
Abstract
Sepsis-associated encephalopathy (SAE) is a diffuse brain dysfunction without overt central nervous system infection. Caffeine citrate has therapeutic effect on different brain diseases, while its role in SAE remains unclear. The expression levels of interleukin (IL)-18 and IL-1β were upregulated in the cerebrospinal fluid of the subjects. In this study, a rat model of SAE was established by cecal ligation and puncture. Caffeine citrate inhibited SAE-induced neuronal apoptosis and astrocytic activation, decreased reactive oxygen species (ROS) generation, and elevated mitochondrial membrane potential (MMP) level in the cerebral cortex. In vitro, primary astrocytes were isolated from rat cerebral cortex and incubated with lipopolysaccharide (LPS) and interferon-γ (IFN-γ). Caffeine citrate reduced ROS and MMP levels and mitochondrial complex enzyme activities in LPS plus IFN-γ-induced astrocytes. Moreover, caffeine citrate inhibited the activation of nucleotide-binding and oligomerization domain (NOD)-like receptor (NLRP3) inflammasome and decreased the production of IL-1β and IL-18 in vivo and in vitro. Notably, caffeine citrate promoted UCP2 expression in astrocytes. The neuroprotective role of UCP2 has been reported in several experimental brain diseases. These results suggest that caffeine citrate inhibits neuronal apoptosis, astrocytic activation, mitochondrial dysfunction in rat cerebral cortex, thereby alleviating SAE. The protection of caffeine citrate against SAE may be achieved by the UCP2-mediated NLRP3 pathway inhibition in astrocytes.
Introduction
Sepsis is an acute systemic inflammatory response caused by bacterial toxin. It has high clinical mortality and may cause multiple organ injury and failure (Taccone and others 2013; Ning and others 2017). During sepsis, the central nervous system (CNS) is considered to be one of the first affected organs, clinically manifested as sepsis-associated encephalopathy (SAE).
SAE is manifested with diffuse cerebral dysfunction and cognitive dysfunction, and is related to increased mortality and morbidity. The mortality rate was reported to reach 70% in SAE patients (Gofton and Young 2012; Widmann and Heneka 2014). The pathogenesis of SAE is not yet known. Previous researches have shown that oxidative stress, inflammatory injury, cerebrovascular dysfunction, and mitochondrial dysfunction were associated with the pathogenesis of SAE (Gu and others 2021). Nevertheless, the precise molecular mechanism of SAE has not been thoroughly studied, and there is lack of targeted treatment.
Astrocytes, accounting for 90% of the total brain cells, are homeostatic cells of the CNS with diverse functions, including ion homeostasis, neurogenesis, neurotransmitter metabolism, and the maintenance of synaptic connectivity and plasticity (Verkhratsky and Nedergaard 2018). It participates in brain injury related to neuroinflammation by releasing proinflammatory cytokines and toxic molecules. In addition, the activation of astrocytes adversely affected neuronal function and led to harmful neurological sequelae (Fan and others 2014; Sharma and others 2016).
Shulyatnikova and Verkhratsky (2020) reported that astrocytes could promote neuroinflammation and neuronal death to aggravate the adverse process of SAE. Moreover, the integrity of mitochondrial function in astrocytes plays an important role in the function of CNS. The damage of mitochondrial function in astrocytes could aggravate the loss of neurons by affecting neuronal survival and metabolism after brain injury (Bambrick and others 2004; Dugan and Kim-Han 2004). Study has shown that in the experiment model of sepsis in astrocytes, the function and morphology of mitochondria in astrocytes were both damaged (Peng and others 2019).
Inflammatory responses, especially the activation of inflammasome, are important for the development of SAE (Yende and others 2008; Annane and Sharshar 2015). Nucleotide-binding and oligomerization domain (NOD)-like receptor (NLRP3) inflammasome is the most well-characterized inflammasome, which is composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1 (Hise and others 2009). Fu and others (2019) reported that inhibition of NLRP3 inflammasome formation positively contributes to the reduction of neurological and cognitive impairment in septic animals (Fu and others 2019).
Meanwhile, evidence indicated that uncoupling protein 2 (UCP2) is related to NLRP3 activation (Danielski and others 2020). UCP2, one of the members of UCP family, is extensively expressed in neurons and immune cells (Mattiasson and Sullivan 2006; Degasperi and others 2008). The main role of UCP2 is to regulate mitochondrial potential and reactive oxygen species (ROS) production (Brand and Esteves 2005). Previous researches demonstrated that UCP2 might be involved in sepsis and sepsis-induced diseases (Moon and others 2015; Ding and others 2019), and its protective function may be mediated by limiting ROS production and inhibiting inflammation and cell death.
Caffeine citrate is a kind of caffeine, belonging to methylxanthine drugs, which is used in the treatment of primary apnea in preterm infants. Caffeine has a relieving effect on different brain diseases, such as showing antioxidant stress, and anti-inflammatory and antiapoptotic effects in neonatal brain injury caused by hyperoxia (Endesfelder and others 2014, 2017). It reduced apoptosis of neurons in the developing brain caused by ischemia and hypoxia (Kilicdag and others 2014a). It is worth noting that caffeine could improve behavior changes and neurocognitive deficits in septic rats (Assis and others 2018). However, the role of caffeine citrate in SAE has not been reported yet. In this study, we set out to evaluate the hypothesis that UCP2 might regulate NLRP3 inflammasome activation and participate in the protective mechanism of caffeine citrate against SAE.
