
Abstract
Immunoglobulin A (IgA) nephropathy is the most common primary glomerulonephritis worldwide, with no disease-specific treatment and up to 40% of patients progressing to kidney failure. IgA nephropathy (IgAN), characterized by IgA1-containing immunodeposits in the glomeruli, is considered to be an autoimmune disease in which the kidneys are injured as innocent bystanders. Glomerular immunodeposits are thought to originate from the circulating immune complexes that contain aberrantly O-glycosylated IgA1, the main autoantigen in IgAN, bound by IgG autoantibodies. A common clinical manifestation associated with IgAN includes synpharyngitic hematuria at disease onset or during disease activity. This observation suggests a connection of disease pathogenesis with an activated mucosal immune system of the upper-respiratory and/or gastrointestinal tract and IgA1 glycosylation. In fact, some cytokines can enhance production of aberrantly O-glycosylated IgA1. This process involves abnormal cytokine signaling in IgA1-producing cells from patients with IgAN. In this article, we present our view of pathogenesis of IgAN and review how some cytokines can contribute to the disease process by enhancing production of aberrantly glycosylated IgA1. We also review current clinical trials of IgAN based on cytokine-targeting therapeutic approaches.
Introduction
Immunoglobulin A (IgA) nephropathy (IgAN), initially described in 1968 by Drs. Jean Berger and Nicole Hinglais (Berger and Hinglais 1968), is the most common primary glomerulonephritis worldwide (Galla 1995) and an important cause of kidney failure (Galla 1995; Wyatt and Julian 2013; Lai and others 2016). Diagnosis of IgAN requires evaluation of renal biopsy tissue and is confirmed by mesangial deposition of IgA, often with variable codeposits of IgG and/or IgM. Complement C3 is usually present but C1q is typically not detected (Jennette 1988; Miyamoto and others 1988; Wyatt and Julian 1988). Electron microscopy usually shows electron-dense glomerular immunodeposits and variable podocyte injury or loss. The findings by light microscopy show features of mesangioproliferative glomerulonephritis with variable tubulointerstitial injury (Roberts 2014; Trimarchi and others 2017).
IgAN affects individuals of all ages, but is diagnosed most frequently in young adults. Male to female ratio is 2–3:1 in patients of European descent, while both sexes are equally affected in east Asia (Lai and others 2016). Clinical presentation is variable, ranging from mild to severe. Asymptomatic proteinuria and painless hematuria are the most common presentations. A painless episode of macroscopic hematuria often coincides with mucosal infections of the upper-respiratory tract or gastrointestinal tract at disease onset or at periods of disease activity flares (Wyatt and Julian 2013). This synpharyngitic hematuria suggests an involvement of mucosal inflammation in the disease process (Knoppova and others 2016). Prevalence of IgAN varies between different parts of the world, being highest in east Asia and lowest in central Africa (Kiryluk and others 2012).
There is currently no disease-specific therapy for IgAN and up to 40% of patients progress to kidney failure within 20 years since diagnosis (Berthoux and others 2008; Wyatt and Julian 2013; Lai and others 2016). Consequently, diagnosis of IgAN reduces life expectancy by a decade on average (Hastings and others 2018). Risk factors for disease progression include reduced renal function at diagnosis, high blood pressure, IgG codeposits by routine immunofluorescence (Coppo and others 2014; Knoppova and others 2016, 2021; Trimarchi and others 2017; Rizk and others 2019), as well as some histopathological features, namely the MEST-C classification by light microscopy, initially known as the Oxford classification (Cattran and others 2009; Roberts and others 2009).
Standard of care includes aggressive control of blood pressure, as well as proteinuria, to a goal <0.5 g/day using primarily inhibitors of the renin angiotensin aldosterone system (RAAS) (Rovin and others 2021). New therapies targeting the disease pathogenesis are emerging and are being tested in clinical trials (Cheung and others 2021). Renal replacement therapy (ie, dialysis, transplantation) is a life-saving treatment, although IgAN recurs in many transplant recipients (Berger 1988; Canaud and others 2012; Rajasekaran and others 2021; Uffing and others 2021).
In IgAN, the kidneys are thought to be injured as “innocent bystanders,” as the cause of injury is due to deposition of pathogenic IgA1-containing immune complexes formed in the circulation. It is hypothesized that aberrantly O-glycosylated IgA1, recognized as an autoantigen, provides a “seeding” event that initiates the immune-complex formation (Suzuki and others 2011). In this article, we will review current understanding of the pathogenesis of IgAN and discuss how some cytokines are involved in the overproduction of aberrantly O-glycosylated IgA1, the main autoantigen in the disease. We will also provide an update on clinical trials that utilize some of these cytokines as therapeutic targets in IgAN.
Pathogenesis of IgAN
IgAN is characterized by IgA-containing glomerular deposits. This IgA is exclusively of IgA1 subclass (Conley and others 1980) and is enriched for glycoforms with some hinge-region O-glycans deficient in galactose (Conley and others 1980; Allen and others 2001; Hiki and others 2001). The current hypothesis for immunological pathogenesis of IgAN postulates that IgA1 molecules with hinge-region O-glycans deficient in galactose (Gd-IgA1) are recognized by autoantibodies, mostly of IgG isotype, resulting in the formation of immune complexes in the circulation (Fig. 1) (Suzuki and others 2011). Additional proteins, such as complement, in the blood can bind to these IgG-IgA1 circulating immune complexes, and some of these complexes deposit in the kidneys.
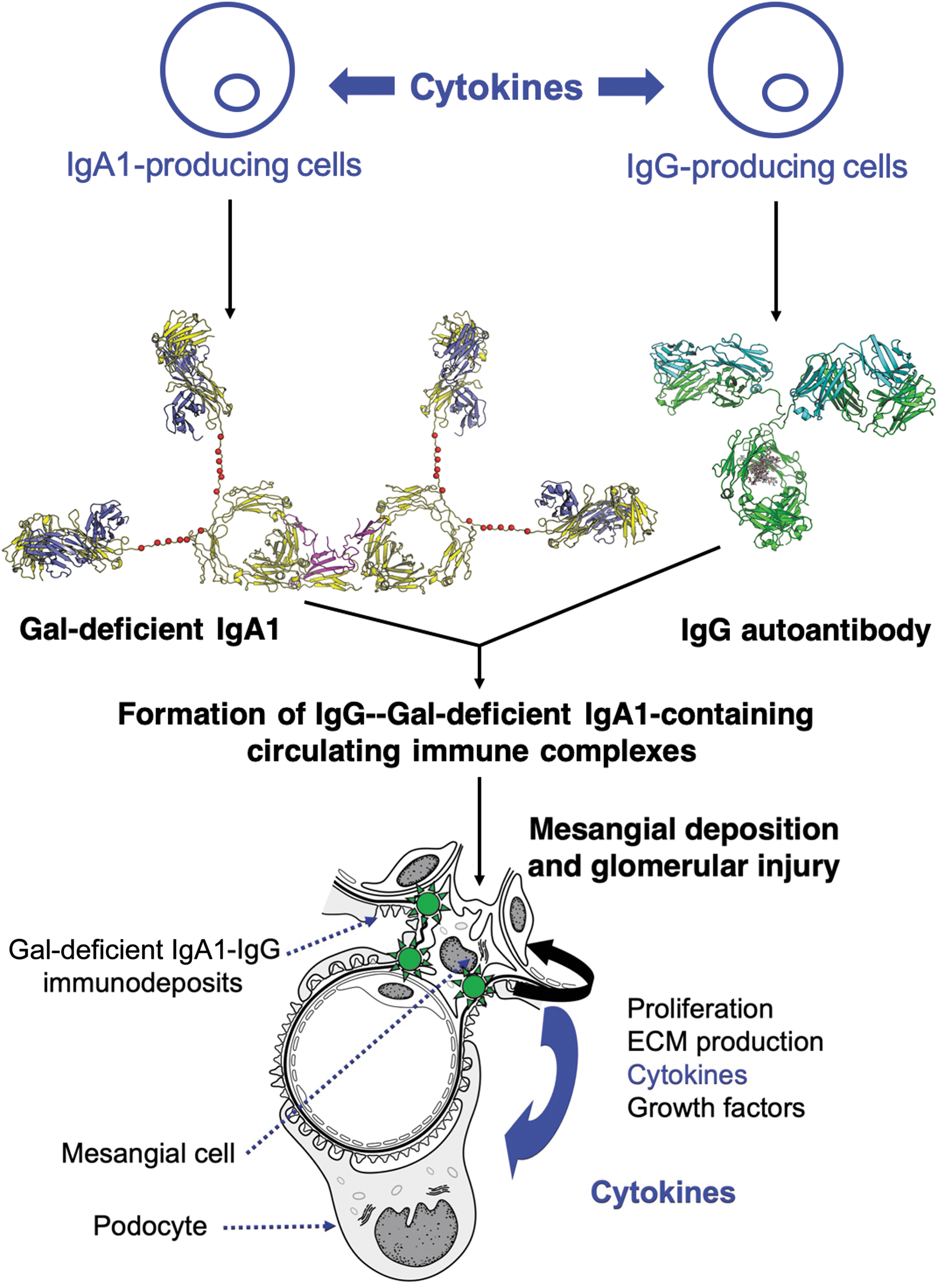
