
Abstract
Natural killer (NK) cells have shown good application prospects in adoptive cellular immunotherapy against cancer. However, due to its insufficient infiltration and low activity, the therapeutic effect of infused NK cells has been limited in solid tumors, such as colorectal cancer. It has been proved that tumor-produced chemokines regulate the migration of NK cells expressing corresponding chemokine receptors, and cytokines could enhance the antitumor activity of NK cells. In this study, we innovatively upregulated the expression of chemokine receptor CXC chemokine receptor 2 (CXCR2) and cytokine interleukin (IL)-2 on NK-92 cells using CRISPR-Cas9 gene-editing technology. We demonstrated that overexpressing CXCR2 and IL-2 promotes NK-92 cells to increasingly transfer into tumor sites and achieve stronger cell-killing and proliferation activity. Moreover, the inhibitory effects of gene-edited NK-92 cells on the growth of human colon cancer in vivo were also improved. The tumor burden of tumor-bearing mice was reduced, and their survival time was significantly prolonged. Gene-editing modification NK cells are expected to become a novel and promising tumor treatment strategy.
Introduction
Adoptive cellular immunotherapy has increasingly become an important means for augmenting classical treatment of cancer (Boyiadzis and others 2018; Laskowski and Rezvani 2020). Natural killer (NK) cells have attracted extensive interest due to their innate immunity. NK cells are not restricted by major histocompatibility complex (MHC) and have the ability to identify and attack a broad range of tumor cells without prior sensitization. Activated NK cells are able to lyse malignant cells through direct cytotoxicity and indirectly kill target cells by secreting cytokines to regulate related immune cells (Fang and others 2017).
So far, the therapeutic efficacy of adoptively transferred NK cells in patients with leukemia and other hematological cancers has been impressive (Chengand others 2013; Gonzalez-Rodriguez and others 2019). However, the insufficient infiltration of infused NK cells to the tumor sites has made it challenging to obtain ideal therapeutic effects in solid tumors (Kremer and others 2017; Reid and others 2020), such as colorectal cancer (CRC). We reasoned that another potential limiting factor is the low activity of NK cells in tumor patients (Peng and others 2013).
Multiple pieces of research have been worked on chemokine/cytokine-activated NK cells. However, there are few studies on chemokine receptors of NK cells. Tumor cells express chemokines in the tumor microenvironment, and NK cells express corresponding chemokine receptors so that they could move directionally along the concentration gradient and gather into the tumor sites (Yao and Matosevic 2021). Upregulating the expression of chemokine receptors on the surface of NK cells to make them better homing to the tumors expressing corresponding ligands could effectively enhance the antitumor effects of NK cells.
The chemokine receptor CXC chemokine receptor 2 (CXCR2) is considered to be one of the most important receptors. Its ligands such as CXCL1 and CXCL8 are readily secreted by a variety of solid tumors, including CRC (Li and others 2004; Lee and others 2008; Yamamoto and others 2019). Kremer and others (2017) discovered that genetic modification of human primary NK cells to highly express CXCR2 improves their ability to specifically migrate along a chemokine gradient, increased killing of target cells, and increased adhesion properties.
NK-92 cell line is an interleukin (IL)-2-dependent NK cell line derived from a patient with rapidly progressive non-Hodgkin's lymphoma. It lacks almost all killer cell inhibitory receptors and highly expresses a series of activation receptors, making it a strong candidate for clinical application in a variety of different cancers. NK-92 is the only cell line that has been infused into patients with advanced cancer with clinical benefit and minimal side effects (Klingemann and others 2016). NK-92 cells grow well in culture and can be markedly activated and propagated by IL-2, showing increased cytotoxic activity, wider tumor-killing spectrum, increased secretion of cytokines, and so on (Wu and others 2017).
Technical strategy commonly used in the past to stimulate NK cells is to add exogenous cytokine IL-2. However, the adverse effects of high-dose IL-2 infusions could be severe, including fever, nausea, vomiting, renal insufficiency, edema, and hypotension (Lundqvist and others 2011; Pol and others 2020), while low-dose IL-2 efficacy is limited by its short half-life in vivo and depletion by other lymphoid cells (Shevach 2000). Another attempt is to use transgenic technology to transfect IL-2 gene into NK-92 cells to express endogenous IL-2 (Nagashima and others 1998). Several strains of IL-2-independent NK-92 have been generated, such as NK-92 MI and NK-92 CI, using retroviral or nonviral gene transduction. Yet the application might be suppressed by MHC molecule restriction, and the transfected gene might be lost after cell subculture.
Therefore, given the limitations of NK immunotherapy mentioned above, we intend to use CRISPR-Cas9, a technology that could precisely locate DNA for gene editing under the guidance of single guide RNA (sgRNA) and directly upregulate the transcription level of endogenous genes combined with VP64 transcription activation domain (Jinekand others 2013), to make NK cells continuously and steadily overexpress chemokine receptor CXCR2 and cytokine IL-2. This project is expected to successfully develop novel efficient antitumor NK cells and provide important data for other similar studies. Genetic modification NK cells could hopefully become a novel and promising tumor immunotherapy strategy and improve the clinical efficacy of NK cells in the treatment of solid tumors.