Materials and Methods
Patient samples
The procedure of the research was reviewed and approved by the Human Ethics Committee of The Second Affiliated Hospital of Anhui Medical University [Clinical Ethics Approval No. YX2021-102 (F1)]. The study protocol was according to the Declaration of Helsinki ethical guidelines. All patients and their legal designees signed written informed consent to take part in this study.
The inclusion criteria for this study were based on the latest definition of SAE (Chaudhry and Duggal 2014). Inclusion criteria were children (<14 years old) with the diagnosis of sepsis as determined by fever (body temperature >38°C) and serum biochemistry for inflammatory parameters, such as white blood cell, C-reactive protein, procalcitonin, and encephalopathy as determined by cerebrospinal infection, abnormal electroencephalography, or cranial magnetic resonance imaging (MRI).
Exclusion criteria included non-SAE, such as metabolic encephalopathy, drug-induced encephalopathy and intracranial causes of encephalopathy, and malignancy. Children with nervous system diseases who met the following criteria served as controls: no neuroinflammatory or neurodegenerative diseases, no sepsis and encephalopathy, no brain injury on MRI, and no infection on cerebrospinal fluid analysis. A total of 13 children patients were studied after screening. At the same time, 13 control volunteers were recruited. After cerebrospinal fluid collection, samples were stored at −80°C until further processing.
Animals and treatment
All experiments were approved by the Animal Ethics Committee of Anhui Medical University (Approval No. LLSC20211514). The male Wistar rats (100–120 g) were provided by Liaoning Changsheng Biotechnology Co., Ltd. Rats were housed in standardized conditions with 22°C ± 1°C, 50% ± 5% controlled humidity environment on 12-h light/12-h dark cycle and free access to water and food.
Rats were randomly divided into sham operation group and SAE group. Rats were subjected to cecal ligation and puncture (CLP) to induce SAE as described by Rittirsch and others (2009). Briefly, anesthetized rats underwent laparotomy to expose the cecum. The cecum was further ligated at half the distance to the end and punctured twice with needle. The intestinal contents were excreted. Sham-operated rats underwent the same procedure without ligation and puncture.
Twelve hours after CLP, rats were diagnosed with SAE if their scores on neurobehavioral tests decreased. Subsequently, the rats with SAE were randomly divided into the SAE group, 10 mg/kg caffeine citrate-treated group and 20 mg/kg caffeine citrate-treated group. In the 10 and 20 mg/kg caffeine citrate-treated group (Kilicdag and others 2014b), rats were intraperitoneally injected with corresponding doses of caffeine citrate once a day for 3 days. Rats in the sham and SAE groups were injected with saline of equal volume. After last administration, rats were sacrificed and brain tissues were collected for later analysis. The experiment is shown in Fig. 1.

The diagram of animal experiments. Schematic protocol of SAE injury and caffeine citrate treatment scheme in vivo. SAE, sepsis-associated encephalopathy.
Neurobehavioral test
After CLP treatment for 12 h, the rats were tested for neurobehavior. Neurobehavioral test was scored according to the report of Zhou and others (2019) including pinna reflex, corneal reflex, paw or tail flexion reflex, righting reflex, and escape reflex. Zero is no reflection; 1 point means weak reflex, 2 points for normal reflection, and the highest score is 10.
Primary astrocyte cultures and treatment
Rat primary astrocytes were prepared as Chen and others (2018) described. In short, rats were sacrificed; the cerebral cortex was removed and separated from blood vessels and meninges. The tissue was dissociated with tryptase (Sigma, St. Louis, MO) at 37°C and terminated by Dulbecco's modified Eagle's medium (DMEM) (Service Biotechnology, Wuhan, China) containing 10% fetal bovine serum (Sijiqing, Hangzhou, China). Dissociated cells were plated on poly-lysine-treated plates (Shanghai Yuanye Bio-Technology Co., Ltd, Shanghai, China) at a density of 2 × 105/mL and cultured at 37°C in a 5% CO2 air atmosphere in DMEM. Glial fibrillary acidic protein (GFAP) immunofluorescence was used to determine the purity of astrocytes.
The primary astrocytes were randomly divided into 4 groups: control group, lipopolysaccharide (LPS)+interferon-γ (IFN-γ) group, LPS+IFN-γ+100 μM caffeine citrate group, and LPS+IFN-γ+200 μM caffeine citrate group. Astrocytes in all groups, except control group were stimulated for 24 h with LPS (100 ng/mL) (Solarbio, Beijing, China) and IFN-γ (200 U/mL) (Sino Biological, Inc., Beijing, China) (Chen and others 2018) and the treatment groups were cocultured with different concentrations of caffeine citrate for 24 h.