Model of pathogenesis of IgAN. IgAN is considered an autoimmune disease with a genetically and environmentally codetermined multistep process (Suzuki and others 2011). Polymeric IgA1 with some O-glycans deficient in galactose [dimeric galactose (Gal)-deficient IgA1 is shown], often elevated in the circulation of patients with IgAN, is recognized by IgG autoantibodies specific for Gal-deficient IgA1. Development of cells producing Gal-deficient IgA1 and IgG autoantibodies is affected by multiple cytokines during their differentiation and maturation from B cells into the corresponding plasma cells. Some of these cytokines may affect glycosylation processes and further enhance production of Gal-deficient IgA1. IgG bound to Gal-deficient IgA1 forms an immune complex to which additional serum proteins are added (eg, complement). Some of these immune complexes deposit in the kidneys and activate mesangial cells. The activated mesangial cells proliferate and overproduce cytokines, some of which activate podocytes, leading to glomerular injury. The composite dimeric Gal-deficient IgA1 was generated based on the structures of the dimeric Fc/J chain complex [PDB ID:6UE7, (Kumar and others 2020)] and solution structure of IgA1 [PDB ID:1IGA, (Boehm and others 1999)]. Most-often utilized O-glycosylation sites in the hinge region of IgA1 are noted with red spheres (Ser/Thr residues at 225, 228, 230, 232, 233, 236). J chain is colored magenta, while heavy and light chains of IgA1 are colored yellow and slate. The model of IgG was generated with PDB coordinates from PDBID:1IGT (Harris and others 1997). IgG heavy and light chains are colored green and cyan, respectively. Adjustments of hinge orientation were made to avoid steric clash. IgAN, IgA nephropathy.
Mesangial cells, thought to be the primary target cell type in the glomeruli, are activated by the soluble IgA1-IgG immune complexes (Ebefors and others 2016). The activated mesangial cells start to proliferate and produce components of extracellular matrix, as well as growth factors and cytokines, that, in turn, activate podocytes. The mesangial secretion of cytokines, such as TGF-β, tumor necrosis factor (TNF)-α, and other cytokines, such as IL-6 and IL-1, may cause podocyte injury, leading to foot-process retraction and/or podocyte detachment and, consequently, proteinuria (Suzuki and others 2011; Wyatt and Julian 2013; Lai and others 2016; Liu and others 2017). Their secretion may contribute to the overall increase of these cytokines that are found in the circulation of IgAN patients.
One major question involving the pathogenesis of IgAN is the origin of B cells that produce Gd-IgA1. IgA is the primary antibody produced in mucosal tissues. However, it is not known where specific subsets of Gd-IgA1 producing B cells in IgAN patients originate. In the literature, it is suggested that these B cells are migratory and can localize in different germinal centers in mucosal tissues around the human body. Nasopharyngeal associated lymphoid tissue and tonsillar derived B cells have been shown to migrate from the mucosal tissues where they differentiated to other sites of inflammation, including the bone marrow (Muto and others 2017). In addition, bone marrow transplantation studies between IgAN donors and healthy control donors in humans and mice have shown that transplantation from an IgAN donor causes disease recurrence in recipients (Rodas and others 2020; Maixnerova and others 2021). This suggests that any Gd-IgA1 producing B cells may be able to localize in the bone marrow, and the source of Gd-IgA1 may be coming from that location (Muto and others 2017).
Two new studies have shown that B cells in IgAN patients are enriched for homing markers of specific mucosal tissues. A 2021 study demonstrated that B cells with gut homing markers CCR9+ β7 integrin+ cells were increased in IgAN patients (Sallustio and others 2021). Those findings support the role of an intestinal-microbial-mucosal axis in the pathogenesis of IgAN, which will be discussed in a later section. In addition, a very recent 2022 study demonstrated that the peripheral blood of IgAN patients was enriched for the marker of gut homing (CCR9+), but also the upper respiratory tract as well (CCR10+) (Zachova and others 2022). Together, these findings suggest that B cells in IgAN patients may be migratory and, during times of infection, can migrate from the bone marrow to other mucosal tissues and produce Gd-IgA1.
There is also evidence of B cells residing in the kidneys. Cytokines that are induced in mesangial cells by IgA or IgA containing immunodeposits include IL-6 and may act on the B cells that are present in the kidneys or may contribute to the overall increase of IL-6 in the circulation of IgAN patients (Schmitt and others 2014; Zhang and others 2020).
Early studies led to the discovery and detailed characterization of IgG autoantibodies that bind aberrantly glycosylated IgA1 and lead to immune-complex formation (Jackson 1988; Tomana and others 1997, 1999; Novak and others 2002, 2005; Suzuki and others 2009). The hypothesis for the pathogenesis of IgAN is based on the production of the 2 key elements, the autoantigen (Gd-IgA1) and the corresponding IgG autoantibodies (Suzuki and others 2011). Multiple lines of evidence provide support for this hypothesis, such as a correlation of serum autoantigen levels, IgG-autoantibodies, and IgA1-IgG immune complexes, with disease progression, disease severity, as well as disease recurrence after transplantation (Placzek and others 2018; Maixnerova and others 2019; Knoppova and others 2021). In addition, IgG codeposits are found by routine immunofluorescence in around 50% of patients with IgAN who have elevated serum levels of the autoantigen and are associated with poor outcomes (Eison and others 2012; Wyatt and Julian 2013; Knoppova and others 2016, 2021).
A more recent study revealed that IgG extracted from glomerular immunodeposits of patients with IgAN is enriched for autoantibodies specific for aberrantly glycosylated IgA1 (Rizk and others 2019). Moreover, the same study detected IgG in glomerular immunodeposits of all patients, including those without detectable IgG by routine immunofluorescence microscopy. IgA and IgG were colocalized in the glomeruli, supporting the conclusion that these immunoglobulins comprised immune complexes (Rizk and others 2019). Further support for the pathogenic potentials of IgG autoantibodies and IgA1 with hinge-region O-glycans deficient in galactose came from experiments in an animal model (Moldoveanu and others 2021). Specifically, immune complexes formed in vitro from IgG autoantibodies and Gd-IgA1 were injected into mice and assessed for their nephritogenic potential. The immune complexes induced glomerular pathological changes, whereas individual immunoglobulins (IgA1 alone or IgG alone) did not (Moldoveanu and others 2021).
