
Abstract
Inflammation is a natural immune defense mechanism of the body's response to injury, infection, and other damaging triggers. Uncontrolled inflammation may become chronic and contribute to a range of chronic inflammatory diseases. Signal transducer and activator of transcription 2 (STAT2) is an essential transcription factor exclusive to type I and type III interferon (IFN) signaling pathways. Both pathways are involved in multiple biological processes, including powering the immune system as a means of controlling infection that must be tightly regulated to offset the development of persistent inflammation. While studies depict STAT2 as protective in promoting host defense, new evidence is accumulating that exposes the deleterious side of STAT2 when inappropriately regulated, thus prompting its reevaluation as a signaling molecule with detrimental effects in human disease. This review aims to provide a comprehensive summary of the findings based on literature regarding the inflammatory behavior of STAT2 in microbial infections, cancer, autoimmune, and inflammatory diseases. In conveying the extent of our knowledge of STAT2 as a proinflammatory mediator, the aim of this review is to stimulate further investigations into the role of STAT2 in diseases characterized by deregulated inflammation and the mechanisms responsible for triggering severe responses.
Introduction
STAT2
IFNs are classified into 3 classes: Type I, Type II (IFN-γ), and Type III (IFN-λ1-4 also referred to as IL-28A, IL-28B, and IL-29). The largest group is type I IFN, which is composed of 13 IFN-α subtypes, a single IFN-β, and poorly characterized IFN-τ, IFN-κ, IFN-ω, IFN-σ, and IFN-ζ (Pestka and others 2004). Of note, IFN-α and IFN-β are the 2 forms of type I IFN used often for the study of type I IFN signaling. Activation of the pathway begins with type I IFN binding to its cognate receptor, which consists of 2 transmembrane subunits, IFNAR1 and IFNAR2, preassociated with Janus kinases TYK2 and JAK1, respectively. TYK2 and JAK1 are activated by transphosphorylation and then phosphorylate the intracellular chains of IFNAR1 and IFNAR2. In the case of type III IFN signaling, a different heterodimer receptor complex consisting of IFNLR1 and IL10R2 is engaged, which leads to STAT1 and STAT2 phosphorylation by JAK kinases in a similar manner as type I IFN.
As STAT1/STAT2 heterodimers assemble, they associate with interferon regulatory factor 9 (IRF9), resulting in the formation of a transcriptional complex termed interferon-stimulated gene factor-3 (ISGF3). The ISGF3 complex translocates to the nucleus and binds to the IFN-stimulated response element (ISRE) located in the promoters of IFN-stimulated genes (ISGs). While the major transcriptional complex is ISGF3, both type I IFN and type III IFNs can also induce the formation of STAT1 and STAT3 homodimers that bind to the IFN-γ-activated sequence (GAS) motif within target gene promoters.
The classical view of ISGF3 as a mediator within most of the transcriptional responses to type I IFN, however, is shifting. It has been known for some time that, in the absence of type I IFN, STAT2 and IRF9 can form a complex independently of STAT1 (Martinez-Moczygemba and others 1997), which shuttles between the cytoplasm and the nucleus (Banninger and Reich 2004). Additionally, unphosphorylated STAT2 is found bound to a subset ISG promoters before type I IFN stimulation takes place (Testoni and others 2011).
Without STAT1, type I IFN can activate an alternative signaling pathway mediated by the STAT2/IRF9 complex consisting of STAT2 homodimers bound to IRF9. This complex induces a delayed yet prolonged transcriptional response and antiviral effects in a manner analogous to ISGF3 (Blaszczyk and others 2015, 2016). These studies also indicated that increased expression of STAT2 and/or IRF9 was required to activate robust expression of shared ISGs. Increased levels of unphosphorylated ISGF3 following type I IFN stimulation has been proposed as a secondary response to prolonged transcription of ISGs (Wang and others 2017a). This information is illustrated in Fig. 1.