Materials and Methods
Cell lines
Human NK cells NK-92 were kindly provided by Dr. Zhenghao Piao from Hangzhou Normal University (Hangzhou, China) (purchased from ATCC, Manassas, VA) and were cultured in α Minimum Essential Medium (Gibco, Grand Island, NY) containing 20% fetal bovine serum (FBS) (Gibco) supplemented with 100 U IL-2 and 1% penicillin-streptomycin (Gibco). Human colon cancer cells HT29 and human chronic myeloid leukemia cells K562 were purchased from Shanghai Zhongqiao Xinzhou Biotechnology (Shanghai, China) and were cultured in RPMI 1640 (Gibco) containing 10% FBS (Gibco) supplemented with 1% penicillin-streptomycin (Gibco). Human embryonic kidney cells 293T were previously preserved in our laboratory and were cultured in Dulbecco's Modified Eagle Medium (Gibco) containing 10% FBS (Gibco) supplemented with 1% penicillin-streptomycin (Gibco). All cell lines were cultivated at 37°C and 5% CO2 in a humidified incubator. All procedures carried out in studies involving animals have passed the ethical review of the Experimental Animal Ethic Committee of Hangzhou Normal University (Ethic Number: 2020048).
Methods
CXCR2-IL-2 sgRNA vector plasmid construction
The CXCR2 target gene (GenBank number: NM_001557) and IL-2 target gene (GenBank number: NM_000586) were obtained from NCBI. Activated sgRNA sequences with better effectivity, specificity, and lower off-target effect were selected using the guide RNA design web tool CRISPR-ERA. We delegated the Syngentech (Beijing, China) to synthesize CXCR2-IL-2 sgRNA vector plasmid (we called LW649) based on our designed sgRNA. And we purchased dCas9-VP64 vector plasmid (we called LW415) which contains a transcriptional activation domain VP64 and an inactive Cas9 from Syngentech at the same time.
Lentivirus production
293T cells were used as lentivirus packaging cells. The lentiviral vector plasmids LW649 and LW415 4 μg, respectively, and the packaging plasmids psPAX2 3 μg, pMD2.G 1 μg were transiently cotransfected into 293T cells, using Lipofectamine 3000 (Invitrogen, Carlsbad, CA). After 48 and 96 h, the transfection efficiency was observed under a fluorescence microscope. The culture medium was collected and centrifuged at 4°C 3,000 g for 20 min, and then the supernatant was taken. The virus solution was concentrated with PEG8000 (Macklin, Shanghai, China).
Infection of NK-92 cells
Virus solution 3 mL and virus concentration solution 50 μL were added to infect NK-92 cells. The solution was replaced by complete medium 5 mL after 12–24 h. After 48 and 72 h, the cell infection efficiency was observed under a fluorescence microscope. Polybrene was added to improve the infection efficiency (10 μg/mL). To enhance the positive rate, the transduced NK-92 cells were selected with 2 μg/mL Puromycin (Invitrogen, Karlsruhe, Germany) for 24 h. The NK-92 cells transfected with LW649 and LW415 plasmids (CXCR2-IL-2-NK-92 cells) were the experimental group, and the untreated wild-type NK-92 cells were the control group.
Quantitative real-time reverse transcription-polymerase chain reaction
The mRNA of NK-92 cells was extracted from whole NK-92 cell lysates using RNAiso Plus (Takara, Japan). And a reverse transcription reaction was conducted using PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan) to synthesize cDNA. The polymerase chain reaction was carried out with specific primers CXCR2 (PF-5′-GTACAGTGCTATTCTGCCTAGA-3′, PR-5′-TTAAATCCTGACTGGGTCGC-3′), IL-2 (PF-5′-TACAAGAACCCGAAACTGACTCG-3′, PR-5′-ACATGAAGGTAGTCTCACTGCC-3′), and GADPH (PF-5′-GAAGGTCGGTGTGAACGGAT-3′, PR5′-TTCCCATTCTCGGCCTTGAC-3′). GADPH was used as endogenous control. All primers were designed and synthesized by Sangon Biotech (Shanghai, China). After normalization against GADPH, the relative expression levels of CXCR2 and IL-2 mRNA were calculated by 2−ΔΔ Ct method.
Enzyme-linked immunosorbent assay
Change to IL-2-free medium to continue culturing CXCR2-IL-2-NK-92 cells and wild type NK-92 cells. The medium volume was fixed to 5 mL, containing 1 × 107 cells. After being incubated for 24 h, the supernatants of each group were collected for detection of secreted IL-2 using human IL-2 ELISA Kit (Multi Sciences, Hangzhou, China). Blank culture medium group (IL-2-free) was set up as negative control.
Transwell assay
Transwell assay was used to compare the chemotactic capacity of NK-92 cells. CXCR2-IL-2-NK-92 cells and wild type NK-92 cells (5 × 105), respectively, were starved for 48 h and added in 100 μL serum-free medium to the upper chambers of a 24-well transwell plate (8 μm pore size; Corning Costar, Corning, NY). Conditioned serum-free medium (supernatants of HT29 cells incubated for 24 h) was collected and placed in the lower chambers with 50 ng/mL CXCL1. To verify the specificity of NK cell chemotaxis, NK cells were preincubated with the selective nonpeptide CXCR2 inhibitor SB225002 (1 μM; Selleck) for 1 h at 37°C. After being incubated at 37°C for 24 h, the number of NK-92 cells that migrated through the porous membrane was evaluated by counting the visible cells on the membrane with 3 mirror images randomly selected for each group under an optical microscope ( × 40; Olympus, Tokyo, Japan).
Detection of cell-killing activity
K562 cells were used as target cells (T, 2 × 104/mL). CXCR2-IL-2-NK-92 cells and wild type NK-92 cells were cultured as effector cells (E), adjusting cell density according to different effector-target ratios (25: 1, 50: 1, 100: 1). The cell-killing activity of NK-92 cells was detected by Cell Counting Kit 8 (CCK8) (Boster, Pleasanton, CA). Each experimental group (E+T) contained E (100 μL) and T (100 μL). Three control groups: E, T, and blank medium were set up simultaneously. After incubation for 24 h, 20 μL CCK8 was added to each well and incubated for another 2 h. Then analyze the OD value at A450 nm. NK-92 killing rate (%) = [1−(AE+T−AE)/AT] × 100%, “A” represents OD value of detection.
