
Abstract
Cytokines are powerful mediators of immune responses and some, such as interleukin-2 (IL-2), have achieved dramatic responses as cancer immunotherapies. Unfortunately, systemic administration often results in deleterious side effects, prompting exploration of strategies to localize cytokine activity to the tumor microenvironment (TME). To this end, we constructed an IL-2/IL2Ra fusion protein (IL-2FP) with an MMP2/9-specific cleavage site, designed to exploit the dysregulated protease activity in the TME to selectively activate IL-2 in the tumor. To determine if TME protease activity is sufficient to cleave the FP and if FP activity is due to specific cleavage, we created Colon 38 tumor cell lines expressing similar levels of IL-2FPs with either a functional cleavage site [H11(cs-1FP)] or a scrambled, noncleavable sequence [H2(scramFP)]. H11(cs-1FP) tumors demonstrated reduced tumor growth, characterized by regressions not observed in H2(scramFP) tumors. Analysis through qRT-PCR, flow cytometry, and immunohistochemistry indicate robust CD8 responses in the H11(cs-1FP) tumors. Interferon gamma (IFNg) knockout mice revealed that the immune effects of the cleavable FP are mediated through both IFNg-dependent and IFNg-independent mechanisms. Collectively, these data suggest that matrix metalloproteinases (MMPs) in the TME can cleave the IL-2FP specifically, thus enhancing an antitumor response, and provide a rationale for further developing this approach.
Introduction
Cytokines have long been recognized as key factors in immune responses. Consequently, there have been many attempts at using cytokines for tumor immunotherapy, leading to significant successes resulting in FDA-approved treatments with alpha interferon for hairy cell leukemia and interleukin-2 (IL-2) for melanoma and kidney cancer (Conlon and others 2019). Unfortunately, while there have been dramatic successes with systemic IL-2 treatment, only a fraction of patients respond, and significant side effects limit its utility (Conlon and others 2019).
To avoid deleterious side effects, there have been many approaches to increase the level of cytokines preferentially within tumors. The benefits of local cytokine expression for antitumor responses have been shown experimentally by intratumoral injection with engineered viruses that express cytokines, the delivery of encapsulated microspheres containing cytokines, as well as by gene transfection techniques to genetically modify tumors (Gansbacher and others 1990a, 1990b; Pulaski and others 1993; McAdam and others 1994; Egilmez and others 2000; Rehman and others 2016; Mills and others 2019).
These studies illustrated that altering levels of key cytokines in the tumor microenvironment (TME) can dramatically enhance antitumor responses in the treated tumors. Conceptually, it was hoped that generating a vigorous immune response would subsequently cause regression of even untreated tumors. Unfortunately, the treated tumor is almost always more affected than distal untreated tumors. While limiting in terms of clinical utility, these observations nevertheless also offer strong rationale for developing approaches for altering cytokine levels in the TME.
One of the best-studied systemic approaches for increasing cytokine levels at metastatic tumor sites used cytokines coupled to antibodies. The idea was that the antibody–cytokine complex would circulate throughout the body, eventually accumulating at the tumor site due to specific binding of the antibody portion to an antigen on the tumor, thus increasing the local concentration of the cytokine [reviewed in (List and Neri 2013)]. This conceptually attractive strategy has shown promise (Hank and others 2009; Buhtoiarov and others 2011; Koehn and others 2012; Yang and others 2013). However, since the cytokine is constitutively active, it is accessible to any cells in the blood or tissues with the high-affinity-specific cytokine receptors, which can limit its efficacy and result in unanticipated side effects [discussed in (Skrombolas and Frelinger 2014)].
To minimize unwanted potential side effects of cytokines, we are exploring a fundamentally different strategy to increase the functional activity of cytokines preferentially in the TME. We have created cytokine fusion proteins (FPs) designed to be initially quiescent but functionally activated by proteases (Puskas and others 2011; Skrombolas and others 2019). This approach exploits the dysregulated expression of proteases in tumors that has long been associated, and may even be essential, for their growth and progression (Liotta and Kohn 2001; Kessenbrock and others 2010; Liotta 2016; Vizovisek and others 2021). Because of their potent pleiotropic and irreversible effects, protease activity is normally tightly regulated. However, in tumors, the dysregulated activity of proteases can contribute in multiple ways to the growth of tumors. For example, they can not only degrade basement, but also release growth factors from the extracellular matrix and even activate receptors (Egeblad and Werb 2002; Kessenbrock and others 2010).
One of the most extensively characterized family of tumor-associated proteases are the matrix metalloproteinases (MMPs) (Kessenbrock and others 2010; Vizovisek and others 2021). Much of what is known about the biochemistry of MMPs relies on in vitro studies that have led to the extensive characterization of the catalytic action of the proteases and the definition of a number of cleavage motifs (Kridel and others 2001; Turk and others 2001; Chen and others 2002; Ratnikov and others 2014). Nevertheless, precisely how these studies translate to the cleavage of proteins in vivo requires experimentation, due to the complex network of proteases and inhibitors, the accessibility of cleavage sites in 3-dimensional structures, and the relative concentrations of the enzymes and the substrates (Mason and Joyce 2011; Vasiljeva and others 2019; Vizovisek and others 2021).
We set out to develop an experimental system to test the ability of cleavable IL-2 FPs to be specifically activated by MMPs in the TME and generate antitumor immune responses. We took advantage of previous work that used fluorescent reporters to validate a short peptide sequence that could be cleaved by MMP2/9 as well as a scrambled sequence that was not effectively cleaved (Bremer and others 2001). Extending these results, we incorporated these sequences into IL-2FP expression vector constructs and generated tumor cell lines that produce equivalent amounts of the 2 FPs. Using these matched tumor cell lines, we showed that an IL-2FP can be activated specifically by MMPs to enhance antitumor immune responses. Interestingly, this response has 2 components, one that is dependent upon IFNg and one that is not. These results illustrate one key step in developing an immunotherapy approach that takes advantage of the dysregulated expression of proteases, such as MMPs that are intrinsic to the growth and progression of many tumors.
Materials and Methods
Mice
BALB/cJ and C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and used at 8–10 weeks of age. B6.129S7-Ifngtm1Ts /J (IFNg knockout) mice were a generous gift from Dr. Edith Lord.
Ethical statement
All animal experiments were performed in accordance with guidelines established by the National Institutes of Health and approved by the University Committee on Animal Resources. No patient or human samples were used in these studies.
Construction of mouse IL-2 FPs
IL-2FP (cs-1 and scram) constructs were shuttled into the phβ-APr-1-neo vector (Gunning and others 1987) using standard molecular biology techniques into the SalI and BamHI restriction sites to facilitate transfections into mammalian tumor cells. The Gibson assembly strategy (Gibson 2009) was used to construct a mouse IL-2 FP containing 5 consecutive MMPcs1 protease cleavage sites. A 4 fragment Gibson reaction was performed using the HiFi DNA Assembly Kit (NEB).
