
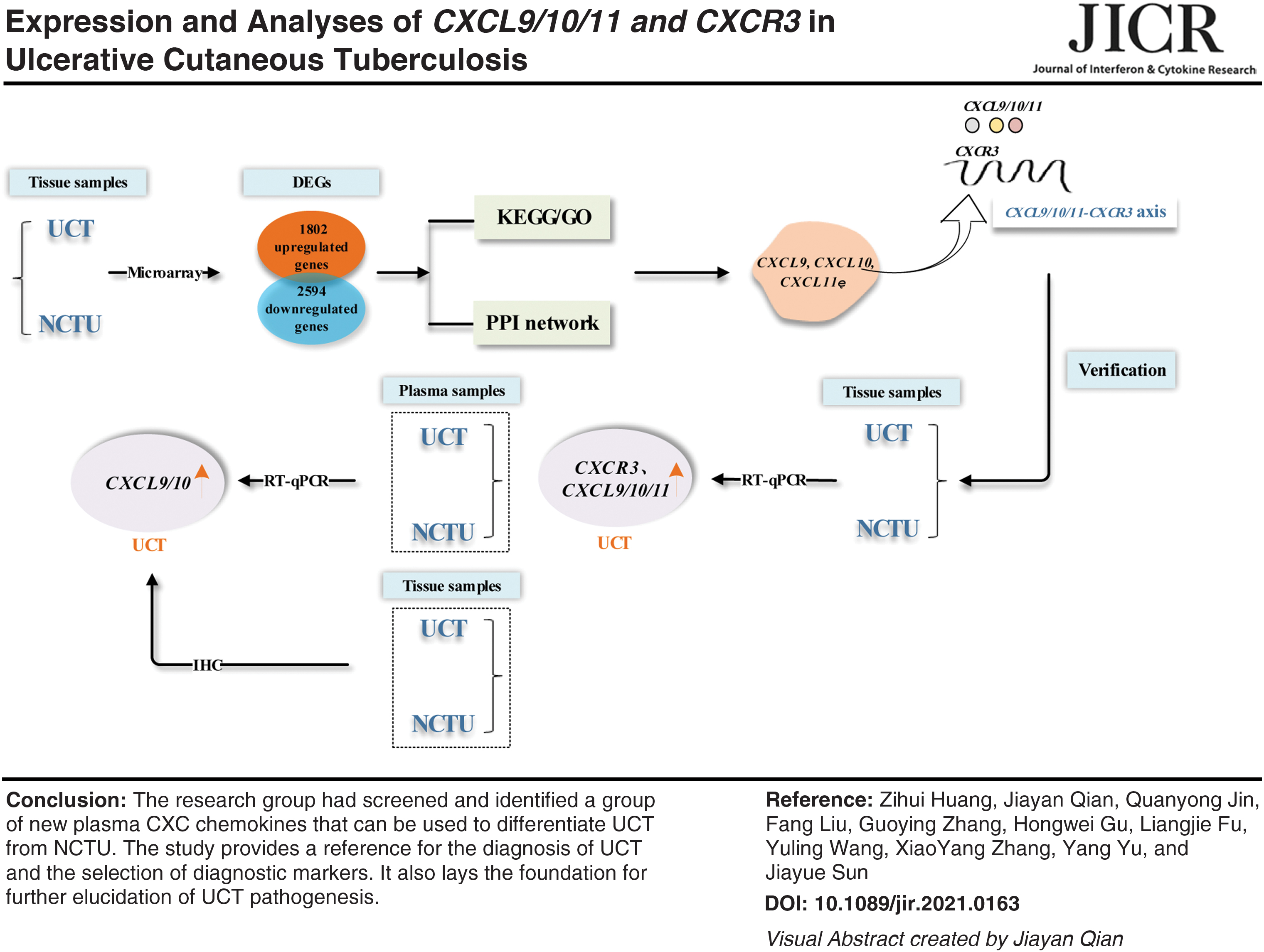
Abstract
We explored the biological functions, signaling pathways, potential inflammation, and immune biomarkers involved in ulcerative cutaneous tuberculosis (UCT). Mycobacterium tuberculosis-infected tissues from UCT patients and patients with noncutaneous tuberculous ulcers (NCTUs) were studied using transcriptomic analysis. Functional enrichment determined using the Gene Ontology database and enrichment of signaling pathways was ascertained using the Kyoto Encyclopedia of Genes and Genomes database. Protein–protein interaction (PPI) networks were analyzed to determine the hub genes. A total of 4,396 differentially expressed genes (DEGs) were identified. DEGs were enriched in CXCR3 chemokine receptor binding, chemokine activity, and cytokine–cytokine receptor interaction and other aspects. Analyses of PPI networks identified 15 hub genes. Expression of chemokine (C-X-C motif) ligand 9 (CXCL9)/10/11 messenger RNA (mRNA) and C-X-C motif chemokine receptor 3 (CXCR3) mRNA in the lesions of patients with UCT increased compared with that in NCTU cases. Expression of CXCL9 mRNA and CXCL10 mRNA in plasma also increased, which was consistent with other test results. We discovered a novel plasma CXC chemokine signature that could be used to differentiate UCT from NCTU. Our study (1) provides a reference for UCT diagnosis and selection of diagnostic markers and (2) lays the foundation for further elucidation of UCT pathogenesis.
Color images are available online
Introduction
Tuberculosis is a major global health concern, particularly in Asia and Africa. The incidence of tuberculosis in Asia is declining every year. However, worldwide, tuberculosis remains one of the top 10 causes of mortality and the leading cause of death by infectious disease (World Health Organization 2020; Nagu and others 2021). According to the 2020 Global Tuberculosis Report, there are ∼7.1 million new cases of tuberculosis worldwide each year, with China accounting for ∼11% of cases (World Health Organization 2020).
Ulcerative cutaneous tuberculosis (UCT) is an extrapulmonary form of tuberculosis and is more common in developing countries. UCT is the most common specific infectious ulcer of extrapulmonary tuberculosis caused by Mycobacterium tuberculosis and presents as necrosis, liquefaction, and ulceration under the skin (Kizny and others 2021). The neck, underarms, and crissum are common sites of cutaneous tuberculosis, which may be transmitted from endogenous tuberculosis (Sherif and Schauer 2015) and exogenous M. tuberculosis infection (Lindi van Zyl and others 2015) or induced by a strong reaction of bacillus Calmette–Guérin (Jacobs Ruschca and others 2016).