Detection of cell proliferation ability
CCK8 assay was also used to test the cell proliferation ability of NK-92 cells. The cell density of CXCR2-IL-2-NK-92 cells (E) and wild type NK-92 cells (C) was adjusted to 1 × 105/mL. All cells were seeded in a 96-well plate with 100 μL medium in each well. Blank medium group (B) was set up simultaneously. After incubation for 6, 12, 24, and 48 h, 10 μL of CCK8 was added to each well and incubated for another 2 h. Then analyze the OD value at A450 nm. NK-92 proliferation activity (%) = (AE–AB)/(AC–AB) × 100%, “A” represents OD value of detection.
Effects of CXCR2-IL-2-NK-92 cells on human colon cancer
In vivo xenograft tumor models
Male severe combined immunodeficient (SCID) mice aged 4–6 weeks were purchased from GemPharmatech (Jiangsu, China). All mice were held under standardized pathogen-free conditions with aseptic food and water provided. To establish human colon cancer models, 200 μL phosphate-buffered saline (PBS) containing 2 × 106 HT29 cells was subcutaneously injected into the left side below armpit of each mouse. These HT29 cells could express red fluorescent protein after using a Red Fluorescent Protein Kit. After tumor formation, the tumor size of each mouse was measured twice a week. Tumors were measured in 2 dimensions: the longest diameter L and the transverse diameter W. Tumor volume was calculated according to the formula (V = 0.52 × L × W2). All procedures carried out in studies involving animals comply with the ethical standards of the institution conducting the study. Once the tumor ruptured or mice suffered great pain, they will be euthanized.
Antitumor effects of NK-92 cells in vivo
When the tumor diameter reached about 5 mm, the mice were randomly divided into 3 groups: experimental group (CXCR2-IL-2-NK-92 cell group), control group (wild type NK-92 cell group), and negative control group (PBS group) (n = 8 per group). Each mouse in the experimental group and the control group was injected with 100 μL PBS containing 4 × 106 CXCR2-IL-2-NK-92 cells and wild type NK-92 cells through the tail vein, respectively, and the negative control group was injected with 100 μL blank PBS. The injection was repeated every 48 h over a period of 2 weeks. The tumor volume was measured, and the survival time of tumor-bearing mice was observed.
To track the distribution of NK cells in vivo, a Green Fluorescent Protein Kit was used to label wild type NK-92 cells in advance, so that both CXCR2-IL-2-NK-92 cells and wild type NK-92 cells could express green fluorescence. Then the fluorescence signals were detected and imaged by the IVIS spectrum imaging system (Carestream, Toronto, Canada) to evaluate the chemotaxis and proliferation of NK-92 cells in vivo.
Immunohistochemistry
The xenograft tumors that expired from tumor-bearing mice were subjected to immunohistochemical staining to evaluate the number of tumor-infiltrating NK-92 cells and CXCR2 expression level. For immunohistochemistry (IHC), tumor specimens were fixed in 4% paraformaldehyde for 24 h. Paraffin-embedded tumor sample sections were cut at 3 μm and then deparaffinized. Endogenous peroxidase was inactivated by incubating the sections in 3% hydrogen peroxide for 10 min at room temperature. Antigen retrieval was performed by heating the slides in sodium citrate buffer. The slides were then subjected to incubation with primary antibodies (anti-CD56, ZM-0057; ZSGB-BIO, Beijing, China; anti-CXCR2, 1:50, ab14935; Abcam, Cambridge, UK) at 4°C overnight in a moist chamber and labeled with a secondary antibody conjugated with horseradish peroxidase (Elabscience, Wuhan, China). The labeled slides were incubated with DAB to visualize proteins. Negative controls used all reagents except the primary antibodies.
Statistical analyses
Each experiment was performed in triplicate. All experimental data in this study were presented as means ± standard deviations. Student's t-test was performed to compare the data between 2 groups after confirming the normal distribution. One-way analysis of variance and Univariate Analysis of Variance were applied for multiple group comparisons. Survival analysis was performed using a log-rank test. P < 0.05 was considered statistically significant. All data were analyzed using SPSS 21.0 statistical software (SPSS, Chicago, IL).
Results
Generation of NK-92 cells simultaneously upregulating chemokine receptor CXCR2 and cytokine IL-2
We designed the CXCR2 and IL-2 sgRNA sequences as described below: CXCR2-sgRNA-F-5′GCGAGTTCAGACAAGTCCGT3′, CXCR2-sgRNA-R-5′ACGGACTTGTCTGAACTCGC3′, IL-2-sgRNA-F-5′GTGGGCTAATGTAACAAAGA 3′, and IL-2-sgRNA-R-5′TCTTTGTTACATTAGCCCAC3′. Then we synthesized the CXCR2-IL-2 sgRNA vector plasmid (we called LW649) and dCas9-VP64 vector plasmid (we called LW415). The structures of plasmid LW649 and LW415 are depicted in Fig. 1a. Sequencing confirmed that the CXCR2-IL-2 sgRNA vector plasmid was successfully constructed. The sequencing results are described in Supplementary Figure S1.
