λ4 expression could blunt the antiviral activity of IFN-α (Fan and others 2016). Whether this occurs in the context of actual HCV infection is unclear.
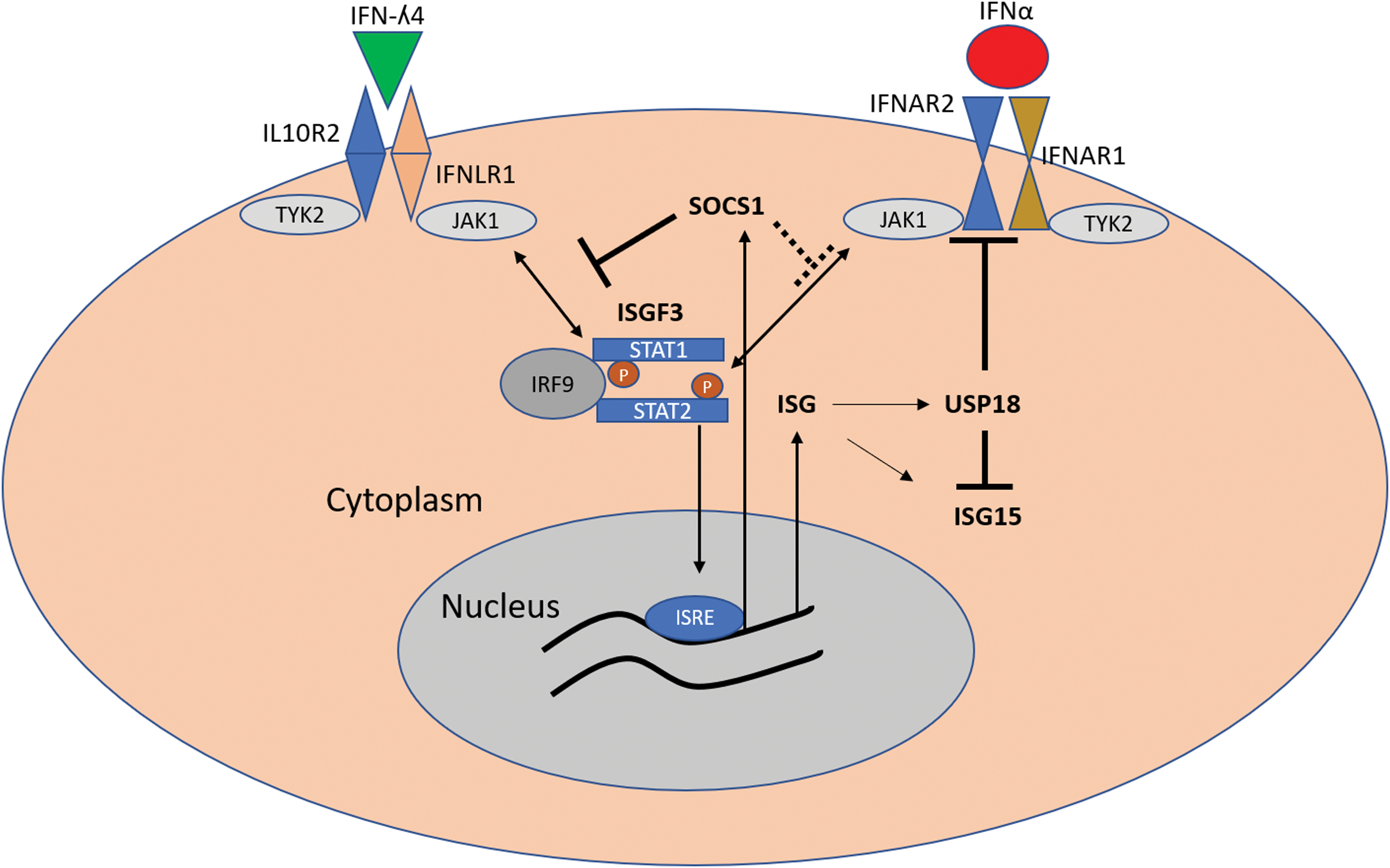
Proposed mechanisms of negative regulation of IFN responses by IFN-λ4. Binding of IFN-λ4 to its receptors IFNLR1 and IL10R2 leads to phosphorylation and dimerization of STAT1 and STAT2, recruitment of IRF9, and formation of ISGF3 complex. ISGF3 complex activates ISRE, leading to expression of multiple ISGs, including USP18 and ISG15. SOCS1 expression is also induced by IFN-λ4, although the role of the ISGF3 complex in SOCS1 induction is unclear. SOCS1 inhibits the formation of ISGF3 complex by inhibiting STAT1 phosphorylation by type III IFNs but to a lesser extent than by IFN-α. In contrast, USP18 inhibits binding of JAK1 to IFNAR2, thus reducing the effectiveness of IFN-α in activating the JAK/STAT pathway. USP18 also targets ISG15 for ubiquitination, thus inhibiting the process of ISGlyation. IL10R2, interleukin 10 receptor 2; ISGs, IFN-stimulated genes; ISRE, interferon-sensitive response element; SOCS1, suppressor of cytokine signaling. Color images are available online.