Although the importance and specific steps of immune-complex formation and mesangial-cell activation have been studied in the last 2 decades, there are many gaps in our knowledge as to their composition and biological activity (Knoppova and others 2016). In addition, little is known about the biochemical processes leading to the production of Gd-IgA1. The factors that drive development and maturation of naive B cells into IgA1-secreting cells that produce IgA1 with aberrant O-glycans remain unknown (Fig. 1). Also unknown is whether the processes are T cell-dependent or independent or both. We will focus our review on the cells producing aberrantly glycosylated IgA1 and the cytokine-mediated effects contributing to the production of these IgA1 glycoforms.
Human IgA Structure and Glycosylation
IgA in humans and higher primates exists in 2 subclasses, IgA1 and IgA2 (Reily and others 2019). Both subclasses are found in the circulation and mucosal secretions. IgA in peripheral blood is predominantly of IgA1 subclass (∼90%) with a minor contribution of IgA2 (10%). Representation of the IgA subclasses in mucosal secretions is more variable, with IgA2 reaching 50% of total IgA in some mucosal sites. Humans produce ∼70 mg of IgA per kg of body weight per day, reaching average IgA concentration of 1–3 mg per mL of serum. Circulatory IgA has a half-life of ∼5–6 days, due to its fast catabolism in the liver (de Sousa-Pereira and Woof 2019).
IgA in the circulation is predominantly in the monomeric form (∼90%), consisting of 2 heavy chains and 2 light chains connected by disulfide bridges. A small proportion of circulatory IgA (∼10%) is dimeric (also called polymeric), in which a joining chain (J chain) connects 2 monomers through disulfide bridges with their tail pieces. Another molecular form of IgA in the circulation is IgA bound in immune complexes (<1%) (Knoppova and others 2016).
IgA in secretions, called secretory IgA, is polymeric (dimeric or trimeric J-chain-containing molecules) with an additional protein attached, termed secretory component. The secretory component of secretory IgA (as well as of secretory IgM) is derived from the polymeric immunoglobulin receptor and attached during transcytosis of polymeric IgA through mucosal epithelial cells onto the mucosal surface layer (de Sousa-Pereira and Woof 2019).
It is thought that circulatory IgA is produced mainly by IgA-secreting cells in the bone marrow as monomeric IgA, whereas secretory IgA is produced as polymeric IgA by IgA-secreting cells in mucosal tissues (de Sousa-Pereira and Woof 2019). This polymeric IgA is mostly produced within lamina propria of the small and large intestines by IgA-secreting cells and binds to polymeric immunoglobulin receptor on the mucosal epithelial cells, initiating the first step of transcytosis of polymeric IgA. Due to the mucosal site of production and required transcytosis for secretory-component attachment to polymeric IgA, the amount of secretory IgA in the circulation is minimal.
IgA1 and IgA2 are ∼80% similar in amino acid sequences; the main differences include the respective hinge-region segments and N-glycosylation sites. IgA1 has an extended hinge region with 9 Ser and Thr residues of which 3–6 carry O-glycans (Fig. 2), whereas IgA2 has a short hinge region without O-glycans. Conversely, IgA2 has 5 N-glycans per heavy chain, whereas IgA1 has only 2. N-glycans can have variable composition, often being bi-antennary sialylated glycans. The hinge-region O-glycans of circulatory IgA1 are core 1 glycans, that is, N-acetylgalactosamine (GalNAc) with β1,3-linked galactose, and one or both sugars may be sialylated. Galactose can contain α2,3-linked sialic acid, whereas GalNAc can have α2,6-linked sialic acid. Some O-glycans can have only terminal GalNAc or sialylated GalNAc; these glycoforms are termed galactose deficient (Suzuki and others 2011; Wyatt and Julian 2013; Reily and others 2014; Knoppova and others 2016).
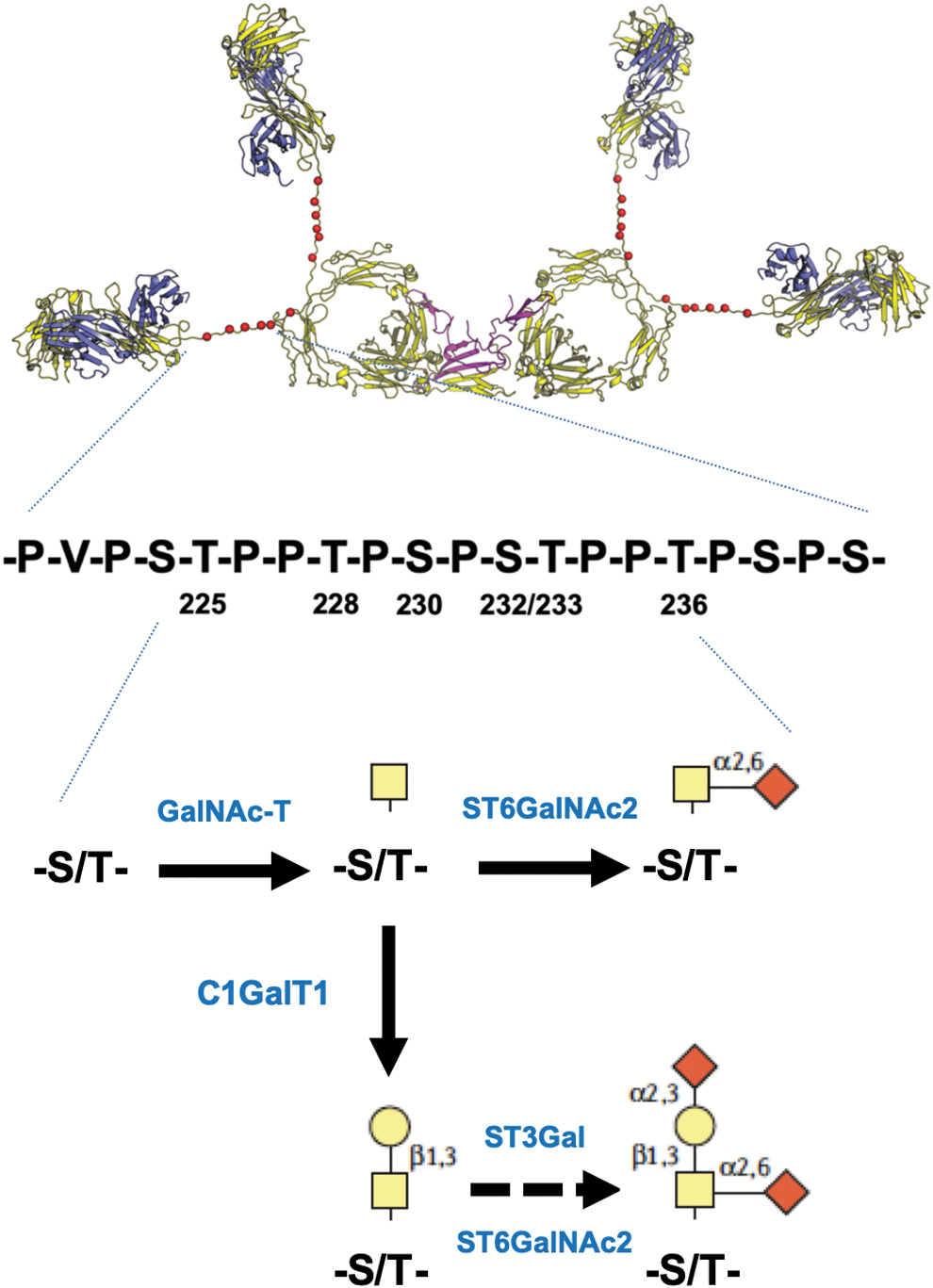
Structure and O-glycosylation of human IgA1. A model shows dimeric IgA1 with the 6 most-often utilized O-glycosylation sites in each hinge region of IgA1 marked with red spheres (Ser/Thr residues 225, 228, 230, 232, 233, 236). Amino-acid sequence below the model of IgA1 shows those residues. Each of those 6 sites can be utilized or remain unglycosylated; hinge-region glycopeptides usually carry 3–6 O-glycans (Reily and others 2019). Each glycosylated site can contain N-acetylgalactosamine (GalNAc; yellow square) without or with α2,6-linked sialic acid (diamond). The latter, sialylated GalNAc, cannot be further modified. However, GalNAc in the hinge region of IgA1 can be modified by β1,3-linked galactose (yellow circle), so called core 1 O-glycan. This disaccharide can be sialylated, by α2,6-linked sialic acid at GalNAC and/or α2,3-linked sialic acid at galactose. Enzymes involved in the step-wise biosynthesis of IgA1 O-glycans in IgA1 producing cells are noted in the figure in blue font. For details, see (Reily and others 2019).