In brief, the phβ-APr-1-neo plasmid was linearized with SalI and KpnI restriction sites. PCR amplification using the forward primer 5′-CGGCTATTCTCGCAGGATCA-3′ and the reverse primer 5′-TTGAGGGCTTGTTGAGATGATGC-3′ were used to generate a mouse IL-2 fragment. Gene blocks were synthesized by Integrated DNA Technologies (IDT) to generate 2 fragments, the first expressing 5 MMPcs1 sites and a (GGGGS)4 linker and the second expressing mouse IL-2 receptor alpha and a 6 × Histidine tag. The IDT Codon Optimization tool was used in the designing of the gene block to avoid repetitive codon usage that could result in premature termination or deletion.
Construction of C38 clones expressing IL-2 FPs and Luminex multiplex for analyte detection
Mouse Colon 38 (C38), a colon adenocarcinoma derived from a C57BL/6 mouse, was obtained from Drs. Lord and Brown URMC. C38 cells were maintained in RPMI medium supplemented with 5% fetal calf serum and 1% penicillin/streptomycin and grown at 37°C in 5% CO2. C38 cells were transfected using Lipofectamine 3000 (Invitrogen) as described by the manufacturer. Twenty-four hours after transfection, 250 μg/mL of G418 was added to growth medium to select for cells expressing the neomycin resistance marker, and subsequently, individual clones were isolated by limited dilution. FP expression in individual clones was determined by measuring the concentration of mouse IL-2 receptor alpha (IL-2Ra) in tissue culture supernatant. Supernatant was collected from 5 × 105 cells plated in 2 mL of growth medium after 48 h.
The Luminex assays were performed according to the manufacturer's protocol (EMD; Millipore). The plate was run on the Luminex BioPlex200 and analysis of the mean fluorescence intensity was determined for each sample. The relative analyte concentrations were calculated from the standard curve. In analogous experiments, the supernatant from the C38/IL2 clone was analyzed using an IL-2 Luminex assay. This clone expressed ∼2,800 pg/mL IL-2 under these conditions.
In vivo tumor growth and measurement
Tumor cells were inoculated subcutaneously at 1e5 cells/mouse. Tumor growth was typically monitored by caliper measurements 3 times weekly. Tumor volume was calculated as (W2 × L)/2. Tumor growth endpoint was generally at 1,000 mm3 unless otherwise stated.
RNA analysis
Tumors were harvested and stored in RNALater Solution (Invitrogen) to preserve high-quality RNA. Tumors were homogenized in TRIzol Reagent (Ambion) and RNA isolated with the Direct-zol RNA MiniPrep Kit (Zymo Research). The RNA Integrity Number (RIN) values to assess the quality of the RNA samples in this study were determined by the University of Rochester Genomics Research Center. Samples with RIN values above 7 were used for analysis. RNA was DNase treated using the “Rigorous Treatment” protocol of the TURBO DNA-free Kit (Invitrogen) and then converted to cDNA using iScript Reverse Transcription Supermix for RT-qPCR (Bio Rad). RT-PCR was run for the following genes: β-actin, Cxcl12, Ifng, Cxcl10, Pd1, Pdl1, Foxp3, Cd4, Cd8b, Nkp46, and Gzma. Primer sequences are listed in Supplementary Table S1.
Fold change was calculated as in Livak's article (Livak and Schmittgen 2001; Schmittgen and Livak 2008). In brief, Ct of 35 was used as the cutoff, such that any Ct above 35 was set to 35 and should thus be interpreted as “at least” 35. Δ CT was calculated by normalizing to β-actin for each sample. ΔΔ CT was then obtained by subtracting Δ Ct of the control sample from the Δ Ct of the sample of interest. Fold change = 2−ΔΔ Ct. Fold changes below 1 were converted to negative numbers by taking the negative inverse.
Flow cytometry on tumors
Tumors were harvested and digested in Collagenase (Sigma) with 1:1,000 BD Golgi Plug for 35 min at 37°C before being passed through a 40 μM filter. All buffers contained 1:1,000 BD Golgi Plug. Cells were stained with the viability dye Ghost Dye Blue 516 (TonboBiosciences) according to the manufacturer's recommendations. Cells were then stained for the following: CD45 (clone 30-F11; BD Bioscience), CD4 (clone GK1.5; BD Horizon), CD8a (clone 53–6.7; BD Horizon), Nkp46 (clone 29A1.4; BioLegend), and CD25 (clone PC61; BD Pharmingen). Cells were fixed and permeabilized using eBioscience Anti-Mouse/Rat Foxp3 Staining Set (Invitrogen) according to manufacturer's recommendations.
Samples were analyzed using the LSRII Fortessa and data evaluated with FlowJo software. Single cells were gated on the basis of Forward and Side Scatter, before gating on live cells, as determined with viability dye. CD45+ cells were then gated from this live population before being further subdivided by CD4+ versus CD8+. NK cells are defined in this study as CD4−CD8−NKp46+. This gating scheme is demonstrated in Supplementary Figure S1.
Histology
Samples were fixed in neutral-buffered formalin and embedded in paraffin. Tissue processing, embedding, sectioning, and staining with Hematoxylin and Eosin were prepared by the Wilmot Cancer Institute Histology Core and additional sample sections were prepared for immunohistochemistry (IHC). In brief, slides were deparaffinized and heat-mediated antigen retrieval was performed using Antigen Retrieval Buffer pH 9.0 (Abcam) before samples were blocked with 2% normal goat serum plus 1% BSA for 2 h at room temperature. Slides were incubated with primary antibodies at 4°C overnight. Secondary antibody was applied for 40 min at room temperature. IHCs were developed using VECTASTAIN ABC-HRP peroxidase RTU and DAB chromogen as described by the manufacturer.
Primary antibodies used were as follows: CD8a (clone: EPR21769; Abcam, 1:2,000), CD4 (clone: EPR19514; Abcam, 1:1,000), Foxp3 (clone: FJK-16s; Invitrogen, 1:100), NCR1 (clone: EPR23097–35; Abcam, 1:500), F4/80 (clone: SP115; Abcam, 1:100), and Rabbit IgG monoclonal isotype (clone: EPR25A; Abcam, 1:500). Secondary antibodies were used as follows: Goat anti-Rabbit IgG (H+L) Biotinylated RTU (Vector Laboratories) and Goat anti-Rat IgG mouse adsorbed (H+L) biotinylated (Vector Laboratories; 1:100).
Statistical analysis
Data were plotted and statistical analysis run using GraphPad Prism software (San Diego, CA). All tests were run as described in figure legends and P-values <0.05 were considered significant.