We also showed that IFN-λ4 induces high levels of suppressor of cytokine signaling (SOCS1) expression, which was shown to be important for the regulation of types I and III IFNs (Blumer and others 2017). SOCS1 belongs to a class of proteins that includes 7 members (SOCS1–7) that act as direct negative regulators of STATs by inhibiting STAT phosphorylation (Starr and others 1997; Song and Shuai 1998; Yoshikawa and others 2001) (Fig. 2), thus blunting the activity of several cytokines, including IFNs. Higher levels of SOCS3 are associated with the development of chronic HCV infection and poor response to therapy (Persico and others 2007; Kim and others 2009; Miyaaki and others 2009). It is possible that high induction of SOCS1 by IFN-λ4 could contribute to the development of chronic HCV infection and resistance to therapy as well (El-Saadany and others 2013; Liu and others 2015). Interestingly there is some dichotomy in the regulation of type I and type III IFNs by both USP18 and SOCS1 (Blumer and others 2017). Conditional SOCS1 knockout mice are much more sensitive to type III IFNs, whereas USP18 knockout mice showed more sensitivity to IFN-α, suggesting that, although both pathways converge on the same JAK-STAT signaling pathway, their negative regulation is different (Blumer and others 2017).
The coordination of type I and type III IFN responses during HCV is not fully understood. HCV appears to have evolved to compromise type I but not type III IFN signaling (Chandra and others 2014), emphasizing the importance of type I IFN signaling in controlling HCV. Interestingly, type III IFNs, including IFN-λ4, appear to have significant anti-HCV activity in vitro (Hamming and others 2013), and recombinant IFN-λ1 has been effective in clinical trials for HCV (Muir and others 2010), suggesting that they may still have an important role to play in the HCV response. Understanding the role of type III IFNs in HCV control may provide additional insight into how IFN-λ4 is associated with negative outcomes of HCV infection.
Concluding Remarks
We are only just beginning to unravel the consequences of IFN-λ4 expression in normal and disease conditions. In part, the challenges have been technical due to the absence of sensitive detection assays that could capture low and transient IFN-λ4 expression. The IFN-λ4 conundrum stems from the significantly impaired viral clearance in individuals who carry the rs368234815-ΔG allele and thus can produce this IFN. Current literature suggests that, mechanistically, this conundrum can be partly explained by increased negative regulation of IFN responses, consequently compromising the antiviral response. These observations, along with stronger antiviral activity observed for IFN-λ4 than for IFN-λ3 (Obajemu and others 2017), were reported using in vitro overexpression systems and thus may not accurately represent biological mechanisms. Functional in vivo studies will be needed to better understand the role of IFN-λ4 in the anti-HCV and other responses.
It is also important to improve our understanding of the IFN-λ4 biology, and analysis of structural and functional differences between IFN-λ4 and other members of the type III IFN family could provide relevant mechanistic insights. The resolving of the IFN-λ4 conundrum will require extensive multidisciplinary work but offers the promise of a better understanding of the genetically determined regulation of the innate and adaptive immune response to HCV and potentially other diseases. The specific impact of IFN-λ4 could also be therapeutically and clinically actionable and impacting human health overall.
Footnotes
Acknowledgments
Members of the Prokunina-Olsson group in the Laboratory of Translational Genomics, National Cancer Institute, are thanked for their comments and discussions. This review is based on the talk presented at the meeting “Interferon Lambda: Disease Impact and Translational Implications” held at the NIH, Bethesda, on October 25–26, 2018. O.O.O. and L.P.-O. are supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health.
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author Disclosure Statement
L.P.-O. is a coinventor on a patent for the IFN-λ4 protein held by the National Cancer Institute.