UCT lesions are difficult to heal or present as recurrent “pseudo-healing” because of the destruction caused by pathogenic bacteria. Furthermore, distinguishing UCT from other ulcerative diseases is challenging. Due to the number of patients with human immunodeficiency virus (HIV) infection (George M Varghese and others 2019) or multidrug-resistant tuberculosis, or the increasing need for immunosuppressive therapy (Gao and others 2020), there has been an increase in UCT incidence and difficulty in making the correct diagnosis and providing appropriate treatment.
Few studies have focused on UCT (Cardona 2018). Although several reports have highlighted the risk factors and cytokine expression in various forms of tuberculosis disease (Hasan and others 2009; Pavan Kumar and others 2013), studies on expression of specific factors of related tissue pathogens in the host have been scarce.
M. tuberculosis is the main pathogen of tuberculosis. M. tuberculosis attacks the main immune cells in the host to aid its replication and spread. Recruitment of immune cells is largely dependent on chemokine release (Martin Rao and others 2019). Chemokines guide the recruitment of immune cells to the infection site to influence disease progression. They are also important mediators of tuberculosis-specific inflammation and play an important part in the pathogenesis of tuberculosis-related diseases. Messenger RNA (mRNA) expression of certain chemokines may be associated with tuberculosis (Moser and Loetscher 2001; Chen and others 2011; Thomas Blauenfeldt and others 2018; Petrone and others 2019).
We aimed to examine global expression profiles and chemokine release in the infected tissue of patients with UCT to better comprehend the pathophysiological changes associated with UCT. Our study provides a scientific basis for selection of biomarkers of patients with UCT and lays the foundation for elucidating UCT pathogenesis.
Methods
Ethical approval of the study protocol
The study protocol was approved (201901001) by the Ethics Committee of Nanjing Hospital of Integrated Traditional Chinese and Western Medicine (Nanjing, China). Patients provided written informed consent for their samples and data to be used in this study.
Exclusion criteria
The exclusion criteria were people with active tuberculosis; suffering from mental illness; with impaired immune function (symptomatic HIV infection, confirmed or suspected HIV infection, leukemia, lymphoma, or systemic malignancy); and receiving immunosuppressive therapy (corticosteroids, alkylating agents, antimetabolites, or radiotherapy).
Study cohort
All patients were from the Nanjing Hospital of Integrated Traditional Chinese and Western Medicine.
We collected diseased tissues from 3 patients with UCT and 3 patients with noncutaneous tuberculous ulcers (NCTUs) for use in the transcriptomic analysis. Then, we collected diseased tissues from 14 patients with UCT and 14 patients with NCTUs to detect RNA to verify the results of the transcriptomic analysis. According to the results of that verification, we collected diseased tissues from 20 patients with UCT and 20 patients with NCTUs for verification using immunohistochemistry. Otherwise, plasma samples from 100 patients with UCT and 29 patients with NCTUs were collected to detect RNA for further verification.
Initially, all patients were assessed by clinical examination, basic laboratory tests, and computed tomography of the chest. Blood samples were collected, and the wound tissue was histopathologically analyzed. Patients had not undergone any form of antituberculosis treatment. There were no significant differences between patients with UCT and NCTUs with respect to age, sex, smoking status, alcohol consumption, or comorbidities.
Microarray analyses and identification of differentially expressed genes
The tissues of patients with UCT and those with NCTUs were selected as samples. Total RNA was quantified by a spectrophotometer (NanoDrop™ ND-2000; Thermo Scientific, Waltham, MA). RNA integrity was assessed using a bioanalyzer (2100; Agilent Technologies, Santa Clara, CA). Sample labeling, microarray hybridization, and washing with phosphate-buffered saline (PBS) were done based on Human lncRNA V6 (4*180K; design ID: 084410) chip standards (Agilent Technologies).
Briefly, total RNA was transcribed to double-stranded complementary (c)DNA, then synthesized into complementary (c)RNA, and labeled with cyanine-3-CTP. Labeled cRNAs were hybridized onto microarrays. After washing, the microarrays were imaged by a scanner (G2505C; Agilent Technologies).
Feature Extraction™ 10.7.1.1 (Agilent Technologies) and GeneSpring™ 13.1 (Agilent Technologies) were used to process the original image and extract the original data. Screening criteria were fold change (FC) ≥2.0 and P ≤ 0.05.
Enrichment analyses of differentially expressed genes
Gene Ontology (GO;
Protein–protein interaction networks and DEG selection
Information on the protein–protein interaction (PPI) networks of DEGs was obtained from the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (
Analyses and verification
According to the screening results for biological function (GO database), signaling pathways (KEGG database), hub genes, and the correlation between key genes and tuberculosis, we selected target genes for further verification and analyses.
Real-time reverse transcription–quantitative polymerase chain reaction for tissue samples
The genes for C-X-C motif chemokine receptor 3 (CXCR3), chemokine (C-X-C motif) ligand 9 (CXCL9), CXCL10, and CXCL11 were selected for real-time reverse transcription–quantitative polymerase chain reaction (RT-qPCR) to further verify the gene sequencing results. Tissue samples were selected from 14 patients with UCT and 14 patients with NCTUs.
Total RNA was extracted with TRIzol® reagent (Thermo Scientific). The RNA yield was determined using a spectrophotometer (NanoDrop 2000; Thermo Scientific). RNA (50 ng) was reverse transcribed into cDNA using HiScript™ II QRT SuperMix (Vazyme Biotech, Nanjing, China) for RT-qPCR.
Immunohistochemical analyses
CXCL10 and CXCL11 were selected for immunohistochemical analyses. We collected diseased tissues from 20 patients with UCT and 20 patients with NCTUs. Paraffin-embedded tissues were sliced to a thickness of 4 μm, deparaffinized, and rehydrated. Tissue sections were placed in a box filled with citric acid (pH 6.0; Servicebio, Wuhan, China) and antigen retrieval buffer. Tissue sections were placed in a solution of 3% hydrogen peroxide for blocking of endogenous peroxidase activity.
Bovine serum albumin (3%) was added to evenly cover tissue sections, and tissue sections were sealed for 30 min at room temperature. The primary antibody prepared with PBS (pH 7.4; Servicebio) at a certain dilution was added to tissue sections, and overnight incubation at 4°C was performed. Then, tissue sections were covered with a secondary antibody (labeled with horseradish peroxidase; 1:200 dilution; Servicebio) from the corresponding species of the primary antibody and incubated for 50 min at room temperature.
Finally, tissue sections were developed with 3,3′-diaminobenzidine (Servicebio) and counterstained with hematoxylin. After sealing tissue sections with neutral resin, they were scanned.