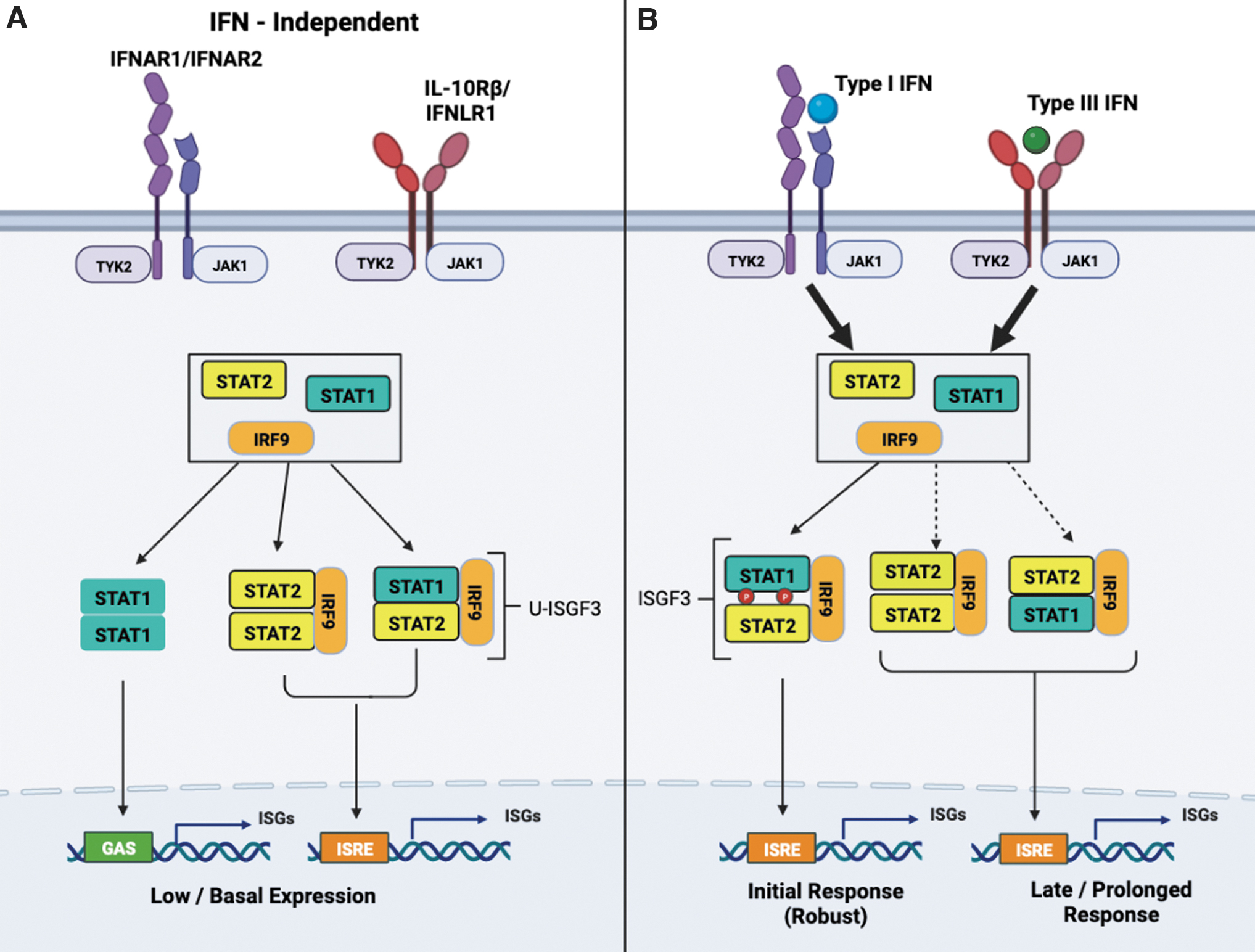
STAT2 is key transcription factor in type I and type III IFN signaling.