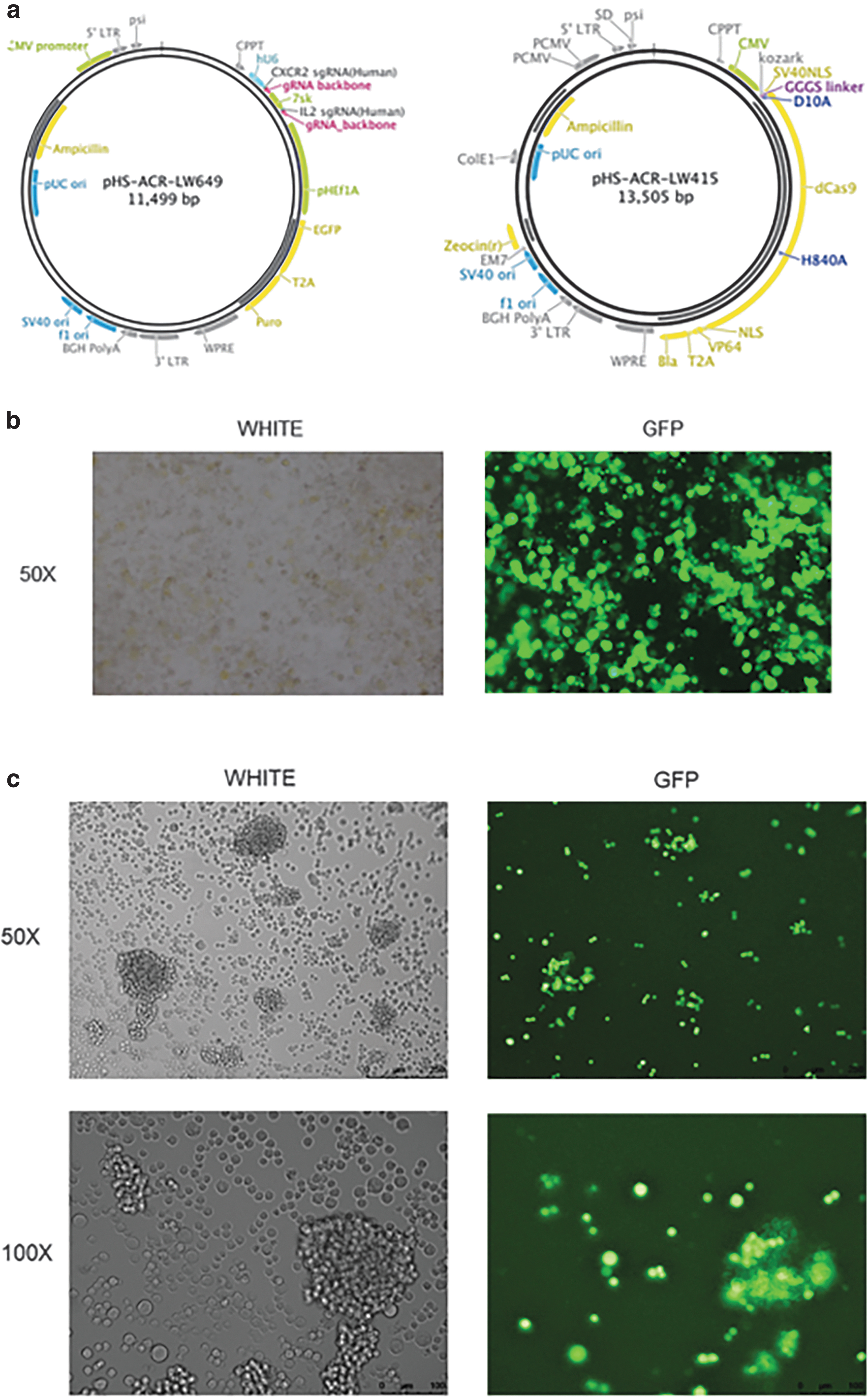
The construction of NK-92 cells simultaneously upregulating chemokine receptor CXCR2 and cytokine IL-2 (CXCR2-IL-2-NK-92 cells).
Then LW649 and LW415 were transiently cotransfected into lentivirus packaging cell line 293T cells. The transfection efficiency was gratifying; the green fluorescence expression rate could reach over 90% as is shown in Fig. 1b. The virus solution was collected and used to infect NK-92 cells. After repeated selection of puromycin, the positive rate could reach 80%–90% when observed under a green fluorescence microscope. The infection efficiency of NK-92 cells is shown in Fig. 1c.
After the gene-edited NK-92 cells were generated, we performed quantitative real-time reverse transcription-polymerase chain reaction and enzyme-linked immunosorbent assay (ELISA) to verify the expression of CXCR2 and IL-2. It was confirmed that NK-92 cells transfected with CXCR2-IL-2 sgRNA plasmid and dCas9-VP64 plasmid successfully activated the upregulation of CXCR2 and IL-2 genes (Fig. 2a, P < 0.05). And it was successfully expressed at the protein level. Higher concentration of IL-2 was detected in the culture supernatant of CXCR2-IL-2-NK-92 cells (Fig. 2b, P < 0.01).