Terminal deoxynucleotidyl transferase dUTP nick end labeling-neuronal nuclei staining
The 5 μM-thick sections from frozen cerebral cortex were incubated with 0.1% Triton X-100 (Beyotime Biotech, Shanghai, China) at room temperature for 8 min and washed in phosphate-buffered saline (PBS) for 3 times, following retrieval with citric acid/sodium citrate solution for 10 min. Slides were incubated with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) solution (Roche, Basel, Switzerland) for 1 h at 37°C in dark area, then with anti-neuronal nuclei (NeuN) antibody (1:200; Abcam, Cambridge, United Kingdom) at 4°C overnight and Cy3-labeled goat anti-mouse IgG (1:200; Beyotime Biotech) for 60 min at room temperature. Nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Aladdin, Shanghai, China). Images were obtained by fluorescence microscopy (Olympus, Tokyo, Japan).
Immunofluorescence assay
The slides of tissues or cells were blocked with goat serum (Solarbio) for 15 min before immunofluorescence staining, and then incubated with primary antibodies for 4°C overnight. After washing with PBS 3 times, sections were incubated with corresponding secondary antibodies. Finally, DAPI was used for nuclei counterstaining. Images were obtained by fluorescence microscopy (Olympus). The primary antibodies were as follows: GFAP antibody (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), and UCP2 (1:200; Affinity, Cincinnati, OH). The secondary antibodies were as follows: Cy3-labeled goat anti-mouse antibody (1:200; Beyotime Biotech) and FITC-labeled goat anti-mouse antibody (1:200; Beyotime Biotech).
Measurement of mitochondrial membrane potential in vitro and in vivo
For in vivo mitochondrial membrane potential (MMP) assay, first, mitochondria were isolated from cerebral cortex by the Tissue Mitochondrial Isolation Kit (Beyotime Biotech). The cerebral cortex tissues were homogenized with mitochondrial lysis buffer on ice, and the homogenate was centrifuged at 600g for 5 min. The upper layer was recovered and centrifuged at 11,000g for 10 min to obtain mitochondrial precipitates. Finally, the mitochondrial sediment was resuspended in PBS solution for MMP assay, which was detected by the MMP Detection Kit according to the manufacturer's instructions (Beyotime Biotech).
For in vitro MMP assay, the cultured astrocytes were stained with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benz-imidazole carbon iodide (JC-1) (Beyotime Biotech) for 20 min at 37°C. Then the cells were collected by centrifugation at 300g for 3 min and washed with JC-1 buffer twice. MMP was measured by flow cytometry.
Western blotting
Total proteins from cerebral cortex tissues or astrocytes were extracted. Aliquots of proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membrane (Thermo Fisher Scientific, Pittsburgh, PA) for 90 min. The membranes were blocked with 5% (M/V) bovine serum albumin (Biosharp, Hefei, China) in tris-buffered saline containing 0.15% (V/V) Tween 20 (TBST) and incubated with antibodies against UCP2 (1:1,000; Affinity, Changzhou, China), NLRP3 (1:1,000), caspase-1 (1:1,000), pro-Interleukin (IL)-1β (1:1,000), IL-1β (1:1,000) (ABclonal, Wuhan, China), or β-actin (1:2,000; ProteinTech Group, Inc., Wuhan, China), respectively, overnight at 4°C.
The membranes were then washed 4 times with TBST, and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (ProteinTech Group, Inc.). Visualization of target proteins was completed by detecting fluorescence generated by the Enhanced Chemiluminescence Kit (7 Sea Pharmtech, Shanghai, China).
ROS detection in vitro and in vivo
ROS levels of tissues were determined using the ROS Determination Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer's printed instructions. The intensity was detected by fluorescence microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
Intracellular ROS was determined by ROS-specific probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (KeyGEN, Nanjing, China). Astrocytes were incubated with DCFH-DA for 20 min at 37°C and washed 3 times with medium without fetal bovine serum to remove excess DCFH-DA. Images were observed under fluorescent microscope (Olympus).
Detection of IL-1β and IL-18 by enzyme-linked immunosorbent assay
IL-1β and IL-18 levels in astrocytes were detected by the Rat IL-1β Enzyme-Linked Immunosorbent Assay (ELISA) Kit (LIANKE Biotech., Co., Ltd., Hangzhou, China) and Rat IL-18 ELISA Kit (Wuhan Fine Biotech Co., Ltd., Wuhan, China), respectively.
Measurement of respiratory chain complexes
The activities of complex I and complex III were measured by the Complex I Assay Kit and Complex III Assay Kit (Solarbio) by ultraviolet-visible spectrophotometer. Data were normalized to the cell protein concentration.
Statistical analyses
All statistical analyses were performed using GraphPad prism 8.0 software (GraphPad Software). All data were expressed as mean ± standard deviation. Student's t-test or 1-way ANOVA were used for the comparisons. P < 0.05 was regarded as statistically significant.
Results
The levels of IL-18 and IL-1β are high in SAE patients
As shown in Fig. 2, the levels of IL-18 (Fig. 2A) and IL-1β (Fig. 2B) in cerebrospinal fluid were significantly upregulated in patients group compared with control subjects.