IgA subclasses and their glycosylation and molecular form impact biological activity and effector functions. For example, IgA2 is more effective than IgA1 in inducing pro-inflammatory responses in neutrophils and macrophages. This difference is related to more extensive sialylation of N-glycans of IgA1 than IgA2 (Steffen and others 2020).
IgA N-glycans and O-glycans are attached and modified in a semistochastic manner in the endoplasmic reticulum and Golgi apparatus of IgA1-producing cells. The resultant glycosylation of IgA depends on expression and activity of specific enzymes, glycosyltransferases (and also glycosidases for N-glycans). Changes in expression and activity of some enzymes can alter the representation of specific glycoforms and can result, for example, in overproduction of galactose-deficient IgA1 glycoforms in patients with IgAN.
Production of galactose-deficient IgA1 occurs due to dysregulated expression and activity of several key glycosylation enzymes in IgA1-secreting cells (Suzuki and others 2008, 2011, 2014). Reduced expression and activity of galactosyltransferase C1GalT1 and elevated expression and activity of sialyltransferase ST6GalNAc2 have been found in the cells from IgAN patients that produced elevated galactose-deficient IgA1 compared to the cells from healthy controlled that produced normally galactosylated IgA1. In addition, the chaperone protein specific for C1GalT1, C1GalT1C1 also called Cosmc, and its expression is also downregulated in these cells (Suzuki and others 2008).
Multiple micro RNAs (miRNAs) are involved in the regulation of B cell and plasma-cell development, as well as in B cell receptor signaling, through regulating downstream signaling pathways (eg, PI3K-AKT, NF-κB, and RAS-MAPK) (Xiao and others 2020; Borbet and others 2021). Those B cell regulatory miRNAs include miR-17–92, miR-29, miR-29b, miR-30a, miR-146a, miR-148a, miR-155, miR-181b, miR-185, miR-210, miR-217, miR-326, and miR-1246 (Xiao and others 2020; Borbet and others 2021). Relevant to this review, some miRNAs are involved in regulation of IgA1 O-glycosylation. For example, upregulation of miR-148b directly correlates with levels of galactose-deficient IgA1 (Serino and others 2012), whereas inhibition of miR-374b increases PTEN (Phosphatase and Tensin Homolog) and COSMC expression and reduces aberrant glycosylation of IgA1 (Hu and others 2015). Multiple miRNAs also regulate COSMC expression, including miR-320, miR-196b, and miR-33a-3p (Li and others 2018; Sun and others 2020).
The miRNA, let-7b, is upregulated in peripheral blood mononuclear cells of patients with IgAN and reduces expression of GALNT2, the gene encoding GalNAc-T2, the main enzyme initiating IgA1 O-glycosylation (Serino and others 2015, 2016). Furthermore, miR-148b modulates the levels of secreted galactose-deficient IgA1 by reducing C1GALT1 expression (Serino and others 2012). Other miRNAs can have their effects mediated by cytokines. For example, miR-98-5p reduced CCL3 expression and thereby increasing IL-6 levels that, in turn, decrease C1GALT1 expression (Liu and others 2020a). miR-630 is overexpressed in palatine tonsillar mononuclear cells from IgAN patients, reducing expression of TLR4 and production of IgA1 while increasing the level of IgA1 galactosylation (Liu and others 2020b). Thus, several miRNAs can have differential effects on IgA1-producing cells and O-glycosylation of IgA1 secreted by these cells.
Genetics of IgAN: Glycosylation of IgA1 and Cytokines
Genetic variability surrounding immune functions in IgAN has implicated a wide variety of processes that are critical for proper maintenance and homeostasis of immune system. We know, from the initial discovery of familial forms of IgAN and other studies, that the disease has genetic components; also, serum levels of galactose-deficient IgA1 are heritable, independent of kidney function (Julian and others 1985; Gharavi and others 2008; Hastings and others 2010; Kiryluk and others 2012; Kiryluk and Novak 2014). In addition, patients with IgAN with blood relatives also with IgAN have a worse 20-year renal survival compared to the patients with sporadic IgAN (41% vs. 90%, P = 0.003) (Schena and others 2002). These studies thus highlight the importance of genetic components in disease susceptibility and progression and underscore the need for further investigations into the relationship of IgAN with immunological function.
Serum levels of galactose-deficient IgA1 represent a heritable trait in patients with familial, as well as sporadic, IgAN (Gharavi and others 2008). More recently, genome-wide association studies (GWAS) identified single-nucleotide polymorphisms (SNPs) in the C1GALT1 and C1GALT1C1 loci that are related to the expression of the respective genes and are associated with serum levels of Gd-IgA1 (Gale and others 2017; Kiryluk and others 2017). Specifically, a SNP associated with a reduced expression of C1GALT1 and C1GALT1C1 would result in decreased C1GalT1 activity and, thus, addition of less galactose to GalNAc in the IgA1 O-glycans.
This SNP-associated differential expression C1GALT1 and C1GALT1C1 genes that encode proteins involved in O-glycosylation of IgA1 explain part of the variability of serum levels of Gd-IgA1. In addition, these genetic mutations can be further impacted by other factors, such as genetic variants of other associated genes (Kiryluk and others 2013; Kiryluk and Novak 2014; Sanchez-Rodriguez and others 2021). Additional effects may come from some cytokines that may further modulate glycosyltransferase expression and, thus, the degree of galactose deficiency of IgA1.
In addition, a recent GWAS study confirmed the association of Gd-IgA1 serum levels with C1GALT1 SNPs and found a link with SNPs in the GALNT12 locus (Wang and others 2021). The GALNT12 protein is an N-acetylgalactosaminyltransferase responsible for addition of GalNAc to Ser/Thr residues. GALNT12 exhibits genetic interactions with C1GALT1 in serum levels of galactose-deficient IgA1 and disease risk in IgAN. GALNT12 expression in patients with IgAN was lower compared with that of healthy controls (Wang and others 2021). This is the first time GALNT12 has been associated with IgAN, but the effect of cytokines on expression of GALNT2 is unknown (Buck and others 2008; Serino and others 2015).
A growing number of GWAS publications revealed associations of IgAN with multiple loci, including genes involving complement regulation, major histocompatibility complex, and various other aspects of innate and acquired immunity. KEGG gene enrichment analysis of these genes (eg, APRIL, CARD9, ITGAM, DEFA, VAV3, LIF, OSM) identified “Intestinal immune network for IgA production” and “Cytokine—cytokine-receptor interactions” and “JAK/STAT signaling” as the top dysregulated pathways (Kiryluk and others 2014).
Among the loci associated with IgAN and serum IgA is the TNFSF13 gene, encoding APRIL (a proliferation-inducing ligand) (Yu and others 2011; Kiryluk and others 2014). APRIL is a cytokine that can activate B cells and induce proliferation and generally maintain homeostasis of B cells and plasma cells (Baert and others 2018). Two other cytokine-related genes are in HORMAD2 locus also associated with IgAN, LIF (leukemia inhibitory factor), and OSM (oncostatin M).