Results
Development and analyses of tumor cell lines expressing IL-2 FPs
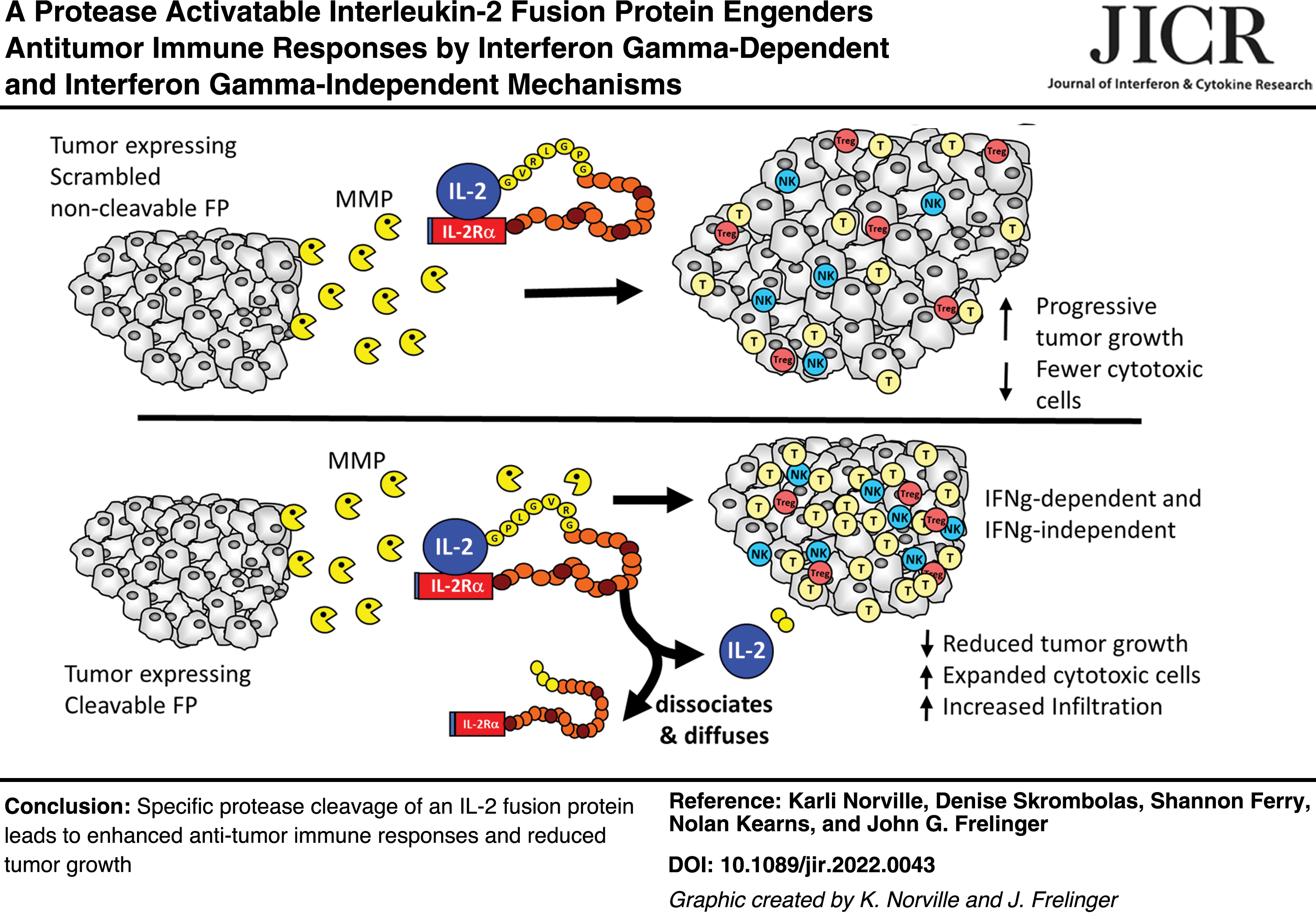
A key question in the development of protease activatable FPs is whether there is sufficient protease activity at the tumor site to specifically cleave and activate the FP in vivo. We created several versions of an IL-2FP employing IL-2 as the active immunomodulator and IL-2Ra as the inhibitory component. These include IL-2 FP containing: a cleavage sequence (cs) recognized by MMP2 and MMP9 (Bremer and others 2001), henceforth called cs-1, a second containing a scrambled version of cs-1 that is noncleavable (called scram), and a third containing 5 cs-1 sites in tandem (called 5mer) as illustrated in Fig. 1A. We employed a gene transfection approach that eliminates any potential delivery issues of the FP to the TME to test directly whether there is sufficient MMP activity at the tumor site to activate the cytokine. Mouse Colon 38 (C38) cells were transfected with expression plasmids encoding IL-2FP with either cs-1, scram, or 5mer sequences, and in vitro expression of the FPs was quantified (Fig. 1B), with cs-1 and scram lines chosen with similar expression levels.
Analysis of C38 transfectants expressing fusion proteins with different cleavage sites. C38 cells were transfected with an IL-2FP containing either a functional MMP cleavage sequence (cs-1)
As a positive control for the biological effects of IL-2 in the TME, we also constructed a C38 cell line (called C38/IL2) that expressed functional IL-2 at levels ∼8-fold higher than the FP levels expressed by the FP-transfected clones. Altogether, these cell lines allow for the direct testing and observation of biological effects of the FP in the local TME in vivo.
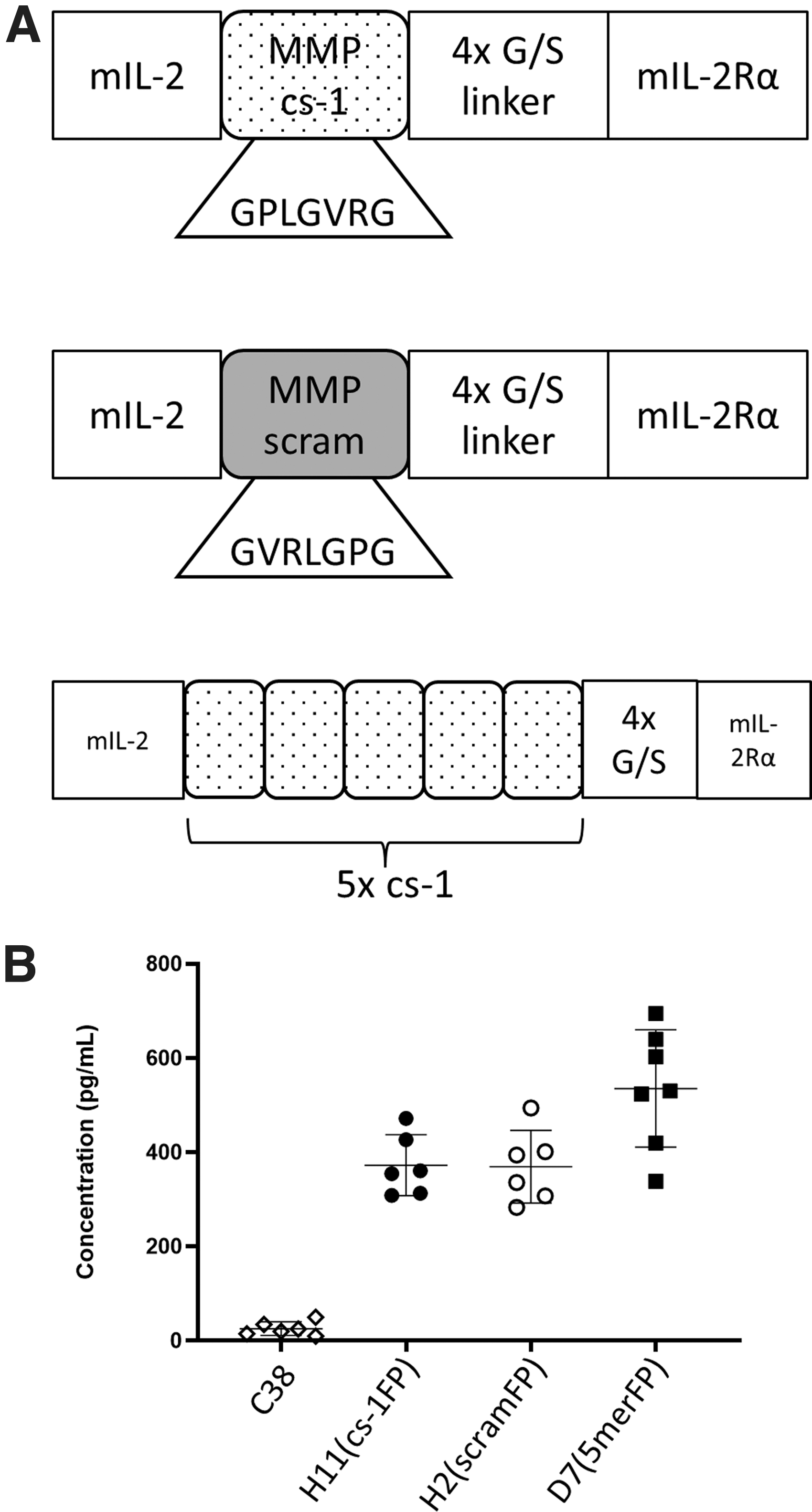
Groups of mice were inoculated with the selected FP-expressing clones, or the C38/IL2-positive control line, and tumor size was measured over time. The H2(scramFP) tumors grew progressively, reaching the 1,000 mm3 endpoint quickly (median 24 days) (Fig. 2A). In contrast, the H11(cs-1FP) tumors exhibited biphasic growth patterns wherein they grew for a short time before exhibiting a period of regression. In this experiment, one tumor rejected fully, whereas the remainder eventually grew to endpoint, although more slowly (median 40 days) than the H2(scramFP) tumors (Fig. 2B).