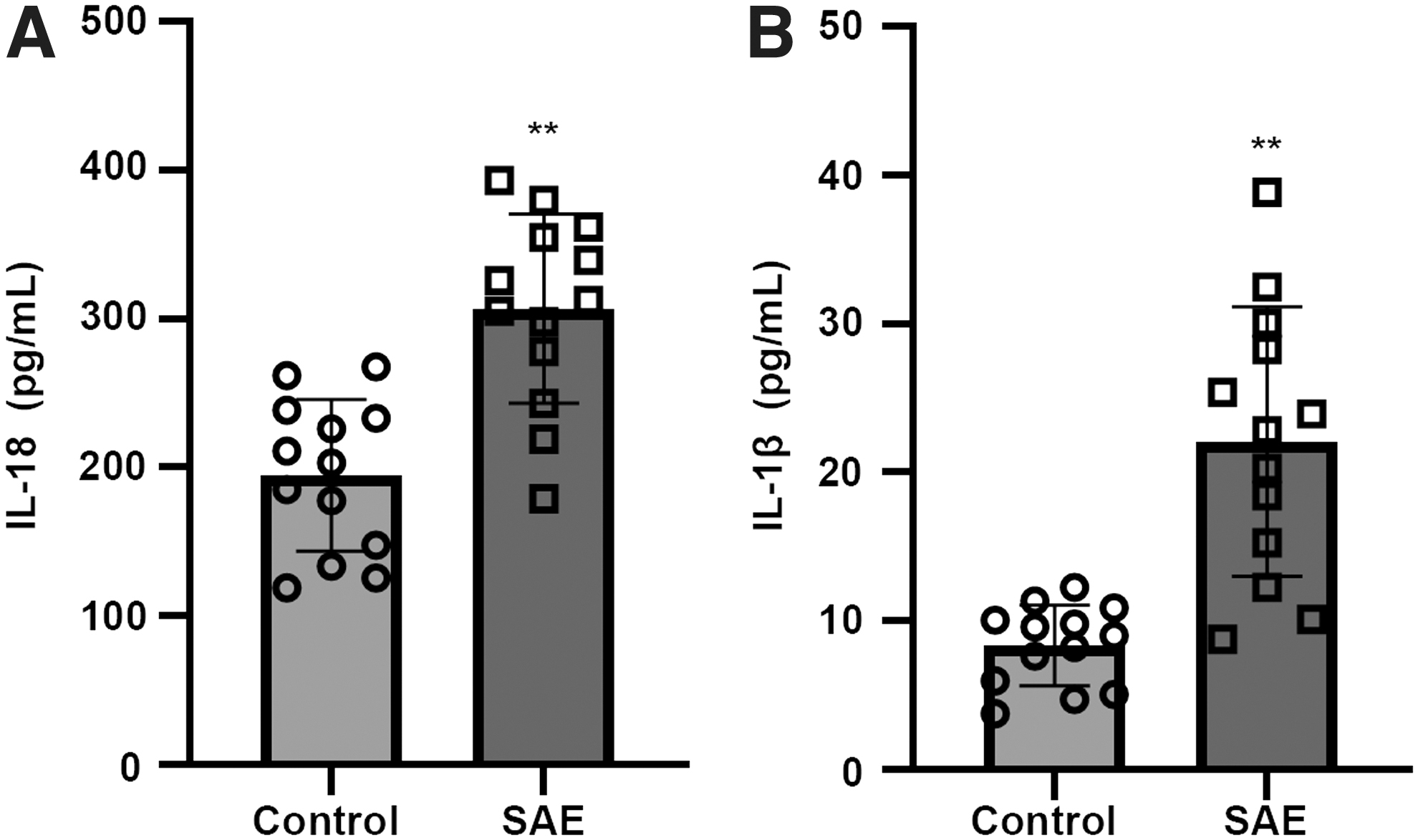
The levels of IL-18 and IL-1β are high in SAE patients.
Rats with SAE show abnormal behavior
To confirm whether rats were suffering from SAE at 12 h after CLP, we assessed the neurobehavioral test scores. CLP rats with low total neurobehavioral test scores would be diagnosed as SAE. Table 1 showed that the neurobehavioral test scores were significantly lower in the SAE group than in the sham group.
Neurobehavioral Test-Scores of 2 Groups
P < 0.01.
SAE, Sepsis-associated encephalopathy.
Caffeine citrate inhibits neuronal apoptosis and astrocytic activation in cerebral cortex of rats with SAE
The apoptosis of cerebral cortex neurons was detected by double staining of TUNEL and NeuN. The results showed a high level of TUNEL/NeuN-positive cells in the cerebral cortex of SAE group in contrast to control group (Fig. 3A, B). With caffeine citrate treatment, the apoptosis was restricted (Fig. 3A, B). GFAP is a marker of activation of astrocyte. GFAP fluorescence intensity, as shown in Fig. 3C, was significantly increased in CLP rats, whereas caffeine citrate reduced the expression of GFAP. These results demonstrated that caffeine citrate might inhibit the CLP-induced neuronal apoptosis and the activation of astrocytes.

Caffeine citrate inhibits SAE-induced neuronal apoptosis and astrocytic activation.