Cytokines in B Cell Development and Differentiation
B cells exist in a spectrum of differentiation states: from the earliest B cell precursors through complex developmental and differentiation checkpoints during immune responses that include formation of long- or short-lived antibody-secreting cells (ie, plasma cells) and memory B cells (Akkaya and others 2020). Like all lymphocytes, B cells are guided through development and differentiation by signals derived from other cells of the adaptive and innate immune system. The interface of B cells with the systemic immune system is complex and involves many cellular communication mechanisms, including adhesion molecules and growth factors; however, cytokines play an outsized role.
Cytokine is a broad term describing soluble proteins that alter cellular behavior through interactions with specific receptors. Several families have been described, categorized by structural similarities of the cytokines themselves, their receptors, as well as the signal transduction cascades that are induced upon receptor binding. Broadly, cytokines include interleukins (ILs), interferons, TNF family, lymphokines, and chemokines.
In the context of B cell fate decisions, terminal differentiation, and antibody production, Type I cytokines and members of the TNF superfamily are critical. Type 1 cytokines include several subgroups (1–5) classified by the composition of the receptor complex. These receptors may be homodimers, in the case of group 1, or heterocomplexes which utilize a common signal transduction subunit, as is the case for the common γ-chain receptors (IL-2R, IL-4R, IL-7R, IL-9R, IL15R, and IL-21R), or the gp130 in the IL-6 family of group 2 type 1 cytokines (Liongue and Ward 2007). In these cases, the signal transduction is mediated by the common chain, while cytokine specificity is determined by the other receptors that comprise the heterocomplex (Ishihara and Hirano 2002).
Type 1 receptor complex with Janus kinases (JAK) results in the phosphorylation of STAT family transcriptional activators (Fig. 3, for example of IL-6). Some of these cytokines have important roles in humoral response, including IL-2, IL-4, IL-6, IL-10, IL-12, IL-13, IL-15, and IL-21 (Moens and Tangye 2014). Signals integrated through these cytokines influence the behavior of activated B cells, impact proliferation and costimulatory receptor expression, induce class-switch recombination, and guide isotype-switch decisions (Xu and others 2012).
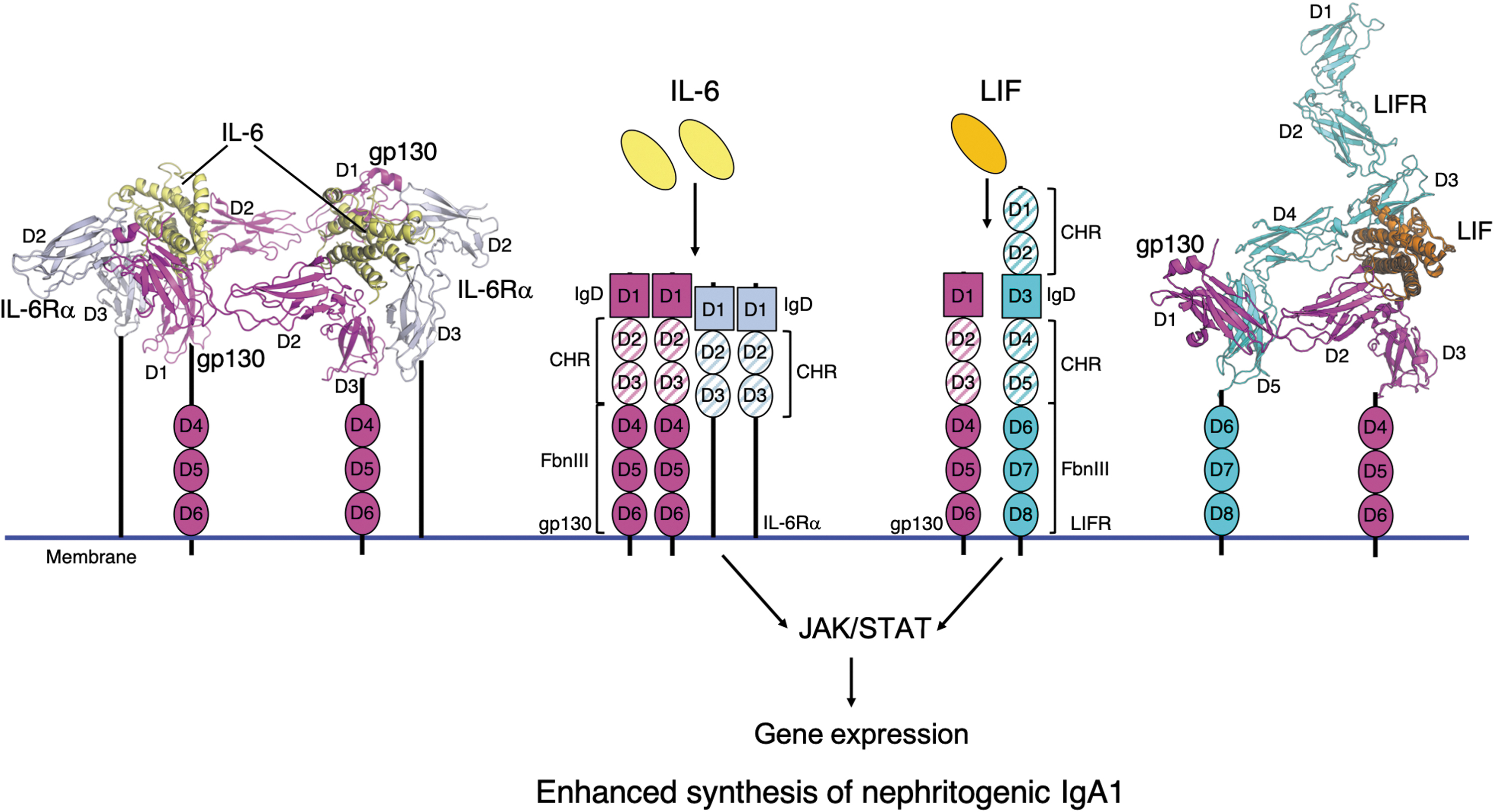
Cytokines IL-6 and LIF in complexes with the corresponding receptors as the initiation of JAK/STAT signaling pathways. Cytokines IL-6 and LIF, upon binding to the receptors, can activate JAK/STAT pathway, leading to altered expression of some genes. In IgA1-producing cells in patients with IgAN, this signaling process is enhanced, leading to dysregulation of specific glycosylation enzymes and overproduction of nephritogenic IgA1 glycoforms. IL-6 and LIF bind to receptor complexes composed of gp130/IL-6Rα and gp130/LIFR, respectively. The center of the figure illustrates the components of each receptor complex. The extracellular portions of gp130, IL-6Rα, and LIFR are composed of evolutionarily conserved fibronectin type III (FbnIII) β-sandwich domains, the number of which varies for each receptor. FbnIII domains that are involved in cytokine binding are denoted as CHR domains. In addition to FbnIII domains, each receptor has an Ig-like domain (IgD). IgD and additional FbnIII domains can contribute to cytokine binding and receptor complex assembly. The IL-6/gp130/IL-6Rα complex is composed of 2 copies of each protein to form a hetero-hexameric complex as shown to the left, with IL-6 (yellow), gp130 (magenta, D1-3), and IL-6Rα (light blue, D2-3). Individual observed domains are labeled, with D3 of gp130 and D3 of IL-6Rα illustrated proximal to the membrane and additional domains leading to the membrane schematically illustrated. To the right, the gp130/LIFR/LIF complex is shown as the ternary complex with each protein shaded magenta, cyan, and orange, respectively. The cytokines, IL-6 and LIF, are formed of α-helical bundles. The following protein coordinates were used to generate models in this figure: hexameric human IL-6/IL-6Rα receptor/gp130 complex [PDB ID: 1P9M, (Boulanger and others 2003a)], LIFR [PDB ID:2Q7N, (Huyton and others 2007)], and LIF/gp130 [PDB ID:1PVH, (Boulanger and others 2003b)]. The composite gp130/LIFR/LIF structure was generated by alignment of LIF in the LIF/LIFR and LIF/gp130 according to methods in reference (Huyton and others 2007). CHR, cytokine-binding homology region; LIF, leukemia inhibitory factor.