Real-time RT-qPCR for plasma samples
CXCR3, CXCL9, CXCL10, and CXCL11 were selected for RT-qPCR. Plasma samples were selected from 100 patients with UCT and 20 patients with NCTUs. Total RNA was extracted with TRIzol reagent (Thermo Scientific). The RNA yield was determined using a spectrophotometer (NanoDrop 2000; Thermo Scientific).
cDNA was synthesized and stored at −20°C. Reaction mixtures were subjected to real-time RT-qPCR using standard 3-step thermal cycling conditions on a fluorescence thermocycler (iCycler iQ™) (Bio-Rad Laboratories, Hercules, CA).
Statistical analyses
Statistical analyses were performed using SPSS 23.0 (IBM, Armonk, NY). Student's t-test, one-way analysis of variance, and chi-squared test were used for intergroup comparison. P < 0.05 was considered significant. Analyses of receiver operating characteristic (ROC) curves were performed, and the area under the ROC curve (AUC) was calculated. The optimal diagnostic threshold was determined by calculating the cutoff value of the ROC curve.
Results
Microarray analysis
Identification of DEGs in UCT and NCTU tissues
The use of 10,261 probes revealed differential expression in genes (adjusted p < 0.05, FC ±1.2 [log FC ±0.27]), including 1,802 upregulated genes and 2,594 downregulated genes. The relationship between gene expression profiles was examined by principal component analysis of the normalized gene expression data of samples and unsupervised hierarchical clustering using the genes with the greatest variance across the dataset. Then, we screened the top 30 upregulated and top 30 downregulated DEGs.
A heatmap and a volcano plot detailing expression of these genes are shown in Fig. 1.
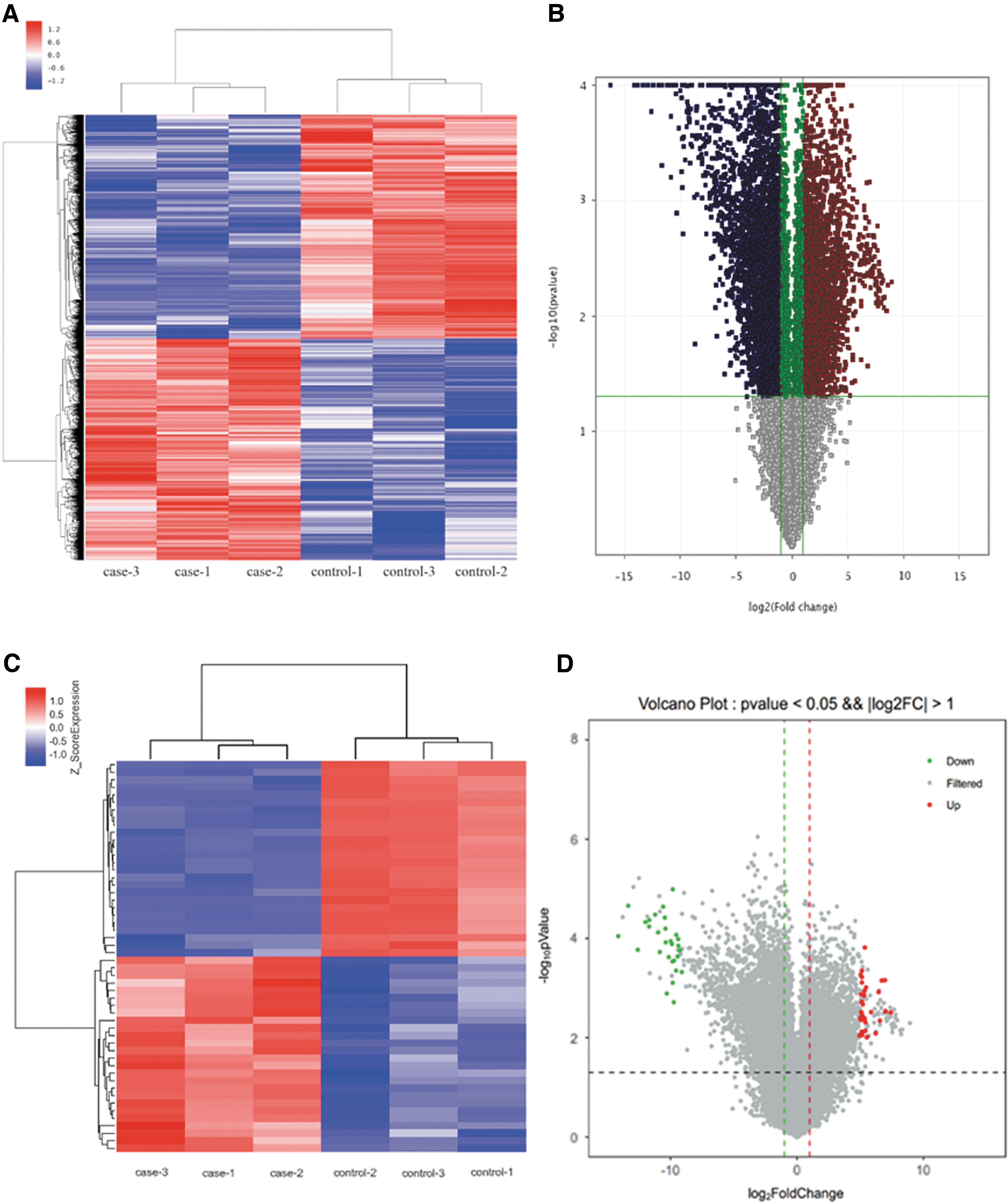
Heatmaps and a volcano plot of DEGs.
Enrichment analyses of DEGs
GO and KEGG databases were used to obtain detailed information about the biological functions and signaling pathways of DEGs.
Functional enrichment of DEGs
Functional analyses using the GO database are based on biological process (BP), cellular component (CC), and molecular function (MF). For DEGs, enrichment of BP was observed in negative regulation of interleukin-4 production, muscle filament sliding, and sarcomere organization. Enrichment of CC was observed in the Z disc and myofibril and external side of the plasma membrane. Enrichment of MF was observed in CXCR3 chemokine receptor binding, CXCR chemokine receptor binding, and chemokine activity (Fig. 2A).