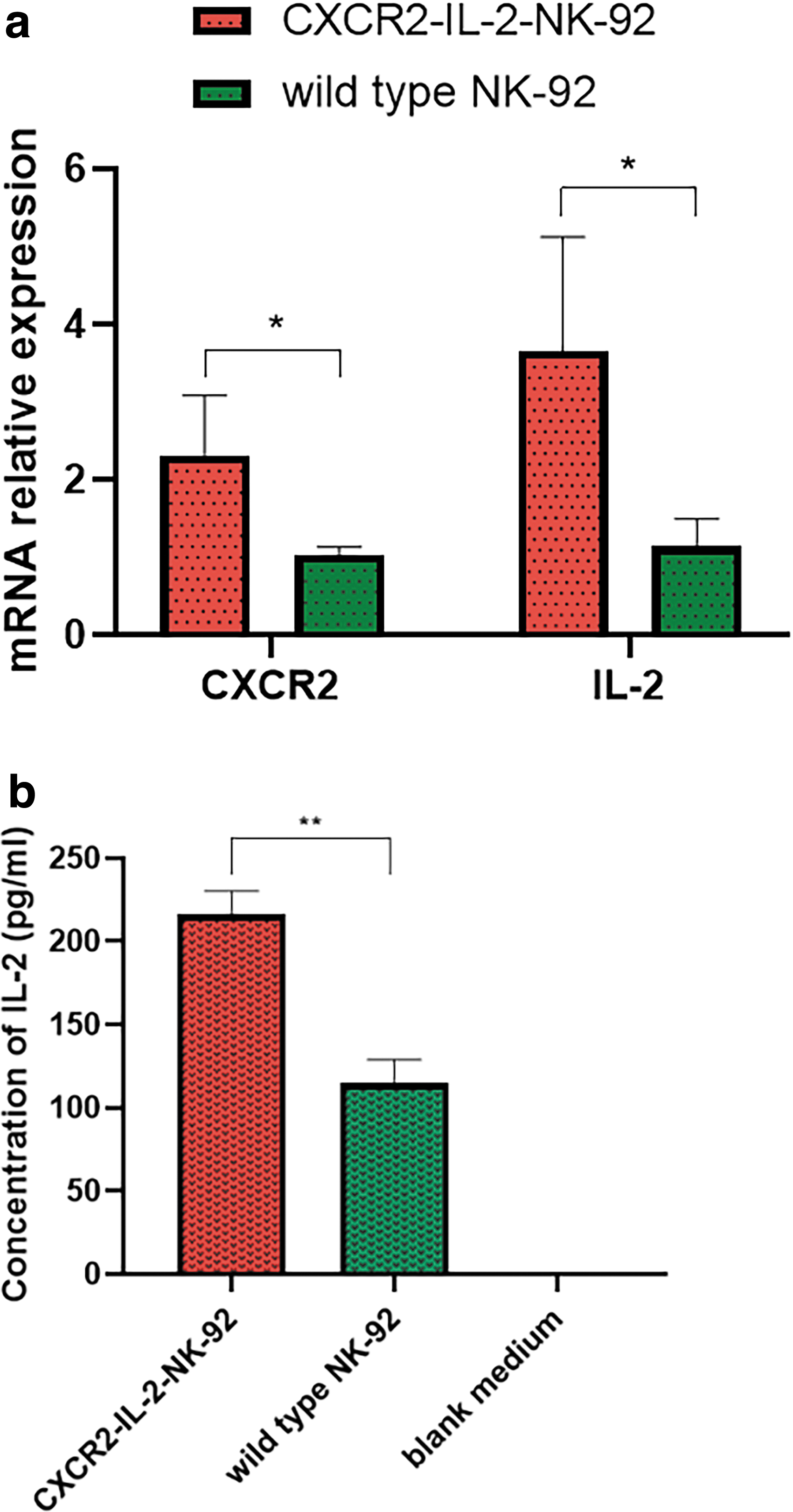
We verified the generation of gene-edited NK-92 cells (CXCR2-IL-2-NK-92 cells).
The CXCR2-IL-2-NK-92 cells enhanced chemotactic capacity toward human colon cancer cells in vitro
We performed a Transwell assay to test the chemotactic capacity of CXCR2-IL-2-NK-92 cells in vitro and used human colon cancer cell line HT29 cells as our target cells. Comparing to wild type NK-92 cells, we observed an increased migration capacity toward CXCL1/GRO-α conditioned medium from HT29 cells of CXCR2-IL-2-NK-92 cells. And we proved the specificity of chemotaxis, using the CXCR2 inhibitor SB225002. The difference is statistically significant (Fig. 3a, P < 0.001).