In contrast to Type 1 cytokines, the TNF superfamily of cytokines includes cell bound, as well as soluble, ligands. These receptors mediate signal transduction through the recruit of death domain adaptors upon ligand binding. Although the first characterized member of this family, tumor necrosis factor, and FAS induce apoptosis by activating caspases, signals derived from most of these receptors, including BAFF-R, CD40, TACI, and BCMA, which are critical in B cell biology, recruit TRAF adaptor proteins and are potent activators of nonconical NF-κB (Wajant and others 2001). The interaction of CD40 and its ligand CD154 expressed on T cell are critical for germinal center B cells and represent a principal mechanism by which B cells receive T cell help (Armitage and others 1993; Kawabe and others 1994; Xu and others 1994; Quezada and others 2004).
In contrast, soluble BAFF (and the related APRIL; Fig. 4) is constitutively required for survival of follicular B cells, and its relative abundance appears to be a rheostat for peripheral B cell compartment (Moore and others 1999; Schneider and others 1999; Batten and others 2000; Thompson and others 2000). Unlike BAFF-R, the BCMA receptor is not expressed by resting B cells, but rather upregulated in germinal-center B cells and is essential for the maintenance of plasma cells (O'Connor and others 2004). Collectively it is the integration of signals derived through the receptors, in addition to B cell receptor mediated activation, that governs the behavior of B cells that ultimately shapes humoral immunity.
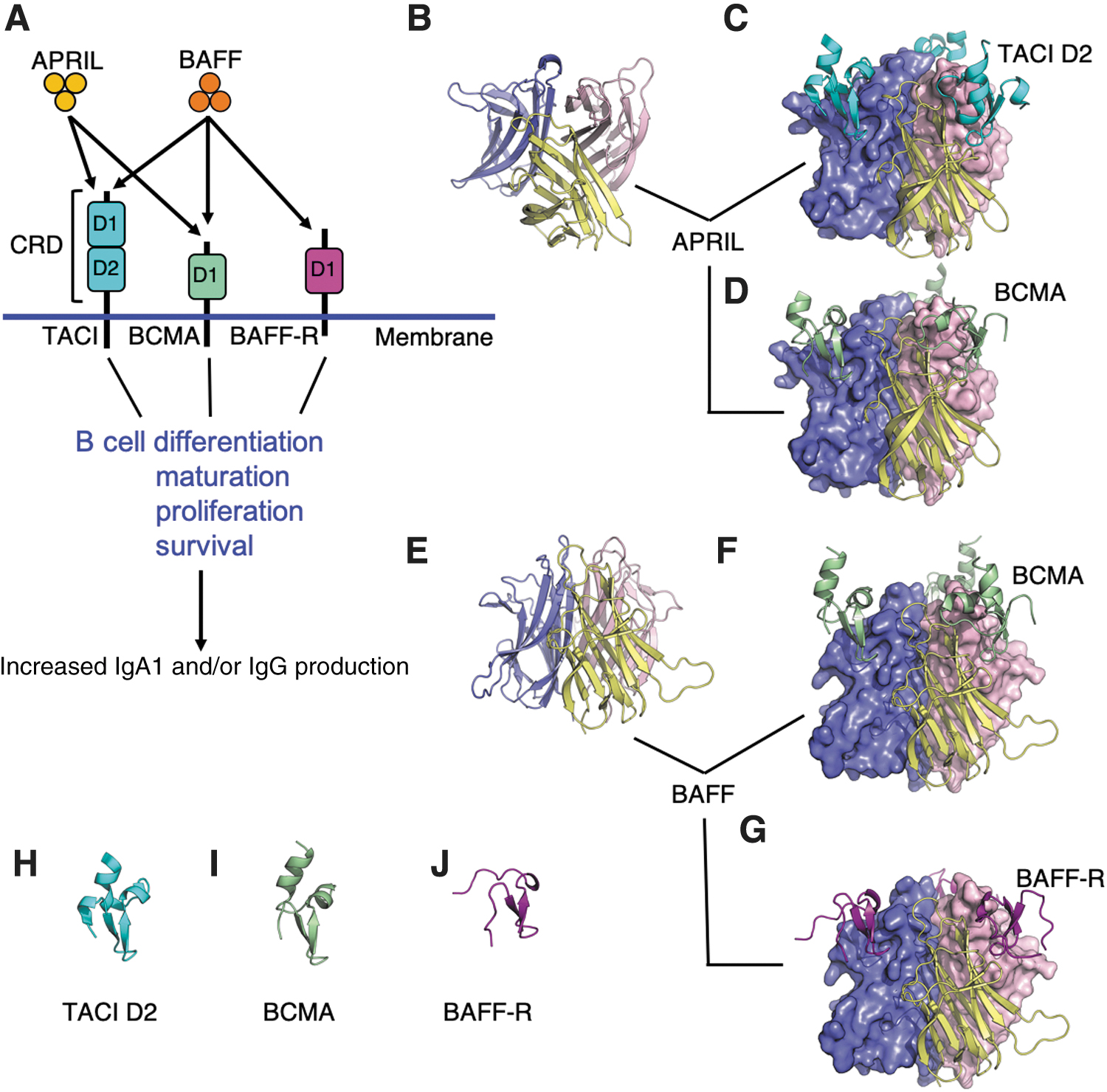
APRIL and BAFF, cytokines and their receptors involved in B cell maintenance. Cytokines, including a proliferation inducing ligand (APRIL) and B cell activating factor (BAFF), are important for B cell maintenance. In
Cytokines and Production of Aberrantly Glycosylated IgA1
Clinically, it is well recognized that one of the first clinical manifestations of IgAN is a mucosal infection, typically an upper-respiratory-tract infection that occurs concurrently with macroscopic hematuria, called synpharyngitic hematuria (Reily and others 2014; Knoppova and others 2016). Synpharyngitic hematuria often recurs and marks the periods of disease activity, with associated increases in serum levels of IgA1 and Gd-IgA1. This observation suggests that the associated pro-inflammatory environment may be contributing to autoantigen production. This further supports a link between the mucosal immune system and enhanced production of Gd-IgA1 that is then bound in the pathogenic circulating immune complexes (Novak and others 2011).
Indeed, it has long been suspected that pro-inflammatory cytokines play a key role in the pathophysiology of IgAN (Rostoker and others 1998). Studies show that there are elevated levels of cytokines in IgAN patients, such as IL-6 and TNFα. Urinary studies showed that patients with elevated IL-6 levels have been associated with worse progression of disease, with higher levels of IL-6 being linked to a higher risk ratio of disease progression to end-stage kidney failure (Harada and others 2002). As noted previously, genomic studies have further established a connection between pro-inflammatory cytokines and IgAN. Specifically, genes encoding LIF and OSM, IL-6 like family of cytokines, are in the disease-associated locus HORMAD2 (Gharavi and others 2011; Kiryluk and others 2014). In vitro studies have revealed that IL-6, as well as LIF, increases production of galactose-deficient IgA1 by IgA1-producing cells from IgAN patients but not from healthy controls.