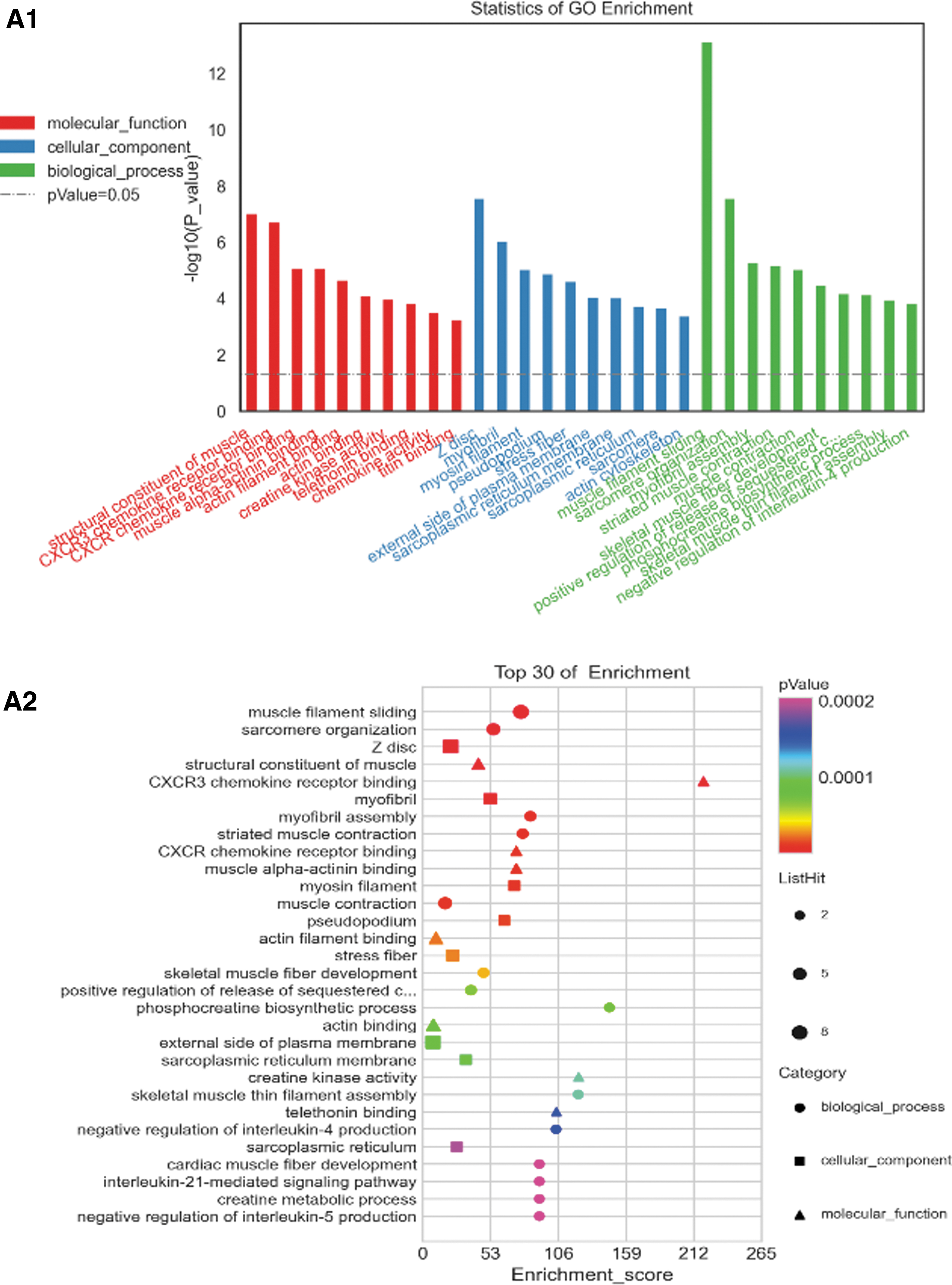
Upregulated and downregulated DEGs with the top 30 enriched GO and KEGG terms.
Enrichment of the signaling pathways of DEGs
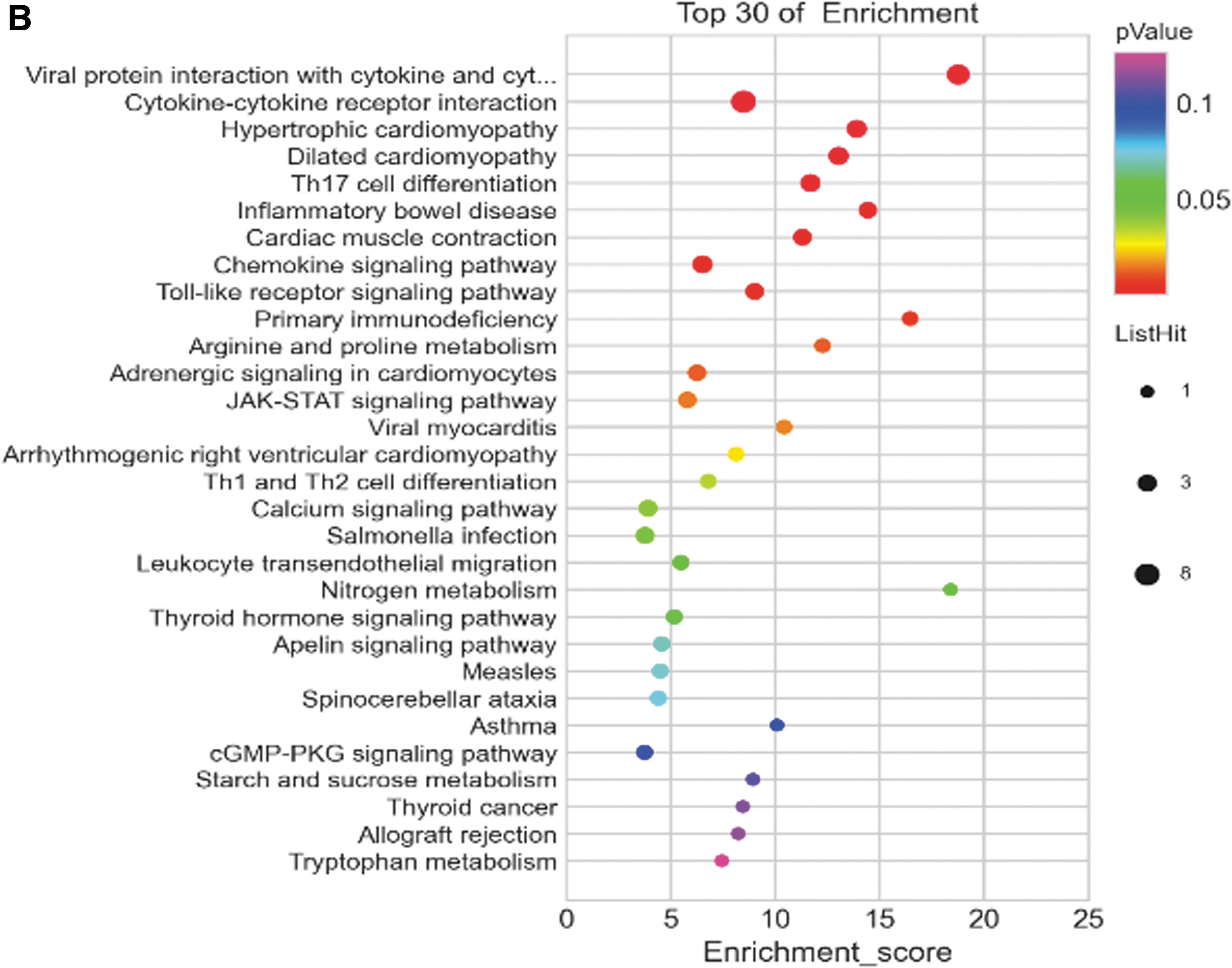
The KEGG database showed 44 pathways with P ≤ 0.05. The top 3 signaling pathways with the smallest P value were cytokine–cytokine receptor interaction (P = 6.5 × 10−19), viral protein interaction with cytokine and cytokine receptor (P = 1.4 × 10−17), and chemokine signaling pathway (P = 3.7 × 10−12). A total of 38 pathways had a false discovery rate ≤0.05 (Fig. 2B and Table 1).