C38 cells expressing IL-2FP with functional cleavage sequences have delayed growth in vivo. Transfected C38 cell lines were injected and monitored by caliper measurement for tumor growth.
Interestingly, the D7 (5merFP) tumors grew in a pattern closely resembling the H11(cs-1FP) tumors (Fig. 2C), indicating that additional css did not result in enhanced FP activity. Both the H11(cs-1FP) and the D7(5merFP) tumor lines had initial growth phases that resemble the growth of the C38/IL2 line that expresses free IL-2 (Fig. 2D), showing early growth followed by regression. Collectively, these data are consistent with the concept that the MMPs in the TME are specifically cleaving the FP within the engineered cleavage site, resulting in decreased tumor growth.
RNA analysis of FP-expressing tumors during regression
We examined mRNA expression of a panel of immune and effector genes in regressing H11(cs-1FP) tumors (Fig. 3A) and size-matched H2(scramFP) tumors (Fig. 3B). Each tumor was analyzed individually and the fold change reported relative to a single H2(scramFP) control tumor, as described in the figure legend, thus allowing for the visualization of heterogeneity in gene expression and of potential coordinate gene expression in a given tumor (Fig. 3C, D). RNA expression of Cd8 and Cd4 was increased in the H11(cs-1FP) tumors as compared with the H2(scramFP) tumors, consistent with the hypothesis that the cleaved FP is driving an effector response. Interestingly, given the biology of IL-2, Foxp3 was only slightly upregulated in the H11(cs-1FP) tumors, suggesting that Treg cells were not selectively expanding. Furthermore, Nkp46 expression was not markedly different in the 2 tumor types, suggesting that NK cells are not the major immune cell subset changing at this time point.
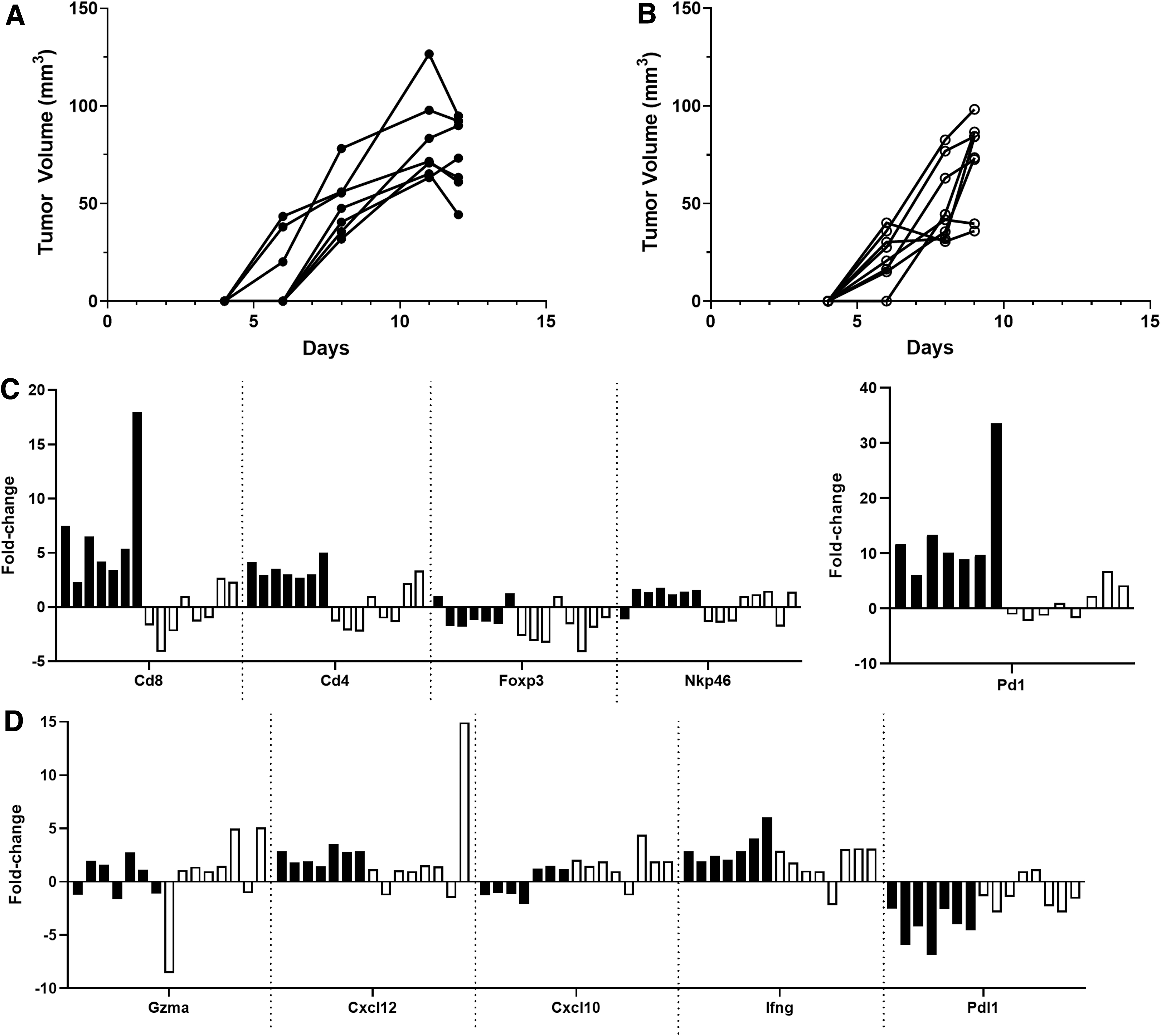
Regressing H11(cs-1FP) tumors more highly upregulate immune cell and effector genes than H2(scramFP) tumors. Regressing H11(cs-1FP)
Pd-1 expression was increased in the H11(cs-1FP) tumors, suggesting a larger population of activated cells, while Pdl1 expression was decreased. Surprisingly, several effector molecules and chemokines were not greatly increased in the H11(cs-1FP) tumors, with even interferon gamma (Ifng) showing only modest upregulation. Similar analyses at an earlier time point revealed a very similar pattern, except Ifng and Pdl1 were more upregulated in the H11(cs-1FP) tumors (Supplementary Fig. S2). Overall, these data suggest a more robust antitumor response occurring in the H11(cs-1FP) tumors than the H2(scramFP) tumors.