The galactose deficiency of IgA1 in patients with IgAN is related to the decreased expression and activity of C1GalT1, the enzyme that adds galactose to GalNAc in the hinge-region of IgA1. The deficiency is also related to an increased expression and activity of ST6GalNAc2 (Suzuki and others 2008, 2014; Reily and others 2014). Increased activity of ST6GalNAc2 leads to over-sialylation of GalNAc, contributing to overall under-galactosylation of IgA1 by inhibiting further glycan additions (Suzuki and others 2014). In addition, IL-6 and IL-4 downregulate COSMC expression, leading to enhanced production of galactose-deficient IgA1. Reducing the expression of C1GALT1 or COSMC by siRNA knockdown results in elevated production of Gd-IgA1 (Suzuki and others 2014; Kiryluk and others 2017), confirming the roles of both genes in O-glycosylation of IgA1. IL-6, but not IL-4, also increased activity of ST6GalNAc2 resulting in elevated production of Gd-IgA1. In concordance with enzyme activity levels, IL-6 reduced expression of C1GALT1, as well as its chaperone protein COSMC, and increased expression of ST6GALNAC2 (Suzuki and others 2014).
Recent studies showed that IgA1-producing cells from patients with IgAN had elevated activation of STAT3 and STAT1 in response to IL-6 and LIF, respectively, leading to overproduction of Gd-IgA1 (Yamada and others 2017, 2020). Upon IL-6 binding to the membrane bound IL-6R, gp130 is added and initiates a cascade of signal transduction through the JAK/STAT signaling pathway (Kishimoto and others 1995; Taga and Kishimoto 1997; Heinrich and others 2003) (Fig. 3). JAK is a family of tyrosine kinases and that phosphorylates STATs, leading to their dimerization and nuclear translocation. STAT3 and STAT1 are canonical transcription factors involved in IL-6 and LIF signaling, respectively (Shuai and Liu 2003; Stark and Darnell 2012).
siRNA knockdown and small molecule inhibitors of JAK confirmed that STAT3 was the IL-6-signal mediator and effector of enhanced production of galactose-deficient IgA1, but only in cells derived from patients with IgAN but not those from healthy controls (Yamada and others 2017, 2020). Western blotting revealed that small-molecule inhibitors of JAK attenuated IL-6-induced phosphorylation of STAT3 and also decreased overproduction of galactose-deficient IgA1. Kinome profiling confirmed these findings. Similar to the IL-6 effects, LIF increased production of galactose-deficient IgA1, again exclusively in the IgA1-producing cells derived from patients with IgAN. Enhanced activation of STAT1, the canonical transcription factor for LIF, was found in the cells derived from patients with IgAN compared to those from healthy controls. siRNA knockdown of STAT1 attenuated LIF-mediated production of galactose-deficient IgA1, confirming that STAT1 was the main signal transducer of LIF signaling. However, inhibition of JAK2 did not block LIF-induced overproduction of galactose-deficient IgA1 by the cells from IgAN patients.
Kinome profiling revealed abnormal signaling in several pathways in cells from patients with IgAN, with a possible role for protein tyrosine kinases of the Src family. These experiments together revealed aberrant regulation of cytokine signaling, and associated overproduction of the main autoantigen in IgAN can stem from multiple cytokine pathways (Yamada and others 2017, 2020).
Another locus associated with IgAN contains the TNFSF13 gene encoding APRIL (Gharavi and others 2011; Kiryluk and others 2014) (Fig. 4). As noted above, APRIL is involved in B cell survival and proliferation and is also an important driver for B cell class switching to IgA, in a T cell-independent manner. BAFF cytokine is also a potent B cell survival factor (Stein and others 2002; Zhai and others 2016). BAFF and APRIL share some features (Fig. 4), including several receptors, TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor) and BCMA (B cell maturation antigen), and they activate downstream NF-κB pathway (Reily and others 2014; Zhai and others 2016). Patients with IgAN have elevated APRIL levels in the circulation, and elevated expression of TNFSF13 was observed in the B lymphocytes of patients with IgAN, concordant with the elevated APRIL levels in serum. Increased levels of APRIL positively correlated with levels of galactose-deficient IgA1 in patients with IgAN.
Application of exogenous APRIL increased production of galactose-deficient IgA1 by cells derived from patients with IgAN compared to those from healthy controls (Zhai and others 2016). In support of the idea that pathogenesis of IgAN involves a system wide dysregulation rather than a single abnormality, it has been shown that APRIL expression is affected by other immune system components. A recent study involving IgAN patient tonsillar B cells demonstrated that APRIL is upregulated in germinal center B cells from IgAN patients. In addition, the innate immune toll-like receptor (TLR) 9 is associated with increased APRIL production in these cells as well (Muto and others 2017). This study demonstrated that ligand activation of TLR9 induced prolonged APRIL secretion in tonsillar B cells in IgAN patients.
It was also shown that TLR9 activation induced upregulation of APRIL receptors BCMA and TACI (Muto and others 2017). They hypothesized from these and other findings that activation of TLR9, perhaps through exogenous antigens, is involved in overexpression in APRIL by germinal center B cells in the tonsils, upregulation of TACI and BCMA, and may be involved in an increase in APRIL sensitivity in B cells as well. Continuing with tonsillar mucosal tissue and cytokine signaling, a more recent study showed that when treated with an APRIL-neutralizing antibody, IgA levels in the cell-culture supernatant of TMCs decreased. In addition, serum levels of APRIL pre- and post-tonsillectomy were assessed; serum APRIL levels decreased after tonsillectomy, which may explain some of the positive effects of tonsillectomy in the Japanese population (Takahara and others 2019).
IgA is produced by mucosa-associated lymphoid tissue (MALT). Gut MALT is thought to produce most of IgA in humans. This is another niche in which interactions between the mucosal immune system and microbiota can play a role in IgAN. APRIL and BAFF are prominent cytokines produced in gut mucosal tissue. In this study, these cytokines are responsible for switching B cells to IgA production in a T cell-independent manner after interaction with present commensal microbiota. They are secreted along with additional cytokines by surrounding intestinal epithelial, dendritic, and stromal cells (Kiryluk and Novak 2014). Several studies have cited a possible link between gut mucosal responses to gut microbial populations (McCarthy and others 2011; Chemouny and others 2019). Mice overexpressing human BAFF develop IgA-associated glomerular immunodeposits, dependent on gut microbiota, with elevated levels of serum IgA (McCarthy and others 2011).
It has been shown that in transgenic humanized mouse models of IgAN, depletion of fecal microbiota through administration of antibiotics significantly decreases IgA deposition in their kidneys (Chemouny and others 2019). This suggests that the microbiota present in mucosal tissue may play a role in production of IgA1 in IgAN patients and its subsequent deposition in the kidneys. In humans, it was revealed that phenotypes of gut microbiota were significantly different between IgAN patients who were defined as having progressive disease, nonprogressors, and healthy control patients (De Angelis and others 2014). Combining these two thoughts, a more recent study aimed to determine if modulating the gut microbial population would influence IgA deposition in the IgAN mouse model. Fecal microbiota transplantation was performed with IgAN patients and healthy controls on the transgenic IgAN mouse model. IgAN patients were reported to have elevated levels of BAFF expression, and when those fecal samples were transferred into mice, those mouse groups showed an increase in BAFF levels and nephritogenic complex deposition.
These studies suggests that differences in the gut microbiome may promote differential cytokine responses in IgAN patients versus healthy controls, such as an increase in BAFF (Lauriero and others 2021). Comparison of tonsillar and stool microbiota in IgAN patients with healthy nonrelated individuals revealed significant differences in microbial populations of tonsil samples. In vivo experimentation in BAFF-overexpressing transgenic mice showed that infection with those microbes led to increased IgA deposition in the kidneys of those mice (Currie and others 2022). In addition, BAFF overexpression has been implicated in the production of autoantibodies in autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, and primary Sjogren's syndrome (Pers and others 2005). Together, clinical observations, in vitro and in vivo studies, and genetic/genomic data implicate several cytokines and their associated aberrant signaling in the onset and/or progression and severity of IgAN, possibly related to overproduction of galactose-deficient IgA1.