Caffeine citrate attenuates mitochondrial dysfunction in cerebral cortex of rats with SAE
We measured the effects of caffeine citrate on ROS levels. As shown in Fig. 4, CLP-induced increases in the levels of ROS were suppressed by the administration of caffeine citrate, suggesting the resumption of mitochondrial functions (Fig. 4A). To test whether caffeine citrate could protect the integrity of mitochondria, we detected MMP. The treatment with caffeine citrate restored MMP, offsetting the effect of CLP (Fig. 4B). These data suggested that caffeine citrate could protect mitochondrial function from SAE damage.
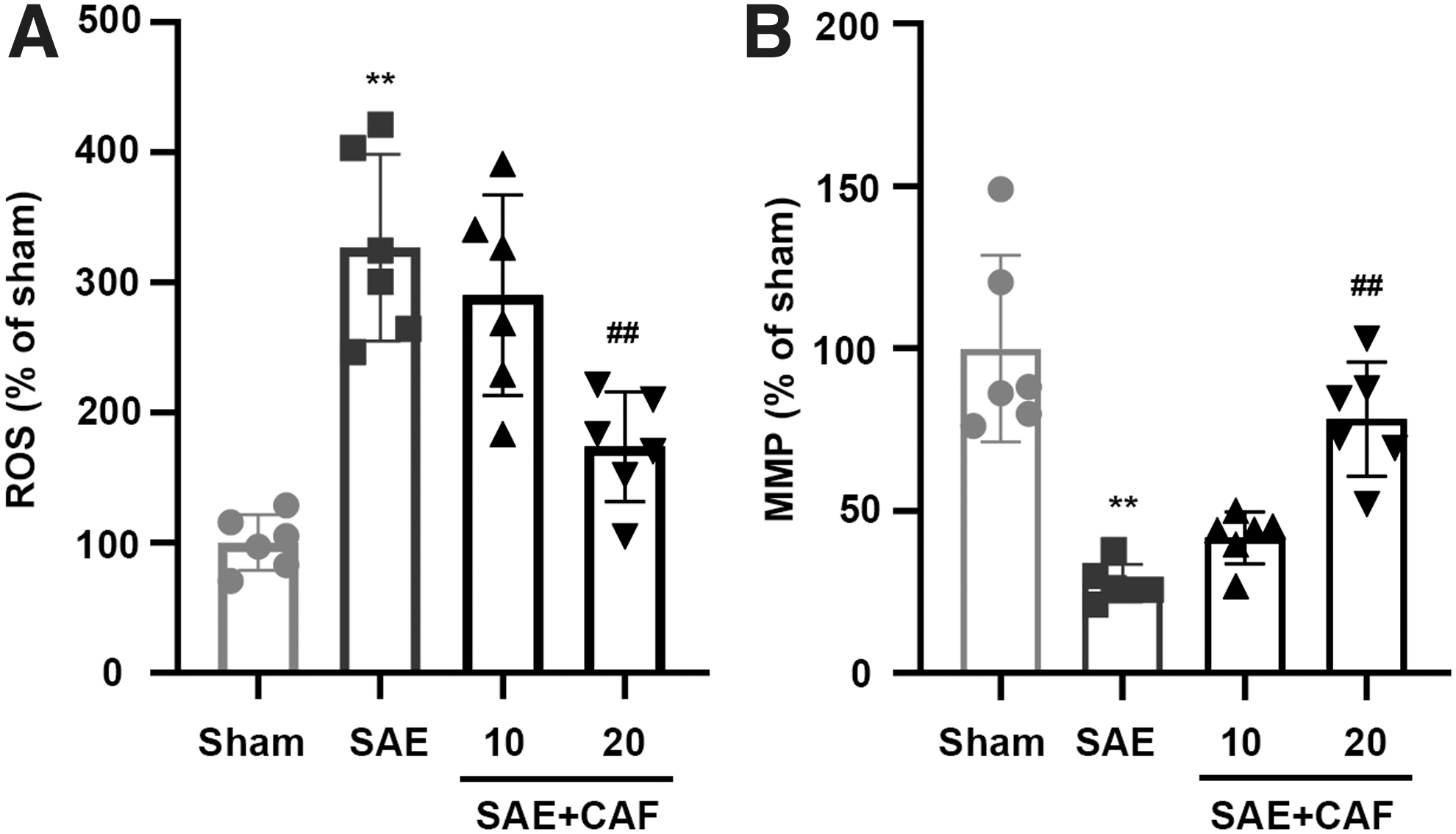
Caffeine citrate improves mitochondrial dysfunction in rat cerebral cortex. The ROS levels
Caffeine citrate activates UCP2/NLRP3 signaling pathway in cerebral cortex of rats with SAE
Furthermore, we detected the protein expression of UCP2/NLRP3 signaling pathway by immunofluorescence and Western blotting. Immunofluorescence and histograms (percentage of cells that stain positive for UCP2 and GFAP) showed low level of UCP2-positive staining in the cerebral cortex from SAE rats but dramatically increased staining from caffeine citrate-treated rats (Fig. 5A). For further validation, Western blotting was also shown in Fig. 5B and C. UCP2 expression was significantly decreased in the cerebral cortex from SAE rats. Treatment with caffeine citrate induced remarkable increase in UCP2 protein expression (Fig. 5B, C).