Activatable IL-2FP-expressing tumors have markedly increased CD8 cells
Flow cytometry analyses were performed on regressing H11(cs-1FP) and size-matched H2(scramFP) tumors to ascertain changes to immune cell infiltration due to the activatable FP. H11(cs-1FP) tumors had more CD45 cells, particularly CD8 cells, and to a lesser extent CD4 cells (Fig. 4A). Unexpectedly, given the responsiveness of NK cells to IL-2, we observed no obvious difference in NK cell density between the 2 tumor types (Fig. 4A). We also calculated frequencies of immune cell subsets within the CD45 population for each tumor, which showed that the H11(cs-1FP) tumors had a marked increase in CD8 cells and a concomitant decrease in the non-CD4, -CD8, -NK cell subset (likely macrophage/myeloid cells) (Fig. 4B). Collectively, these data complement the RNA data from Fig. 3 on a protein level and strongly indicate that the tumor regressions observed in the H11(cs-1FP) tumors are immune-cell mediated.
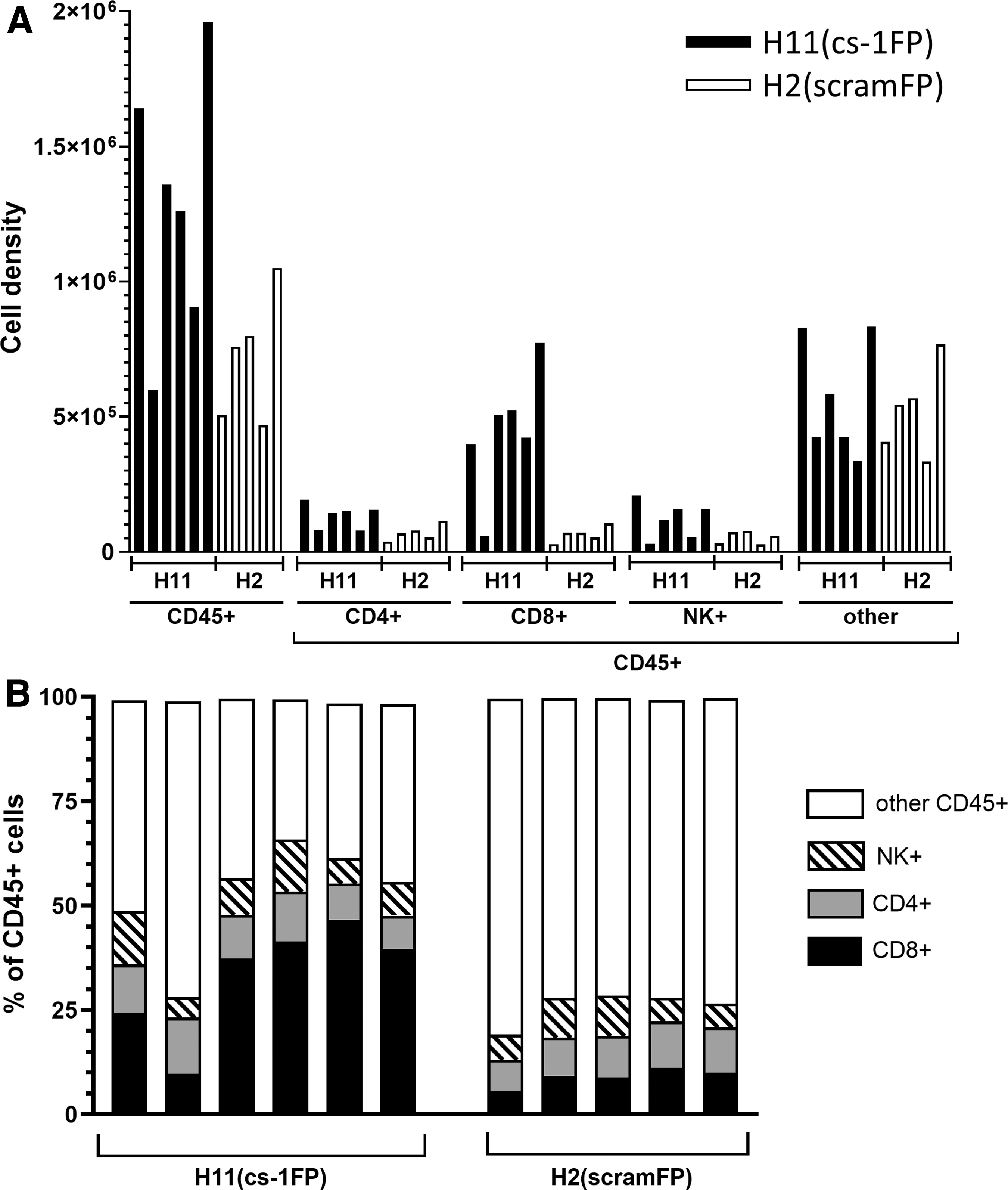
H11(cs-1FP) tumors have increased effector cell populations compared with H2(scramFP) tumors. Flow cytometry analysis of regressing H11(cs-1FP) and size-matched H2(scramFP) tumors. Individual tumors are displayed with largest tumor in a group on the left and the smallest on the right. H11(cs-1FP) tumors n = 6. H2(scramFP) tumors n = 5.
To investigate whether the activatable FP changes the localization of immune cells within the tumors, we performed a histologic analysis of regressing H11(cs-1FP) and size-matched H2(scramFP) tumors (Fig. 5). H&E staining revealed that the H11(cs-1FP) tumors appeared to have more apparent CD45 cells, compared with H2(scramFP) tumors. IHC revealed that there were more CD8 cells in the H11(cs-1FP) tumors compared with the H2(cs-1FP) tumors, and there were more CD8 cells in the interior of the H11(cs-1FP) tumors. In contrast, the presence of the activatable IL2 FP seemed to affect neither the density nor overall location of NK cells. CD4 cells followed a similar trend as the CD8. Interestingly, the activatable FP tumors appeared to have more Foxp3+ cells in the tumor center, but these cells remain only as a fraction of total CD4 cells. F4/80 revealed that both H11(cs-1FP) tumors and H2(scramFP) tumors are heavily infiltrated by macrophages, but that the cleavable IL-2FP has no obvious influence on this cell population (Supplementary Fig. S3).