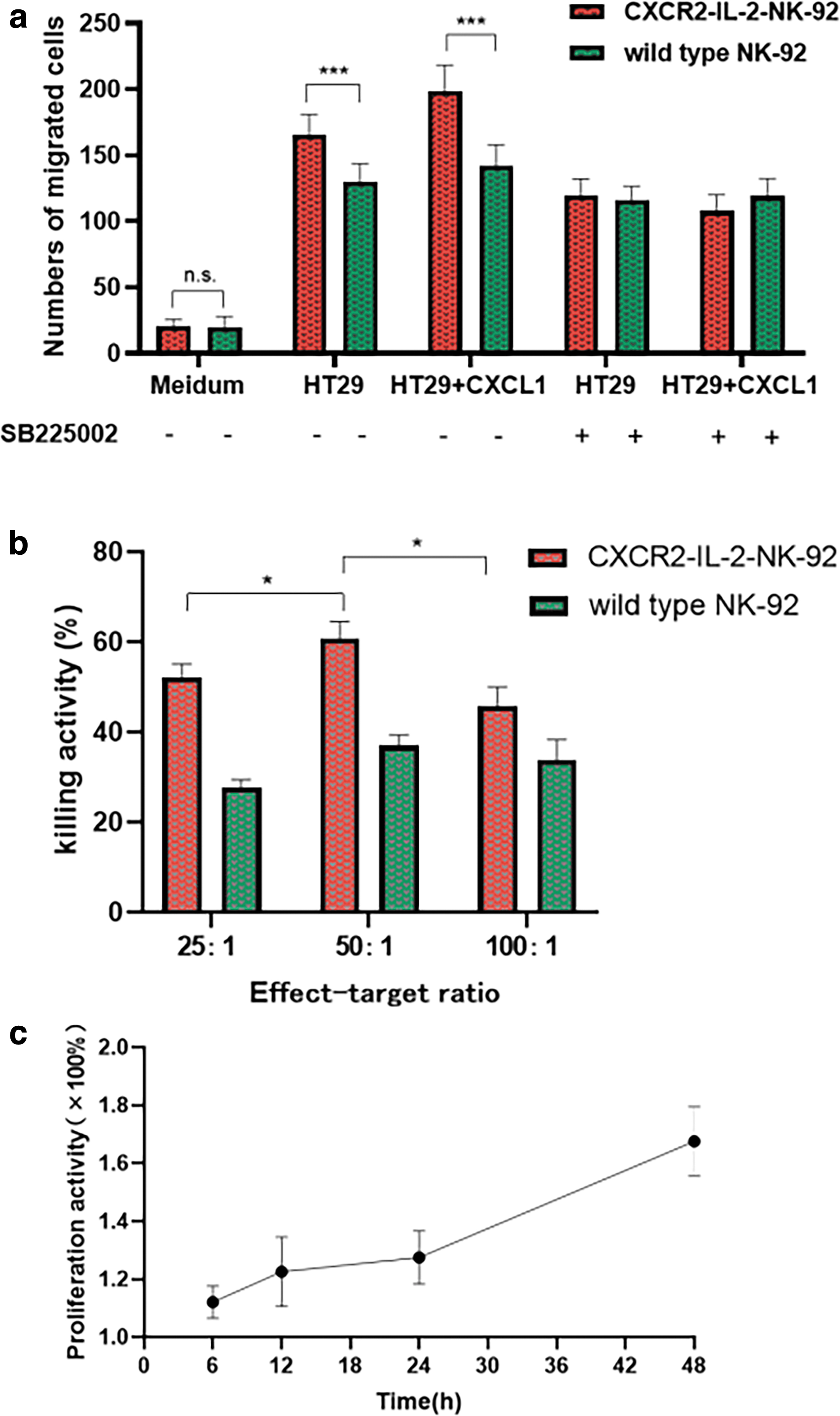
The CXCR2-IL-2-NK-92 cells enhanced cell-killing activity and proliferation ability
We performed 3 effect-target ratios from 25:1, 50:1 to 100:1. It turned out that under the same ratio, CXCR2-IL-2-NK-92 cells displayed a stronger killing activity on K562 tumor target cells compared with wild type NK-92 cells (P < 0.001). And NK-92 cells achieved the best killing activity against K562 cells at an effect-target ratio of 50:1 (Fig. 3b, P < 0.05).
We also used CCK8 assay to test the proliferation ability of NK-92 cells. It can be clearly seen in Fig. 3c that the proliferation ability of CXCR2-IL-2-NK-92 cells was enhanced compared with the control group, since all values were greater than 1. And it gradually increased with incubation time, most amplified at 48 h, about 1.68 times that of the control group.
Adoptively transferred CXCR2-IL-2-NK-92 cells increased infiltration and proliferation in vivo, thus reduced tumor burden and prolonged survival of tumor-bearing SCID mice
Since we have proved the increased chemotactic capacity and cell-killing activity of CXCR2-IL-2-NK-92 cells in vitro, we furthermore investigated whether we could get the same result in vivo. We used SCID mice to establish human colon cancer models. SCID mice were injected with HT29 cells that carried red fluorescent protein. When the tumor diameter reached about 5 mm, CXCR2-IL-2-NK-92 cells and wild type NK-92 cells carrying green fluorescent protein (GFP) were injected through the tail vein. The IVIS spectrum imaging system showed that infiltration of CXCR2-IL-2-NK-92 cells into tumor sites in SCID mice was significantly increased (Fig. 4a) compared with wild type NK-92 cells. The same result was obtained in xenograft tumors by immunohistochemistry using anti-CD56 as the primary antibody (Fig. 5d). Moreover, the expression of CXCR2 on the surface of CXCR2-IL-2-NK-92 cells was stronger compared with the control group, located in the cell membrane and cytoplasm.