Caffeine citrate activates UCP2/NLRP3 signaling pathway in cerebral cortex.
Moreover, Western blotting results showed that the protein expression levels of NLRP3 (Fig. 5D), caspase-1 (Fig. 5F), pro-IL-1β (Fig. 5G), and IL-1β (Fig. 5H) were prominently increased in the SAE group compared with sham-operated group, but there was no significant change in pro-caspase-1 expression (Fig. 5E). Furthermore, an opposite trend was observed for caffeine citrate treatment (Fig. 5A–H). The above data indicated that caffeine citrate could promote UCP2 expression, and inhibit the activation of NLRP3 inflammasome.
Caffeine citrate stimulates UCP2 expression and improves mitochondrial function in LPS plus IFN-γ-stimulated astrocytes
Astrocytes are the major source leading to inflammatory responses in the brain and play a vital role in the pathogenesis of SAE (Shulyatnikova and Verkhratsky 2020). To further explore the protective mechanism of caffeine citrate in SAE, we established a sepsis model in astrocytes for the next study. For experiment accuracy, the identification of primary astrocyte cells was determined by GFAP staining. As shown in Fig. 6A, the primary astrocytes met the requirements of experiment.
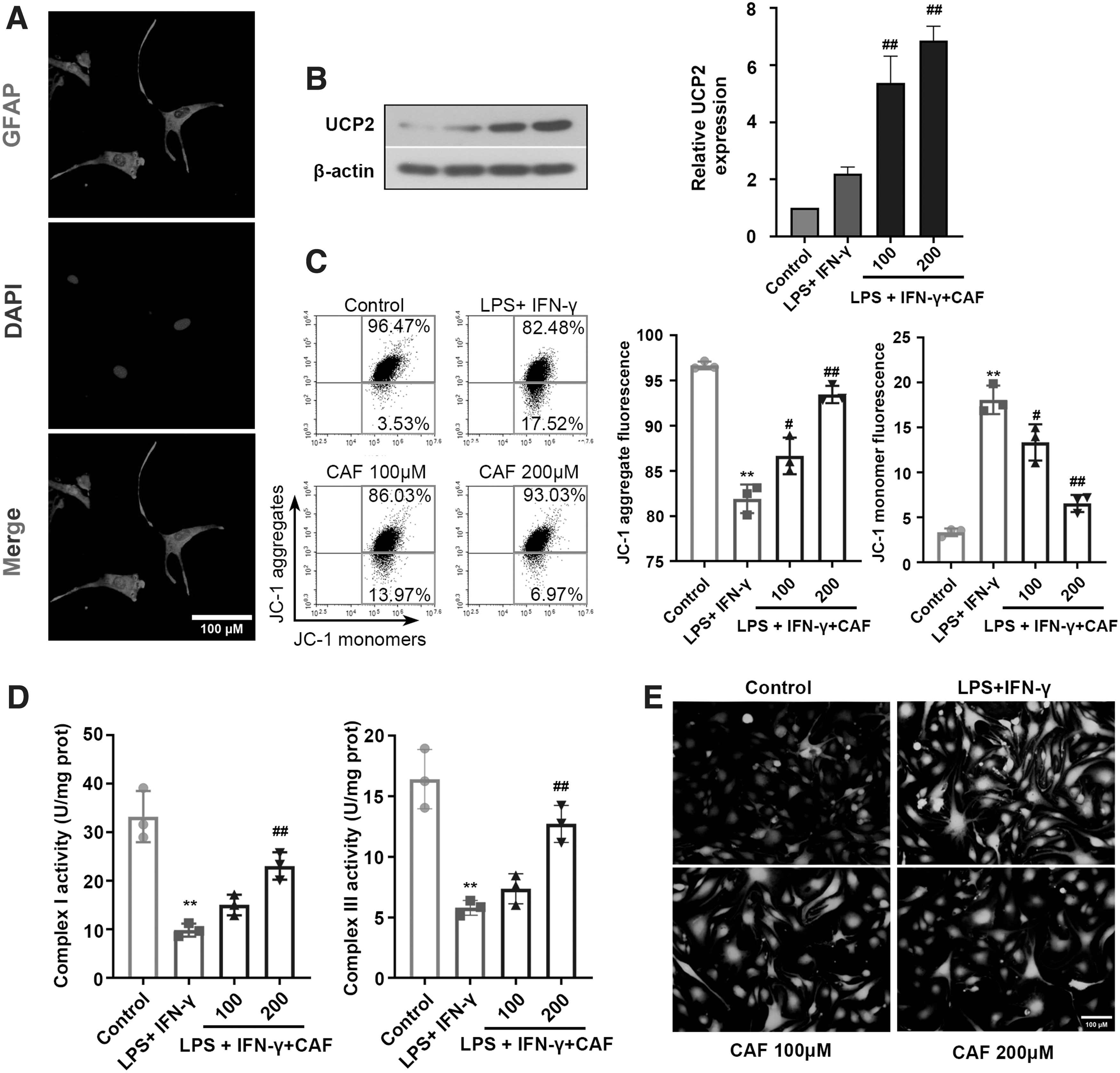
Caffeine citrate stimulates UCP2 expression and improves mitochondrial function in LPS plus IFN-γ-stimulated astrocytes.