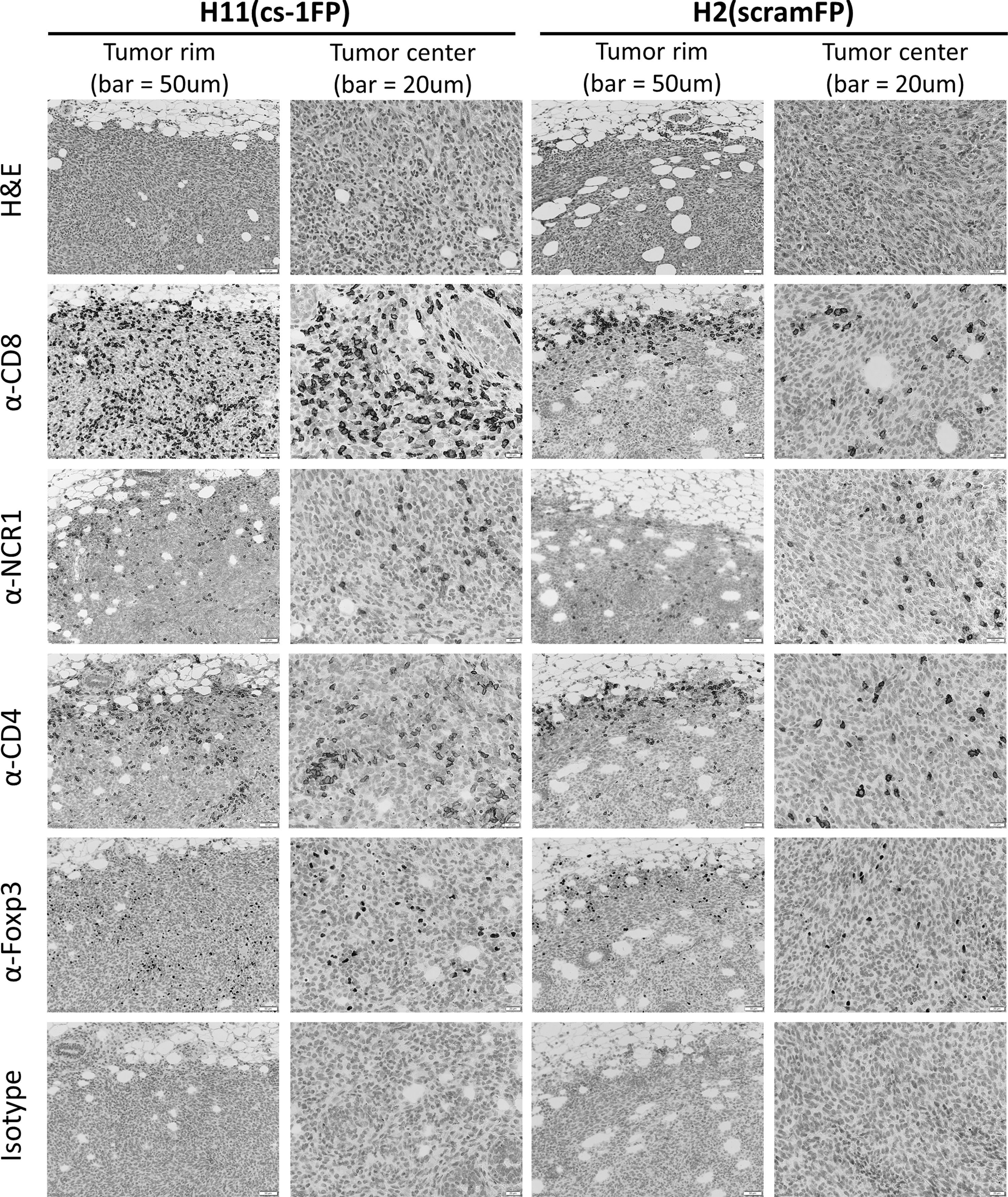
IL-2 FP cleavage results in change in immune cell localization. Representative images of histological analysis of regressing H11(cs-1FP) tumors (columns 1 and 2) and size-matched H2 (scramFP) (columns 3 and 4). A view of tumor rim and center presented for better understanding of immune cell infiltration. Images of tumor rim (columns 1 and 3) have scale bars representing 50 μm. Images of tumor center (columns 2 and 4) have scale bars representing 20 μm.
There are both IFNg-dependent and -independent antitumor immune responses generated by IL-2FP
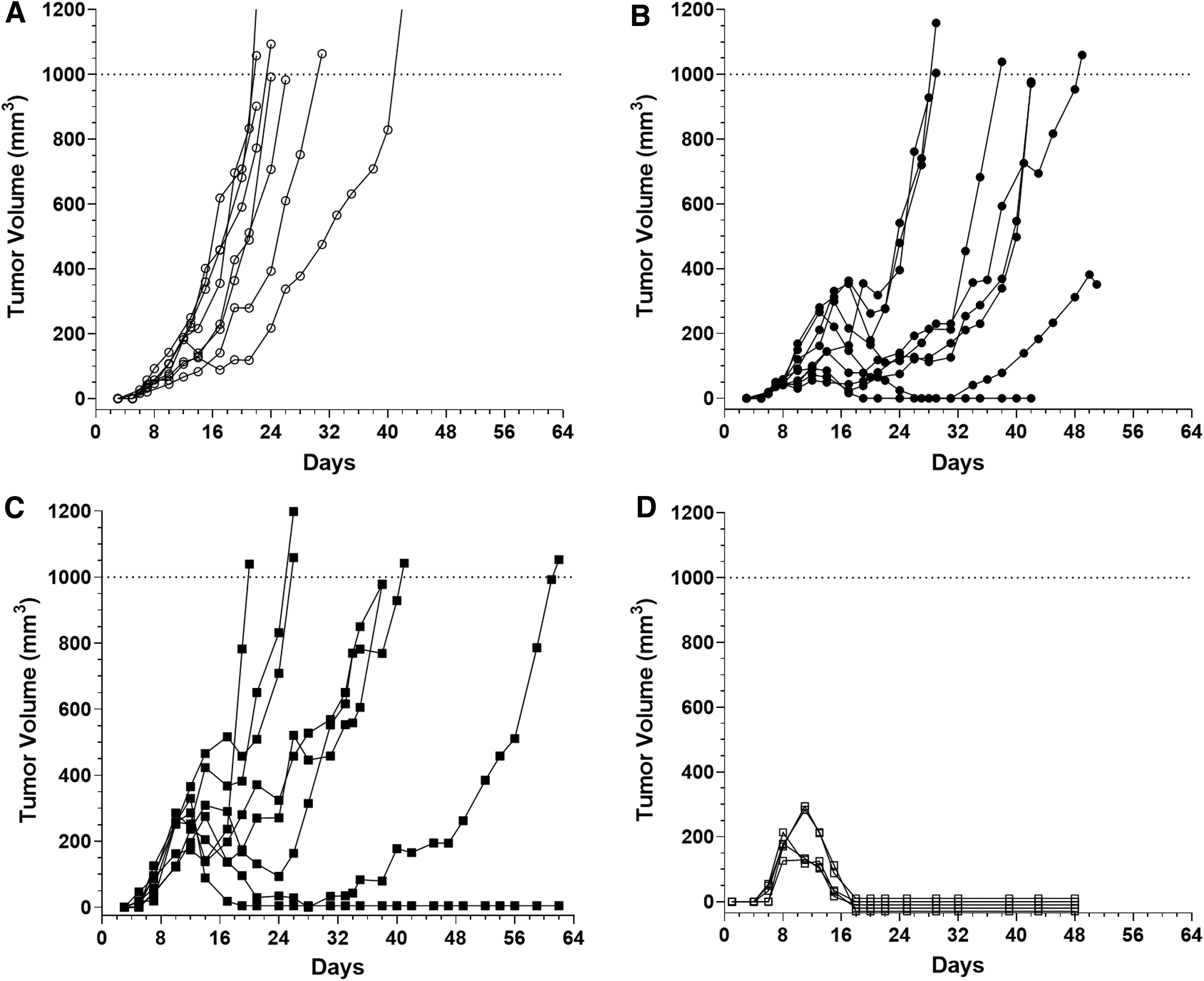
To determine if the differences in tumor growth observed between the H11(cs-1FP) tumor line and the H2(scramFP) tumor line were dependent upon IFNg, we inoculated each tumor line into syngeneic wild-type (WT) mice or IFNg knockout (KO) mice and monitored tumor growth (Fig. 6). As before, in WT mice, the H2(scramFP) tumors grew progressively (Fig. 6A), whereas the H11(cs-1FP) tumors exhibited a biphasic growth pattern with 4/8 eventually rejecting completely in this experiment (Fig. 6B). To better visualize the differences in early tumor growth between the 2 cell lines, the dotted line insets in Fig. 6A and B are expanded in Fig. 6C and D, respectively.