Analyses of Signaling Pathways of Integrated Differentially Expressed Genes Using the Kyoto Encyclopedia of Genes and Genomes Database (P < 0.05)
PPI networks and DEG selection
The top 15 DEGs with high connectivity were selected as hub genes through Cytoscape. These DEGs were CXCL9, CXCL10, CXCL11, interleukin 2 receptor gamma (IL2RG), interleukin 2 receptor beta (IL2RB), troponin C2, fast skeletal type (TNNC2), nebulin (NEB), cardiac alpha-myosin (MYH6), myosin light chain-2 (MYL2), myosin-binding protein-C1 (MYBPC1), MYBPC2, interleukin 21 receptor (IL21R), IKAROS family zinc finger 3 (IKZF3), titin (TTN), and chemokine receptor 7 (CCR7) (Fig. 3).
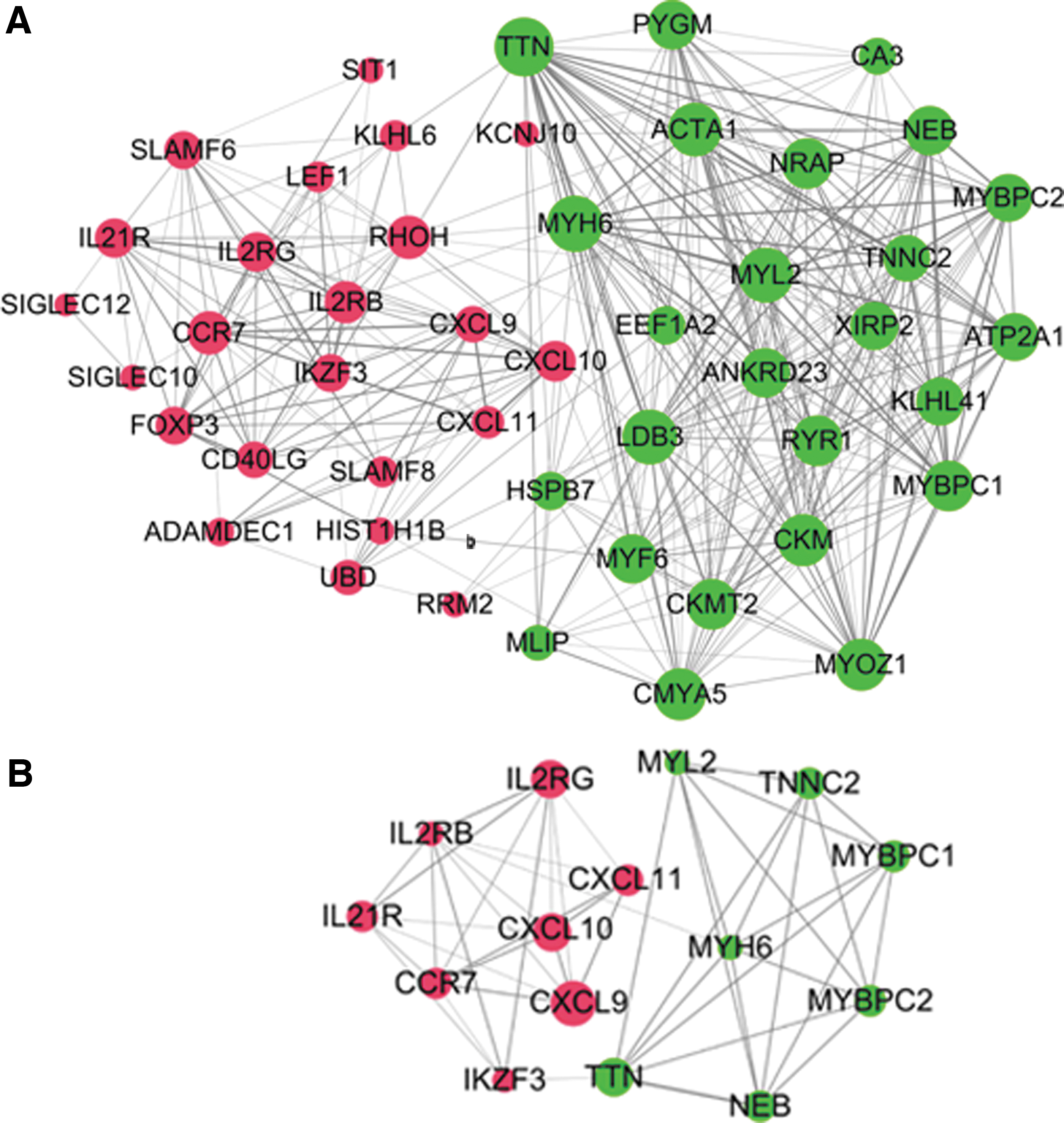
PPI network of DEGs (red represents upregulated genes and green represents downregulated genes).
Verification of key DEGs
Verification for tissue samples using RT-qPCR
The candidates chosen by gene analysis (CXCL9, CXCL10, CXCL11, and CXCR3) were subjected to further validation by RT-qPCR. Expression of CXCL9, CXCL10, CXCL11, and CXCR3 in samples of UCT tissue was higher than that in samples of NCTU tissue (Fig. 4).