The expression levels of UCP2 were significantly increased for caffeine citrate treatment under LPS plus IFN-γ stress (Fig. 6B). Moreover, caffeine citrate markedly restored MMP levels (Fig. 6C), as evidenced by the increase in JC-1 aggregate fluorescence and the decrease in JC-1 monomer fluorescence. In addition, caffeine citrate significantly improved the activities of complex I and complex III (Fig. 6D), and decreased ROS levels (Fig. 6E) in LPS plus IFN-γ-stimulated astrocytes, indicating that caffeine citrate could promote UCP2 expression and alleviate the mitochondrial dysfunction of astrocytes.
Caffeine citrate inhibits the activation of NLRP3 signaling pathway and the release of inflammation factors in LPS plus IFN-γ-stimulated astrocytes
The protein expression of NLRP3 signaling pathway was also assessed in astrocytes. LPS and IFN-γ-treated astrocytes showed an increased expression of NLRP3 (Fig. 7A). In contrast to NLRP3, the expression of pro-caspase-1 was unchanged (Fig. 7B). Moreover, the activated caspase-1, pro-IL-1β, and matured IL-1β were significantly increased in the LPS+IFN-γ-treated astrocytes compared with the control group (Fig. 7B–D). However, the opposite results were observed for the caffeine citrate treatment.

Caffeine citrate inhibits the activation of NLRP3 signaling pathway and the release of inflammation factors in LPS plus IFN-γ-stimulated astrocytes.
Inflammatory factors are the ultimate common pathway in the pathophysiology of SAE. (Xie and others 2021) We next observed the secretion of IL-1β and IL-18. Quantification of IL-1β and IL-18 in the culture medium of LPS+IFN-γ-treated cells revealed dramatical elevation in the release of IL-1β and IL-18; caffeine citrate remarkable inhibited IL-1β and IL-18 release after LPS+IFN-γ challenge (Fig. 7E, F). The above data indicated that, similar to the results of cerebral cortex, caffeine citrate inhibited the activation of NLRP3 inflammasome and the secretion of proinflammatory factor in astrocytes.
Discussion
In this study, we demonstrated that caffeine citrate has a protective effect on SAE. These conclusions were based on the melioration of neuronal apoptosis, astrocytic activation, and mitochondrial dysfunction. Moreover, caffeine citrate was verified to activate UCP2/NLRP3 axis both in vivo and in vitro (Fig. 8).

Schematic presentation of a potential mechanism for the protective role of caffeine citrate against SAE. Caffeine citrate has a protective effect on SAE. SAE triggers ROS generation followed by mitochondrial dysfunction, inflammatory response, and apoptosis, thus aggravating the disease process. Caffeine citrate is likely to inhibit NLRP3 inflammasome pathway by activating UCP2 expression to alleviate SAE process.
After modeling with CLP, we measured neurobehavioral test scores to analyze whether the SAE rat model was successfully established. Based on previous study in relevant literature and our research data (Zhou and others 2019), it could be considered that the SAE rat model was successfully established in this study. Caffeine citrate once enters the organism, it is rapidly metabolized to caffeine. As a nonselective adenosine receptor antagonist, caffeine can cross the blood–brain barrier (BBB) to exert pharmacological effects on brain tissue (Summerfield and others 2007). On this basis, the interventional effects of different doses of caffeine citrate on SAE were studied.
Accumulating researches indicated that the development of SAE involved a number of mechanisms, such as cerebral inflammation, BBB disruption, and the activation of glial cell and neuronal death (Hotchkiss and Karl 2003; Zampieri and others 2011; Michels and others 2015). Wang and others (2018) have reported that apoptosis was observed during the pathogenesis of SAE. Indeed, increased apoptotic death of neurons has been shown in the cerebral cortex in response to SAE in our research, suggesting that CLP-induced neuronal apoptosis might contribute to neurodegeneration and cognitive impairment of SAE.
In addition, astrocytic alterations during sepsis play an important role in the pathogenesis of SAE (Ren and others 2020). The immunofluorescence (IF) assay in our study presented reduced GFAP expression involved in astrocytic activation. Reactive astrocytes could increase expression of complement cascade genes and release unknown neurotoxins that induce the death of neurons and oligodendrocytes, which in turn leads to cognitive impairment (Liddelow and Barres 2017).
As reported in a previous study, 48 h after sepsis induction, astrocytes in the rat brain showed strong activation and it could be alleviated by simvastatin (Catalão and others 2017). In our study, caffeine citrate administration reduced apoptosis and the activation of astrocytes, suggesting it has a protective effect on SAE. Astrocytes serve as an important component matrix of the BBB and its activation leads to the breakdown of BBB. Fredholm and others (1999) reported that caffeine could penetrate the BBB and exert multiple effects at a cellular level on the CNS, proving the therapeutic effect of caffeine citrate on astrocytes in SAE for our study. Therefore, we built experimental sepsis model in astrocytes that costimulated with LPS and IFN-γ to further investigate the protective role of caffeine citrate on astrocytes.