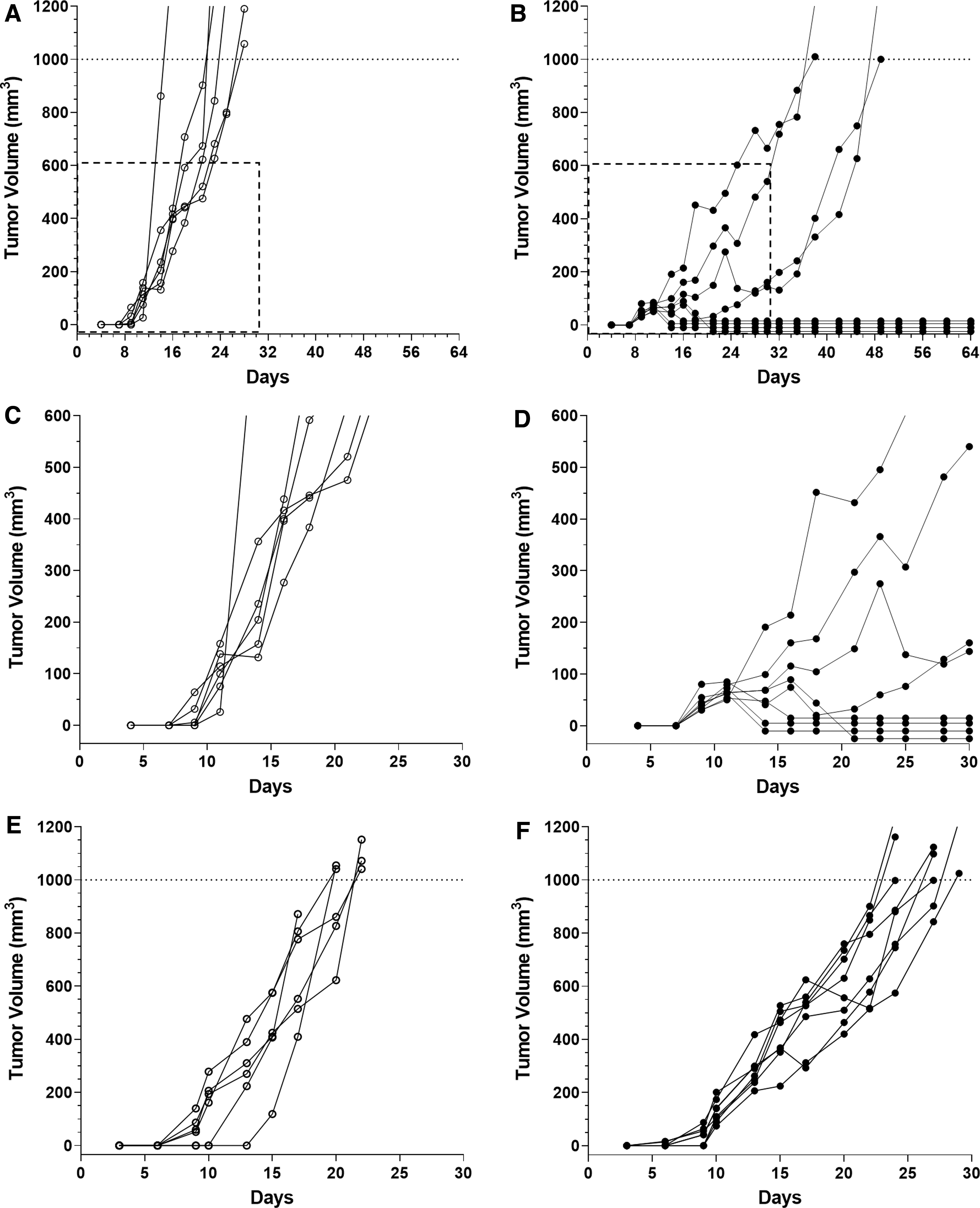
Tumors expressing activateable IL-2FP with a functional cleavage site fail to regress in IFNg KO mice. H2(scramFP) and H11(cs-1FP) cell lines were injected into WT or IFNg KO mice and monitored by caliper measurement for tumor growth.
As expected, the H2(scramFP) tumors grew progressively both in the KO mice and the WT mice (Fig. 6A, E). Notably, in contrast to the growth pattern in WT mice, the H11(cs-1FP) tumors in the KO mice grew progressively, although more slowly than the H2(scramFP) tumors in the KO mice (Fig. 6F). This abrogation of initial tumor growth delay demonstrates that the tumor regression is IFNg-dependent. Nevertheless, there are IFNg-independent antitumor factors that also play a role in delaying tumor growth. Indeed, the IFNg-independent factors can sometimes result in complete regression of the C38/IL2 line in the KO mice (Supplementary Fig. S4), underlying the importance of both pathways in the cleavable FP tumors.