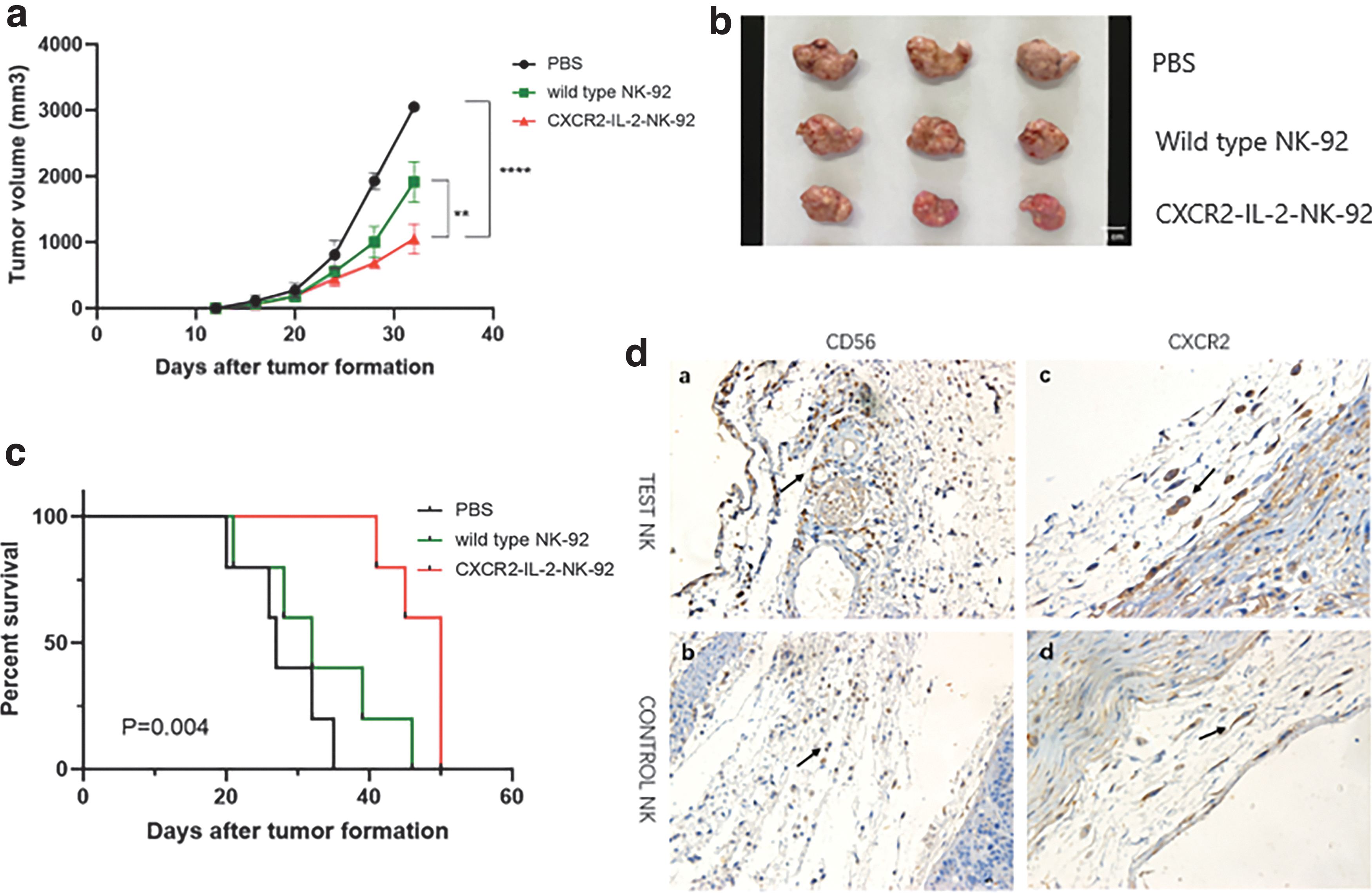
Antitumor effects of adoptively transferred CXCR2-IL-2-NK-92 cells on human colon cancer.
To assess the proliferation ability, we compared the fluorescence of CXCR2-IL-2-NK-92 cells and wild type NK-92 cells at 2 and 4 weeks, respectively. And it showed that CXCR2-IL-2-NK-92 cells were able to achieve self-proliferation in vivo over time, while wild type NK-92 cells began apoptosis (Fig. 4b). Meanwhile, the tumor size of each mouse was measured twice a week to evaluate the antitumor effects of NK-92 cells. And we found that the progress of tumor volume of mice treated with CXCR2-IL-2-NK-92 cells was significantly postponed compared with the wild type NK-92 group and the PBS group (Fig. 5a, b, P < 0.01, P < 0.0001), and the survival time of mice in CXCR2-IL-2-NK-92 group was significantly prolonged (Fig. 5c, P < 0.01). CXCR2-IL-2-NK-92 cells enhanced the inhibitory effects on the growth of tumors. Taken together, our data indicated an improved antitumoral performance of adoptively transferred CXCR2-IL-2-NK-92 cells against human colon cancer.
Discussion
Cellular immunotherapy using donor NK cells is an emerging field that has achieved some remarkable responses and has shown a good safety profile. It could avoid the potentially fatal “cytokine storm” and eliminate the risk of inducing graft-versus-host disease, which usually occurs in many T cell-based immunotherapies (Jamieson and others 2004; Shah and others 2017). Multiple studies have proved the feasibility of adoptive immunotherapy with NK cells (Shah and others 2017; Vela and others 2018), but how to improve its clinical efficacy in the treatment of solid tumors is the problem we wish to solve.
Compared with autologous NK cells, healthy allogeneic NK cells displayed better antitumor effects. The consumption of immune cells is increased in tumor patients. The number of effector cells that can be transformed into NK cells is inadequate, leads to a decrease of NK cells, with its function impaired to varying degrees by hypoxia tumor microenvironment (Sakamoto and others 2015). Besides, only a small part of NK cells can enter the microenvironment of solid tumors through capillaries. Therefore, we believe that making a sufficient number of activated NK cells directly to the tumor sites is also a key step to optimize NK immunotherapy.
In the early stages of immune response, NK cells are activated and recruited to the tissue where they function. This process depends on chemokines and chemokine receptors (Haidl and others 2020). NK cells express a variety of chemokine receptors, which interact with corresponding chemokines to regulate the migration, recruitment, and other biological functions of NK cells. CXCL12 (SDF-1)/CXCR4 axis and CCL21/CCR7 axis are currently the most widely studied chemotaxis models for redirecting NK cells (Müller and others 2015; Zhou and others 2019). Müller and others (2015) genetically modified YTS cells with an EGFRvIII-CAR to overexpress CXCR4. These NK cells conferred a specific chemotaxis to CXCL12/SDF-1α secreting U87-MG glioblastoma cells.