Plenty of studies on sepsis patients and animal models have demonstrated that mitochondrial damage seems to be associated with the progression and outcome of sepsis (Eyenga and others 2018). A latest research reported that mitochondrial disorders could result in SAE, including excessive ROS production and reduced MMP. Studies have shown that caffeine has the ability to directly scavenge free radicals (León-Carmona and Galano 2011), and it could recover mitochondrial function and increase cardiac function during sepsis (Verma and others 2009).
Similar to our results, SAE rats exhibited recovery of mitochondrial dysfunction after caffeine citrate treatment. In addition, in vitro experiments further proved that caffeine citrate alleviated mitochondrial dysfunction in astrocytes by inhibiting ROS overproduction and MMP dissipation, and restoring mitochondrial respiratory chain complex activities. This further suggested that caffeine citrate could protect mitochondrial function of astrocytes against SAE. Astrocytes play a critical role in supporting neuronal activity and maintaining an optimal neuronal environment. These functions require energy and hence depend on the functionality of the mitochondrial network (Kubik and Philbert 2015).
Neuroinflammation is considered to be a possible pathogenic mechanism of SAE with long-term cognitive impairment (Gu and others 2021). NLRP3 inflammasome is the most extensively studied inflammasome. Once activated, NLRP3 will bind to ASC and activate pro-caspase-1. The activated caspase-1 rapidly converts pro-IL-1β and pro-IL-18 to IL-1β and IL-18, respectively. Subsequently, inflammatory factors are released extracellularly, leading to inflammation (Meyers and Zhu 2020). In addition, inflammatory factors, especially high levels of IL-1β, can inhibit the proliferation of neural progenitor cells and the growth of neurospheres, ultimately leading to reduced neurogenesis (Green and others 2012).
It was reported that NLRP3 inflammasome activation induced inflammation and promoted the excessive release of cytokines, resulting in brain injury in SAE (Fu and others 2019). Previous studies found increased levels of IL-18 and IL-1β in multiple organs or the serum of patients with sepsis (Dolinay and others 2012; Wu and others 2019; Shao and others 2020).
The clinical results from our study showed a significant increase in IL-18 and IL-1β secretion in cerebrospinal fluid of patients with SAE, indicating the potential involvement of NLRP3 inflammasome activation and related inflammatory factors in SAE pathogenesis. These findings suggested that NLRP3 inflammasome may serve as therapeutic targets for SAE. Moreover, the usefulness of the inhibition of NLRP3 pathway for the treatment of SAE has been reported (Sui and others 2016). Therefore, on the mechanism, we wondered whether caffeine could act on this pathway and thus alleviate SAE. This promoted us to establish SAE rat models and challenge our hypothesis in vivo.
The experimental data demonstrated the activation of NLRP3 inflammasome and the release of IL-18 and IL-1β in SAE rats. Additional, caffeine citrate could attenuate above changes, suggesting that caffeine citrate may be a potential treatment for SAE. Moreover, the activation of NLRP3 also contributes to the activation of astrocytes. Astrocyte-mediated inflammation plays a crucial role in the pathogenesis of SAE.
In our study, we have further proved that caffeine citrate inhibited SAE-induced activation of NLRP3/caspase-1 pathway and decreased inflammatory factors' release in astrocytes. These data revealed that caffeine citrate might attenuate neuronal apoptosis, astrocyte activation, as well as neuroinflammation by the inhibition of NLRP3/caspase-1 pathway in astrocytes. The inhibitory role of caffeine citrate on the activation of NLRP3 has been demonstrated in several cell or disease models (Zhao and others 2019; Wang and others 2022).
In addition to its ability to induce mitochondrial dysfunction, the production of ROS is considered to be a common activator of NLRP3 inflammasome by promoting the formation of caspase-1/ASC complexes (Heid and others 2013), whereas the upstream mechanisms regulating ROS production remain uncertain. It was reported that ROS production could be regulated by UCP2 in mitochondria (Arsenijevic and others 2000). Inhibition of UCP2 protein can regulate NLRP3 inflammasome through ROS mediation. Peng and others (2009) have reported that UCP2 played a positive role in sepsis, and its silencing aggravated mitochondrial dysfunction in astrocytes under septic condition (Peng and others 2019).
Previous study has demonstrated that UCP2 negatively regulated the activation of NLRP3 in astrocytes in depressed mice model (Du and others 2016). The results of our study revealed that caffeine citrate could promote UCP2 overexpression and negatively regulate the downstream pathway of NLRP3/caspase-1 to decrease the production of IL-18 and IL-1β, thereby alleviating SAE, indicating that astrocytic UCP2 was involved in the protective mechanism of caffeine citrate.
Conclusion
In conclusion, caffeine citrate plays a protective role against SAE by activating UCP2 expression and thus inhibiting the activation of NLRP3 inflammasome in astrocytes, and then attenuates mitochondrial dysfunction, astrocytic activation, and neuronal apoptosis. Our results suggested that UCP2 might be a therapeutic target for the use of caffeine citrate to treat SAE.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.