Discussion
Several lines of evidence suggest that IL-2FP in the TME can be specifically cleaved by MMP and enhance IL-2-mediated immune antitumor responses
First, while the H2(scramFP) clone grew progressively, the H11(cs-1FP) clone grew more slowly and exhibited partial or complete rejection, a growth pattern reminiscent of that observed in tumors expressing free IL-2 (Fig. 1D). Since these tumor cell lines express FPs that are identical, except for a sequence with differential susceptibility to MMP cleavage, these results strongly suggest that tumor growth differences are due to specific cleavage of the FP. Second, as illustrated by the RNA expression and flow cytometry data, there were more IL-2-responsive cells, notably CD8 cells, in the H11(cs-1FP) tumors compared with the H2(scramFP) tumors. Third, the IHC data showed that there were not only more CD8 cells but that they also infiltrated the H11(cs-1FP) tumors more than in the H2(scramFP) tumors, characteristic of more effective antitumor immune responses (Fridman and others 2012; Lanzi and others 2020; Marliot and others 2020). All these immune effects indicate that specific cleavage of IL-2cs-1FP releases IL-2 locally, which activates antitumor effectors in a cascade fashion.
The IL-2FP generates an IFNg-dependent and -independent antitumor response
The H2(scramFP) tumors had rapid, progressive growth in both WT and IFNg KO mice. In contrast, the H11(cs-1FP) tumors exhibited an unusual biphasic growth pattern in WT mice, with an initial period of slow growth, followed by a period of regression, and in some cases, total elimination. This biphasic growth was absent in IFNg KO mice, where the same tumors grew progressively, indicating that IFNg was essential for the regression phase. Intriguingly, despite the loss of the biphasic growth in the IFNg KO mice, the H11(cs-1FP) tumors still grew more slowly than the H2(scramFP) tumors in this host, suggesting additional IFNg-independent antitumor effects, potentially mediated by the cytolytic activity of CD8 cells.
Mechanistically, while IFNg has profound antitumor immune effects, it is also important to note that IFNg can have nonimmunologic antitumor effects, reflecting its complex in vivo functions (Castro and others 2018). Taken together, these data strongly suggest that the IL-2FP stimulates both IFNg-dependent and -independent response pathways, which underscores the potent, pleiotropic nature of IL-2.
IL-2 has complex roles in the immune response to tumors
IL-2 likely acts in several ways to affect antitumor immune responses. The local expression of free IL-2 or a cleavable IL-2FP, results in the enhancement of cytotoxic cells and increases antitumor immune responses. This likely reflects the relatively high local concentration of IL-2 in the TME. Moreover, this is consistent with the finding that high-dose IL-2 is clinically effective (Conlon and others 2019) in part due to its activity within local tumor sites.
In contrast, systemic low-dose IL-2 enhances Tregs and is being explored for the treatment of autoimmune diseases (Skrombolas and Frelinger 2014; Abbas 2020). Although we noted an increase in Tregs in the tumors expressing the IL-2FP with a cleavable sequence, they had an even more dramatic increase in CD8 cells, suggesting that the ratio of CD8 to Tregs may determine the ultimate effect of the immune response. Intriguingly, there is also increasing evidence that IL-2 may enhance the innate-like function of CD8 cells. For example, LCMV-specific CD8 effector or memory T cells can be stimulated in vitro by cytokines, including IL-2 in an innate-like fashion without TCR stimulation to produce IFNg (Freeman and others 2012).
Interestingly, it is widely accepted that many T cells in tumors are not specific for tumor-associated antigens. Indeed, it was recently elegantly demonstrated that CD8 cells specific for virally expressed antigens are present in tumors and can be triggered by their cognate peptides, and are therefore functional, potentially contributing to tumor rejection if stimulated (Rosato and others 2019). Taken together, these results raise the possibility that TCR-independent stimulation of CD8 T cells in the TME by IL-2, regardless of tumor specificity, may contribute to the antitumor response, thus providing a mechanistic explanation for the profound antitumor effects of IL-2 we observed.
While activated effector T cells are essential for eliminating tumors, driving effector T cells to high cytolytic activity and IFNg production is also associated with upregulation of inhibitory receptors, exhaustion, and a short half-life that ultimately limit their function (Blank and others 2019). The duality of IL-2 activity may help explain the ultimate outgrowth of tumors if elimination is not completed before the counter regulatory processes become dominant or if suppression predominates overstimulation (Charych and others 2017).
Novel approaches for enhancing effects of IL-2 based on its biology
There is renewed interest in the use of IL-2 to manipulate immune responses that take advantage of the unique biological properties of IL-2 (Overwijk and others 2021). One strategy is to stimulate either Tregs or CD8/NK cells preferentially (Letourneau and others 2010; Levin and others 2012; Charych and others 2017; Spangler and others 2018; Boyman and Arenas-Ramirez 2019; Pol and others 2020; Sahin and others 2020). We have previously used an IL-2FP constructed with a different cs and demonstrated tumor growth reduction in a peritoneal tumor model, in which the FP was injected intraperitoneally (Puskas and others 2011). Conceptually related, others have shown that a mutein of IL-2 connected by an MMP cleavable linker to the beta chain of the IL-2 receptor and fused to immunoglobulin constant region, also showed efficacy in mouse tumor models (Hsu and others 2021). Paradoxically, others have shown that an IL-2FP very similar to the IL-2FP in the current work, except designed with a noncleavable flexible linker connecting IL-2 and IL-2Ra, formed an unusual dimer and dramatically enhanced the generation of Tregs in vivo.
Importantly, systemic treatment with this IL-2FP inhibited the development of disease in the NOD mouse model and in a preclinical model of systemic lupus erythematosus (Khalili and others 2018; Ward and others 2018; Xie and others 2021). One possibility is that the dose of IL-2/IL-2Ra, much like IL-2 itself, may help determine whether it is stimulatory for effector cells or for T regs (DeOca and others 2020; Hernandez and others 2021). Further experimentation will be required to tease out the critical functional differences between the similar, but not identical, FPs. Nevertheless, these studies collectively underscore that understanding and manipulating the complex and potent biological properties of IL-2 will be critical for its effective clinical use.
In conclusion, we demonstrated that an IL-2FP can be specifically activated by MMP proteases and enhance antitumor immune responses by IFNg-dependent and IFNg-independent mechanisms. While cytokine therapies may be effective by themselves, it is likely they will be even more effective when combined with novel immunization and immunotherapy strategies, such as those employing neoantigens, novel adjuvants, checkpoint inhibitors, or new adoptive CAR T cell or NK cell therapies.(Yadav and others 2014; Delamarre and others 2015; Vormehr and others 2015, 2020; Conlon and others 2019; Gandhapudi and others 2019; Overwijk and others 2021; Portillo and others 2021; Butler and others 2022).
Footnotes
Acknowledgments
The authors would like to thank Ms. Nancy Corson for technical assistance, Dr. Aditi Murthy for advice on Immunohistochemistry, and our colleagues for helpful suggestions to the article. The authors want to also acknowledge generous support from Steven and Alison Krausz and from F.C. Blodgett.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
K.N. was supported by The American Association of Immunologists through a Careers in Immunology Fellowship and in part by the National Institutes of Health Training Grant AI07285. D.S. was supported in part by the National Institutes of Health Training Grant AI07285. The project was supported in part by 1R21CA184433.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1