Cytokine-Targeted Treatments for IgAN in Clinical Trials
The 5 decades since the first description of IgAN (Berger and Hinglais 1968) have witnessed a significant progress in our understanding of the disease pathogenesis. This, in turn, has allowed identification of potential areas for therapeutic interventions in patients at high risk for disease progression. Cytokines that contribute to the formation of precursors of the pathogenic circulating immune complexes are among the considered targets. Of particular interest are two of the cytokines that are important for B cell maintenance, B cell activating factor (BAFF) and a proliferation inducing ligand (APRIL) (Fig. 4). BAFF is also known as TNF ligand superfamily member 13B, B lymphocyte stimulator, and TALL-1. APRIL is also known as TNF ligand superfamily member 13. Both cytokines are critical for the development, maturation, and function of B cells and, thus, have been selected for development of therapeutic inhibitory approaches (Maixnerova and Tesar 2020).
In a phase II randomized double-blind placebo-controlled study (NCT02062684), Blisibimod (a selective BAFF antagonist) was administered subcutaneously to patients with IgAN with persistent proteinuria despite maximally tolerated RAAS blockade. Preliminary interim results presented at the American Society of Nephrology Kidney Week meeting in 2016 showed promising results about Blisibimod leading to a reduction in proteinuria (Huang and Xu 2021). Since then, and although the study has completed enrollment in 2017, no additional results have been reported and the phase 3 trial that was planned has been withdrawn.
VIS649 is a humanized IgG2 monoclonal antibody that binds to and blocks the biological actions of APRIL. A phase I study in healthy volunteers revealed that VIS649 infusions were well-tolerated and safe. VIS649 reversibly suppressed IgA, Gd-IgA1, IgG, and IgM and effectively suppressed free serum APRIL levels (Suzuki and others 2021). The EnVISion Trial (NCT04287985) is a multicenter, randomized, double-blind, placebo-controlled phase II study evaluating the efficacy and safety of multiple doses of VIS649 compared to placebo. Patients enrolled in EnVISion had biopsy-proven IgAN with persistent proteinuria of ≥1 g/d despite RAAS blockade and estimated glomerular filtration rate (eGFR) ≥45 mL/min/1.73 m2. The trial recently completed enrollment. The primary outcome will be the investigational product effect on proteinuria at 12 months. The results of this trial are eagerly awaited and, if successful, the sponsor is gearing to start a phase III trial using a subcutaneous formulation of VIS649 now known as Sibeprenlimab.
BION-1301 is another humanized anti-APRIL IgG4 monoclonal antibody being evaluated in Phase I/II trial (NCT03945318) recruiting healthy volunteers and adults with IgAN, respectively. Eligibility criteria for IgAN patients included eGFR >45 mL/min/1.73 m2, as well as urinary protein ≥0.5 g/24 h, or urinary protein to creatinine ratio (UPCR) ≥0.5 g/g with optimized doses of RAAS blockers. Preliminary results of the trial, presented at the European Renal Association congress in May 2021, confirmed that BION-1301 was well tolerated and led to durable reduction in serum APRIL levels. There was a significant decrease in proteinuria at 12 weeks (Barratt and others 2021). Additional preliminary, although limited, data with subcutaneous formulation of BION-1301 administered every 2 weeks were shared during the ASN Kidney Week meeting in November 2021 and were equally promising.
Atacicept is a recombinant fusion protein designed to inhibit B cells through dual BAFF and APRIL inhibition. A Phase II randomized, double-blind, placebo-controlled study was designed to determine the safety and efficacy of 2 doses of Atacicept (25 and 75 mg) administered subcutaneously in patients with IgAN with persistent proteinuria (NCT02808429). The preliminary interim results for that trial at 24 weeks showed a median reduction in 24-h UPCR of 18.6% and 25.34% with the 25 and 75 mg dosing, respectively. Importantly, this was paralleled by a 25% and 60% drop in the serum galactose-deficient IgA1 during that same timeframe with the 25 mg and 75 mg Atacicept doing, respectively (Barratt and others 2020). With these promising results, a phase IIb trial (NCT04716231) was launched looking at the effect of 3 different weekly doses of Atacicept compared to placebo.
Conclusions and Future Directions
The predominant research areas of IgAN have focused on classification and prognosis of disease based on renal biopsies, biochemical disease pathogenesis, and development of disease-specific biomarkers, such as galactose-deficient IgA1 and the corresponding IgG autoantibodies (Coppo and others 2020; Suzuki and Novak 2021). Over the last decade, as genetic analysis has become more affordable, researchers have made connections between IgAN, inflammation, and other autoimmune diseases related to mucosal immunology (Kiryluk and others 2014). Additional GWAS publications have confirmed the role of some glycosyltransferases in serum levels of Gd-IgA1 (Kiryluk and others 2017). However, in IgAN, biochemical mechanisms that control activity and expression of various glycosyltransferases involved in IgA1 O-glycosylation remain an understudied area. This is also true for research relating to states of inflammation that lead to changes in glycosylation of specific proteins.
Cytokine control of glycosylation is still not well understood, partially due to the difficulties in applications of appropriate analytical techniques (such as mass spectrometry) and to the complexity of the sugar structures. Some of the early work investigating IgA1 glycosylation and cytokines found that Th2 cytokines (IL-4 + IL-5) elicited altered N-glycosylation patterns of IgA in mice (Chintalacharuvu and Emancipator 1997). This use of single versus multicytokine stimulation raises an important question of what the appropriate stimulation should be to better replicate in vivo conditions.
A number of studies using single-cytokine stimulation report alterations in Gd-IgA1 production and important O-glycosylation genes, such as C1GALT1, COSMC, and ST6GALNAC2, but may not accurately reflect what cytokine milieu is critical in enhancing autoantigen production in galactose-deficient IgA1-producing cells (Yamada and others 2010, 2017, 2020; Suzuki and others 2014). A recent study from our laboratory showed that multicytokine stimulation (IL-4, IL-6, IL-21, CD40L) increased Gd-IgA1 production in primary cells from IgAN donors, suggesting that O-glycosylation of the IgA1 hinge region can be modified by cytokine milieu (Reily and others 2018). Better elucidation of the differential mechanisms involved in single versus multicytokine stimulation that control glycosylation will be critical to identifying those targets and pathways that are abnormal in in vivo inflammatory conditions.
One of the major questions in the field of IgAN is the origin of galactose-deficient IgA1 in its primarily polymeric form, a form typically associated with mucosal IgA1. It is not clear whether the polymeric galactose-deficient IgA originates from mucosal-derived IgA1-secreting cells or whether these cells may be relocated in other sites, such as bone marrow. This area is of our acute interest, and new single-cell analytical techniques, such as ATAC-seq with select antigen profiling by sequencing, are being used for ex vivo studies (Mimitou and others 2021). With these single-cell techniques, research in IgAN has started to focus on a few specific glycosyltransferases responsible for aberrant glycosylation in IgA1-secreting cell subpopulations. These different techniques may help determine whether inflammatory stimuli may exert differential signaling depending on the specific subpopulation of B cells or plasma cells, thus leading to enhanced Gd-IgA1 production only in some IgA1-secreting cells (Reily and others 2021).
Further development of disease-specific treatments will need to address specific molecular pathways that are responsible for autoantigen production in those IgA1-producing cells that abnormally respond to pro-inflammatory stimuli and alter expression of critical glycosyltransferases.
Footnotes
Acknowledgment
The authors are grateful to their colleagues and collaborators who have worked with them, as well as to all volunteers and patients with IgAN who provided biospecimens for various studies.
Authors' Contribution
All authors wrote and edited the article. T.J.G. generated the molecular models for figures.
Author Disclosure Statement
J.N. is coinventor on US patent application 14/318,082 (assigned to UAB Research Foundation). D.V.R. and J.N. are cofounders and co-owners of and consultants for Reliant Glycosciences, LLC.
Funding Information
This work was supported, in part, by the National Institutes of Health - DK106341, DK122194, AI149431, DK078244, and DK082753. T.P. was supported, in part, by PRIME T32 DK116672.