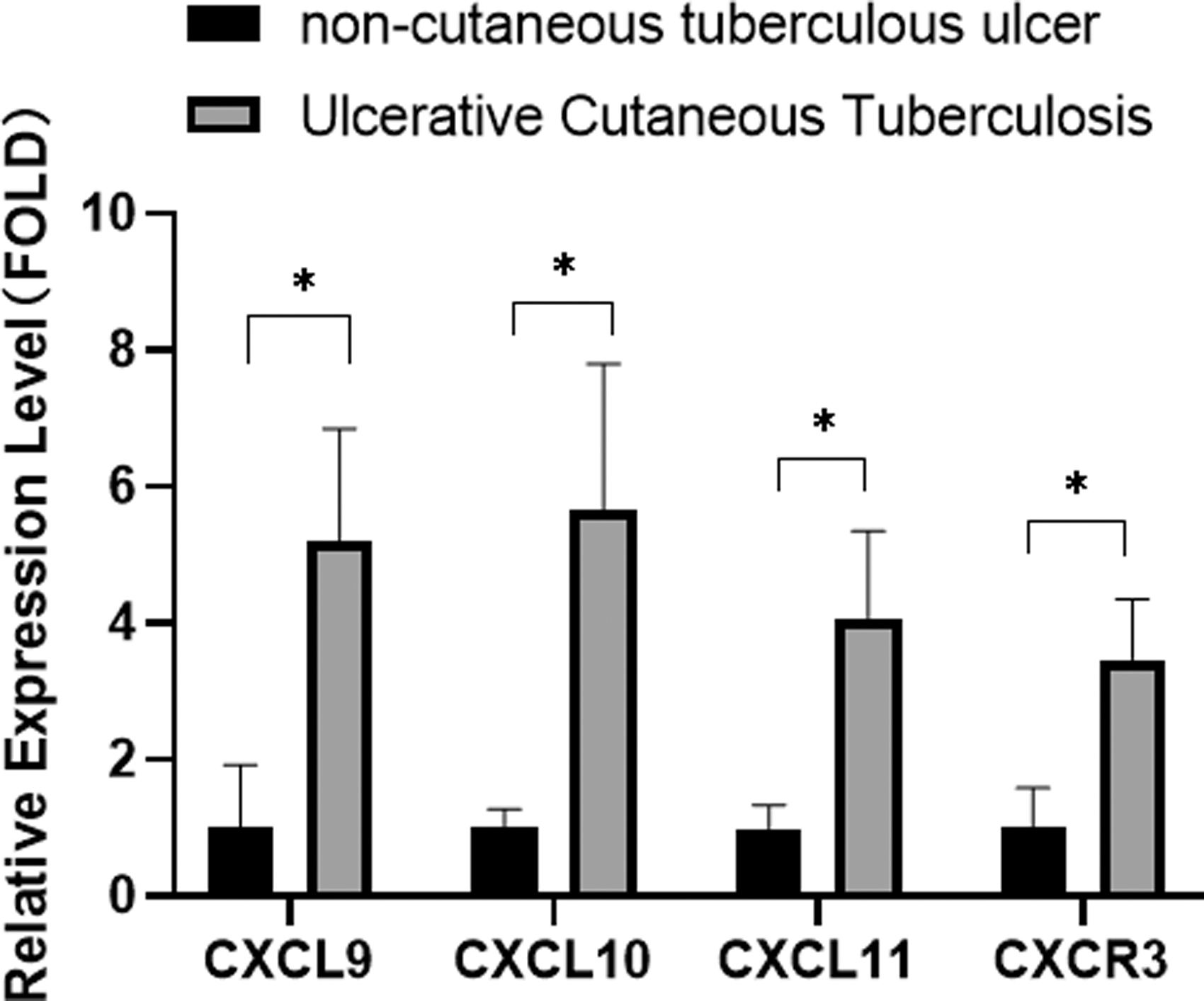
Verification of sequencing results using real-time RT-qPCR. RT-qPCR, real-time reverse transcription–quantitative polymerase chain reaction. The ratio of the expression level of the two groups (P < 0.05).
Verification using immunohistochemistry
Twenty UCT and 20 NCTU tissue samples were selected for immunohistochemical analyses to measure expression of CXCL9 and CXCL10. Expression of CXCL9 and CXCL10 in UCT tissues was higher than that in NCTU tissues (Fig. 5).
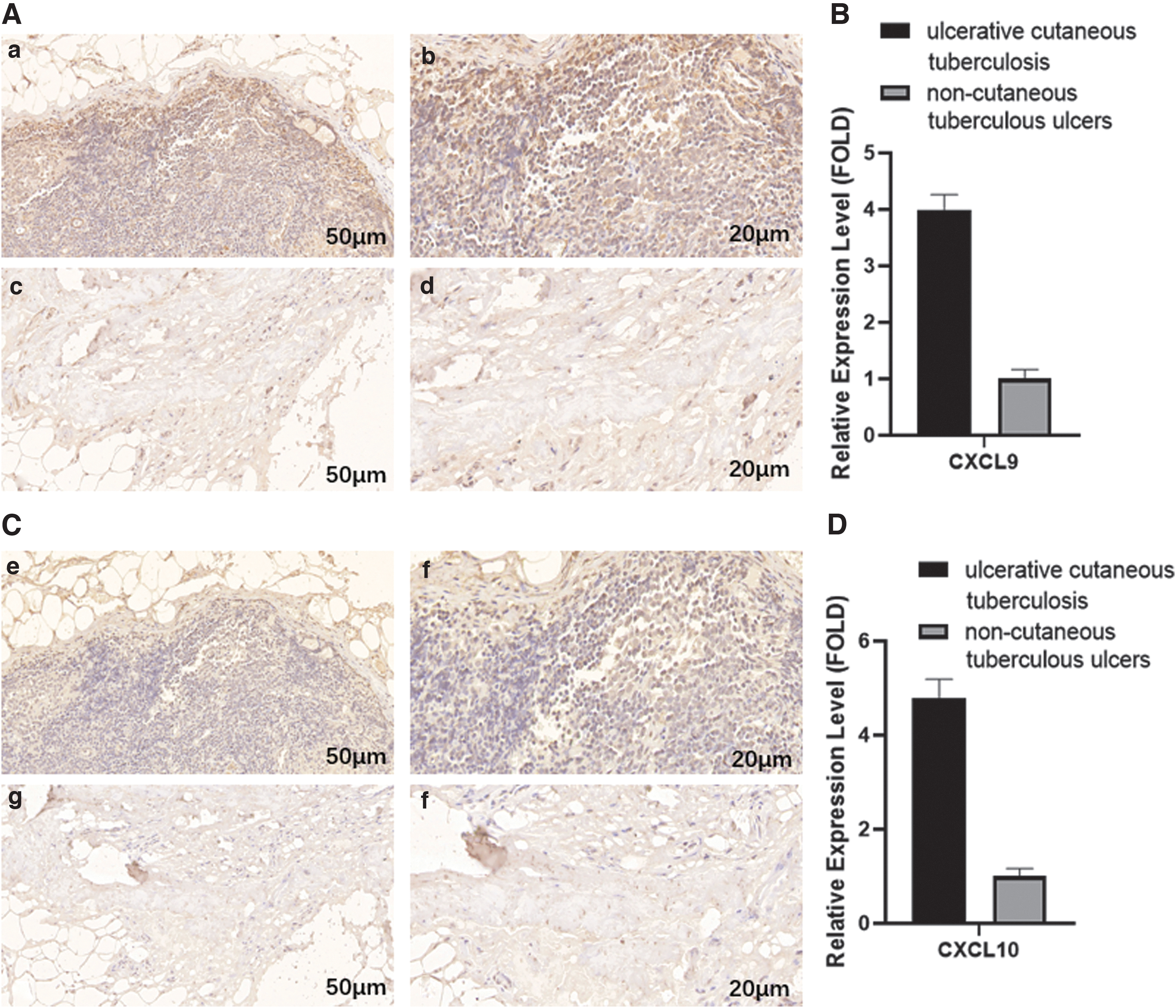
Expression of CXCL9 and CXCL10 by IHC analyses.