IL-2 has been considered to be an essential factor for NK cells and an enhancer of their expansion and cytotoxic potential (Jounaidi and others 2017; Mortara and others 2018). Campbell K and others described that IL-2 in NK-92 cell helps drive gene expression and interaction to enhance the killing against tumor cells (Pazina and others 2019). Numerous attempts have been made to strengthen the antitumor effects mediated by IL-2. Boissel L and others have developed a variant of NK-92 called hank, which comprises an ER trapped IL-2 as selectable marker, expressing high affinity CD16 allele, so as to give better play to the role of antibody-dependent cell-mediated cytotoxicity (Jochems and others 2016). In this study, we innovatively upregulated chemokine receptor CXCR2 and cytokine IL-2 of NK-92 cells simultaneously through CRISPR-Cas9.
CRISPR-Cas9 system relies on guide RNAs to conduct site-specific DNA cleavages facilitated by the Cas endonuclease. Before CRISPR-Cas9, DNA editing was mainly carried out by Zinc-finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) technology. However, the protein motif involved in this specific recognition of DNA sequence is tedious and time consuming. CRISPR-Cas9 has incomparable maneuverability of ZFN and TALEN. Therefore, it is widely used in DNA editing of various species and has been well applied in genetic diseases, infectious diseases, and other research fields (Mali and others 2013; Liao and others 2017). CRISPR-Cas9 can be used to initiate the transcriptional activation of endogenous genes in the off state and is easy to achieve compared with classical exogenous plasmid gene transduction methods.
We transfected NK-92 cells with pLV-hU6-CXCR2 sgRNA(Human)-gRNA_backbone-7SK-IL-2 sgRNA(Human)-gRNA_backbone-hef1a-EGFP-T2A-puro plasmid (pHS-ACR-LW649) and pLV-CMV-NLS-dCas9-NLS-VP64-T2A-bla plasmid (pHS-ACR-LW415), using lentivirus transfection. CXCR2-IL-2-sgRNA could guide the transcriptional activation domain dCas9-VP64 located upstream of CXCR2 and IL-2 gene in nuclear DNA to achieve the upregulation of target chemokine receptor and cytokine. After repeated selection, the positive rate of transduced NK cells can reach 80%–90%, and the related genomic information can be stably transmitted to the offspring cells, so that the overexpression of CXCR2 and IL-2 could be constant and lasting.
CXCL1 (GRO α) is a selective agonist of CXCR2, which is highly expressed in colon cancer cells (Li and others 2004). We performed Transwell assay and discovered that CXCR2-IL-2-NK-92 cells were able to migrate more toward human colon cancer HT29 cells. Moreover, after increasing the content of CXCL1 in the lower chamber, the number of CXCR2-IL-2-NK-92 cells migrating downwards increased again, and this migration can be inhibited by CXCR2 inhibitor. Therefore, we have reason to believe that CXCL1/CXCR2 axis could promote the recruitment of NK cells into tumors. Upregulation of CXCR2 expression could enhance this chemotaxis.
In vitro cytotoxicity and proliferation of NK-92 cells were detected by CCK8 assay. CXCR2-IL-2-NK-92 cells indeed showed better cytotoxicity against tumor cells and achieved sustainable proliferation. The upregulated expression of IL-2 can act on itself and surrounding NK cells continuously through an autocrine or paracrine pathway, allowing for endogenous production of sufficient IL-2 to maintain proliferation, thus realizing continued growth of the cell (Treisman and others 1995).
We also proved that in vivo using SCID tumor-xenografted mice. CXCR2-IL-2-NK-92 cells showed increased infiltration into tumor sites and self-proliferation. We noted a considerable delay of tumor growth in a sizable number of mice treated with CXCR2-IL-2-NK-92 cells compared to control groups. CXCR2-IL-2-NK-92 cells could effectively alleviate tumor progression and extend the survival time of tumor-bearing SCID mice. Some mice still showed a good survival status at the end of the observation period.
In conclusion, our research successfully developed a novel efficient type of NK cells with better tumor chemotaxis, sustainable proliferation, and stronger antitumor effects using CRISPR-Cas9 gene-editing technology. Modification of chemokine receptors and cytokines on NK cells by gene editing may be another powerful tool for adoptive cellular immunotherapy. The prospect of tumor immunotherapy is promising.
Conclusion
The CRISPR-Cas9 gene-editing technology can make NK cells constantly upregulate the expression of specific chemokine receptors and cytokines. Overexpressing CXCR2 and IL-2 promotes NK-92 cells to increasingly transfer into tumor sites, and its antitumor effects have been improved. Gene-editing modification NK cells are expected to become a novel and promising tumor treatment strategy.
Ethical Statement
All procedures performed in studies involving animals were following the ethical standards of the institution conducting the study. This research does not contain any study involving human participants performed by any of the authors.
Consent for Publication
All authors agree to submit the article for publication.
Footnotes
Authors' Contributions
L.G.: Formal analysis, Methodology, Investigation, Writing—original draft. L.Y.: Formal analysis, Investigation, Writing—review and editing. S.Z., M.S., and Z.G.: Formal analysis and Methodology. Y.S. and X.W.: Methodology and Validation. C.H.: Conceptualization, Supervision, Funding acquisition, Project administration, Writing—review and editing.
Acknowledgments
The authors thank Dr. Zhenghao Piao from Hangzhou Normal University for his assistance in providing NK-92 cells and NK-92 cell culture technology. The authors thank all the teachers of translational medicine platform of Affiliated Hospital of Hangzhou Normal University for providing experimental space and technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Major Science and Technology Project of Zhejiang Province, China (Project number: 2017C03053), funded by Zhejiang Provincial Department of Science and Technology.
Supplementary Material
Supplementary Figure S1