Verification in plasma samples using RT-qPCR
Expression of CXCL9 and CXCL10 was markedly increased in patients with UCT compared with that in patients with NCTUs (P < 0.01) (Fig. 6).

Relative expression of CXCL9 mRNA and CXCL10 mRNA in plasma. The ratio of the expression level of the two groups (P < 0.05).
AUC, specificity, negative predictive value, positive predictive value, and accuracy for identification of chemokines
At the optimal cutoff value, the positive predictive value and negative predictive value of a 2-chemokine (CXCL9/CXCL10) panel for RCC was 95.05% and 85.71%, respectively (Table 2).
Risk Scores of the Two Groups
ASC, sensitivity; SPE, specificity; NPV, negative predictive value; PPV, positive predictive value.
We examined ROC curves to estimate the ability of the 2-chemokine (CXCL9/CXCL10) panel to diagnose UCT. The AUC was 0.892 and 0.825, respectively. The cutoff value for CXCL9 and CXCL10 was 0.939 and 2.292, respectively (Fig. 7).
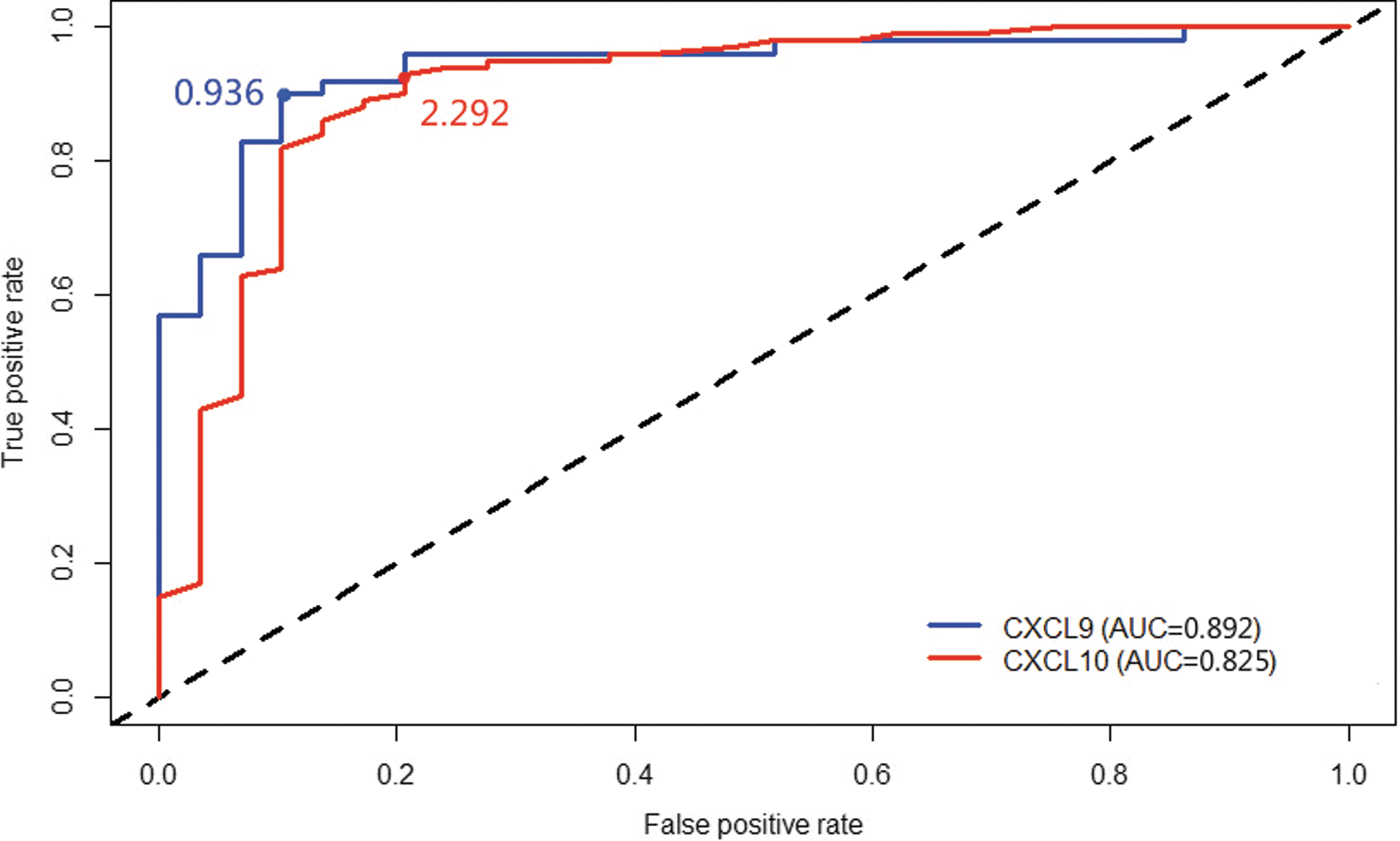
ROC curve and AUCs of CXCL9 and CXCL10. AUC, area under the receiver operating characteristic curve; ROC, receiver operating characteristic.
Discussion
An effective way to shorten the course of UCT is early diagnosis and early treatment. However, the diagnosis and treatment of UCT can be very difficult due to the number and virulence of M. tuberculosis, spread and location of the disease, and shape of cutaneous lesions. The mechanism of the occurrence and development of UCT is relatively complicated and its pathogenesis is not completely understood. Therefore, we analyzed the DEGs in tissues of patients with UCT and those with NCTUs. In addition, the key (or potentially key) genes that could be used to diagnose UCT were screened out. We aimed to lay the foundation for further elucidation of UCT pathogenesis.
Gene sequencing showed that mRNA expression in tissues from UCT patients was different from the mRNA expression in tissues from NCTU patients. There were 4,396 DEGs, of which 2,594 were upregulated and 1,802 were downregulated. Analyses of GO and KEGG databases enabled prediction of chemokine activity and chemokine signaling pathways. Binding of CXCR3 to receptors and interactions of viral proteins with cytokines and cytokine receptors may have important roles in UCT progression. Analyses of PPI networks and screening of key DEGs revealed the top 15 genes with the largest combined scores: CXCL9, CXCL10, CXCL11, IL2RG, TNNC2, NEB, MYH6, MYBPC2, MYL2, MYBPC1, IL21R, IL2RB, IKZF3, CCR7, and TTN.
We found that CXCL9, CXCL10, and CXCL11 selected by the research group from the above genes were encoded by highly DEGs, and according to the analysis of international tuberculosis-related research reports, this group of chemokines also had differential expression. They may be closely related to the pathogenesis of tuberculosis. At present, concerned researchers studying tuberculosis have not conducted research and published reports on chemokines of UCT. Therefore, the research team has considered this group of chemokines in the scope of this study.
CXCL9/10/11 are non-ELR (glutamic acid–leucine–arginine) CXC chemokines. They are CXCR3 ligands and are synthesized by cytokines or Toll-like receptor ligands. CXCL9/10/11 interact with receptors on the surfaces of bacterial membranes in vivo and in vitro to mediate bacterial lysis (Margulieux and others 2016). It was found that the CXCR3-CXCL11 signal axis drives the occurrence and expansion of mycobacterial granulomatous lesions in the establishment of a zebrafish larval tuberculosis model to explore the host–pathogen interaction (Torraca Vincenzo and others 2015).
Therefore, the chemokines, CXCR3 and CXCL9/10/11, should be involved in the infection and spread of M. tuberculosis and other bacteria. They are involved in the infection process of bacteria such as M. tuberculosis.
CXCL9/10/11 are possess the homologous receptor CXCR3 chemotaxis. They can recruit macrophages and T cells to the site of infection and participate in the spread of pathogens (Romagnoli and others 2016). Studies have shown that CXCL9 and CXCL10 are involved in recruitment of T cells from the mucosa to the lamina propria. This action leads to an imbalance in the number of T helper type 1 cells in mucosal immunity, promotion of mucosal damage, and aggravation of the inflammatory response (Romagnoli and others 2015).
UCT can be controlled by limiting M. tuberculosis spread in the infected site and inhibiting the development of wound inflammation. In this regard, we speculate if the formation of UCT wounds is related to chemokines.
Extrapulmonary tuberculosis has been rarely studied, as has UCT. Scholars have conducted related studies on CXCL9/10/11 and CXCR3, but the results have been contradictory. Some scholars have measured expression of CXCR3, CXCL9, CXCL10, and CXCL11 in the serum of patients with pulmonary tuberculosis and found that compared with healthy people, CXCL10 expression in the plasma of patients with pulmonary tuberculosis decreased and CXCL9 expression increased (Chen and others 2016; Novel and others 2016).
Sauty and colleagues documented differences in CXCL9 expression in the blood circulation of patients with pulmonary tuberculosis or extrapulmonary tuberculosis (without cutaneous tuberculosis) (Sauty and others 2001). The results from studies on CXCL10 expression in pulmonary tuberculosis and extrapulmonary tuberculosis have also been controversial. However, the sample size of those studies was relatively small, and samples were blood, not tissue. Therefore, because of the deficiencies in those studies, the present study conducted further research on this group of chemokines (CXCL9/10/11 and CXCR3).
The experiment in this article was to further verify the results of gene sequencing at the molecular protein level. RT-qPCR, Western blotting, and immunohistochemistry revealed that compared with patients with NCTUs, CXCL9/10/11 and CXCR3 had abnormal expression in UCT patients. We determined the cutoff value and AUCs for CXCL9 and CXCL10 in plasma, as detected by RT-qPCR. The AUC values were >0.5 and close to 1.
The high expression regions of CXCL9 and CXCL10 in UCT tissues are mainly found in the cytoplasmic regions of Langhans cells, epithelial cells, fibroblasts, and other cells. It could provide a reference basis for detection of the existing tuberculosis immunohistochemical index and achieve the purpose of helping clinicians with improved and effective diagnosis. In addition, compared with tissue sampling/testing, plasma is relatively easy to obtain. We speculate whether measurement of such chemokines can be used for preliminary screening of UCT.
Patients in the NCTU group mainly had pressure ulcers, venous leg ulcers, diabetic feet, cutaneous and soft-tissue infections, and burns. NCTU can be diagnosed according to the medical history and clinical manifestations. However, later in the disease course, NCTUs can be prone to immunodeficiency, exposed wounds, and M. tuberculosis infection, which ultimately lead to delayed treatment. We showed that CXCL9 and CXCL10 have extremely high specificity for tuberculosis and clinical diagnostic value for UCT.
The regulatory mechanism of CXCL9 and CXCL10 expression in patients with UCT is not clear (Pavan Kumar and others 2013), but analyses of GO and KEGG databases showed that activation of various immune and inflammatory responses may be related to regulation of chemokine genes.
Conclusions
We discovered a novel plasma CXC chemokine signature that could be used to differentiate UCT from NCTU with a high degree of accuracy. Our study provides a reference for the diagnosis of UCT and selection of diagnostic markers. It also lays the foundation for further elucidation of UCT pathogenesis.
Ethical Approval and Consent to Participate
The study protocol was approved (201901001) by the Ethics Committee of Nanjing Hospital of Integrated Traditional Chinese and Western Medicine (Nanjing, China). Patients provided written informed consent for their samples and data to be used in this study. This study was conducted in accordance with the ethical principles enshrined in the 1964 Declaration of Helsinki and its later amendments.
Footnotes
Authors' Contributions
Z.H.H. and Q.Y.J. were major contributors to writing the manuscript. Z.H.H. was responsible for the conception and design of the study. J.Y.Q., H.W.G., Q.Y.J., F.L., G.Y.Z., L.J.F., Y.L.W., X.Y.Z., Y.Y., and J.Y.S. were responsible for the acquisition and analyses of data. J.Y.Q. was in charge of statistical analyses. Z.H.H. and Q.Y.J. drafted the manuscript and revised it. All authors approved the final version of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Nanjing Key Medical Science and Technology Development Project (ZKX18042), Nanjing Health Young Talents Program (QRX17030), and Science and Technology Project of Jiangsu Chinese medicine and Pharmacy Bureau (YB2017042). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.