Under homeostatic conditions, constitutive low levels of type I IFN maintain cells with adequate levels of ISGF3 components (STAT1/STAT2/IRF9). In the absence of type I IFN, however, unphosphorylated STAT2 in complex with IRF9 (STAT2/IRF9) can drive ISG transcription independently of STAT1 (Platanitis and others 2019). Once type I IFN becomes available, ISGF3 replaces STAT2:IRF9 and powers a robust primary transcriptional response. This observation highlights the role of STAT2 as a signaling factor that can function without STAT1 and does not require tyrosine phosphorylation to maintain basal gene transcription levels. The latter finding emphasizes its potential relevance in other biological actions, which may not be entirely dependent on the classical type I IFN signaling pathway.
Our current understanding of the far-reaching effects of STAT2 in mediating the biological effects of type I and type III IFNs appear to be expanding. STAT2 may have a wider sphere of influence than previously assumed, and in fact, have a dual function in disease. As examples, pathological inflammation triggered by persistent activation of type I/III IFN signaling, lack of negative regulation provided by STAT2 deficiency, or excessive production of type I/III IFNs can be seen in inflammatory diseases. Collectively, studies of STAT2 in infectious, autoinflammatory, and autoimmune diseases such as psoriasis, asthma, hemophagocytic lymphohistiocytosis (HLH), and type I interferonopathies, shed new light on these additional functions of STAT2. The following summary describes those findings of current literature with respect to the inflammatory behavior of STAT2 and its role in the pathogenesis of various diseases marked by chronic inflammation.
STAT2 in Viral Infection
STAT2 is an integral downstream effector of type I and type III IFN signaling in restricting the replication of pathogenic viruses. Nonetheless, viruses are naturally equipped with their own armamentarium of genes to subvert the initial protective innate host response initiated by type I IFN. Similar responses are observed with type III IFN-induced activation of ISGF3 (Kotenko and Durbin 2017). Specific viruses inhibit the induction of type I IFN antiviral responses by targeting STAT2 for degradation or preventing its activation by impeding tyrosine phosphorylation. It is important to highlight that some of the IFN target genes or ISGs exert antiviral activity while others contribute to inflammation. The orchestrated activity of all these genes is necessary to eradicate viruses.
Seminal studies in mice lacking Stat2 have shown the importance of STAT2 in mediating the antiviral effects of type I IFN by restricting the replication of vesicular stomatitis virus (VSV) (Park and others 2000). In the context of influenza infection, Stat2-deficient mice are prone to hyperinflammation. Loss of Stat2 increases the levels of proinflammatory cytokines (IL1-α, IL1-β, IL-6, IL-12, IL17-A, and IFN-γ) (Gopal and others 2018). This inflammatory response intended to eradicate viruses was shown to be exacerbated resulting in severe lung injury (Tavares and others 2017).
An interesting phenotypic feature of Stat2 deficiency, first described in murine macrophages lacking STAT2, is the acquired ability to upregulate expression of major histocompatibility complex class II after type I IFN stimulation (Zhao and others 2007; Gothe and others 2022), a response unique to IFN-γ. A change in transcriptional response to type I IFN that mimics IFN-γ could initially be protective but then become potentially deleterious in the setting of persistent viral infection by favoring the activation of proinflammatory genes driven mainly by STAT1. In support of a regulatory role, STAT2 has been reported to inhibit STAT1 in response to several proinflammatory cytokines, including IL-6, IL-27, and IFN-γ (Ho and others 2016). Interestingly, effective control of mouse cytomegalovirus infection requires activation of STAT2 by all 3 types of IFNs (Zimmermann and others 2005; Le-Trilling and others 2018), suggesting that the presence of STAT2 is beneficial in antiviral IFN-γ responses. In the case of dengue virus, STAT2 and STAT1, independently, are protective in restricting infection (Perry and others 2011).
Following dengue infection, Stat2-deficient mice showed similar increase in serum levels of proinflammatory cytokine TNF-α as Stat1-deficient mice when compared with wild-type mice. In the absence of Stat1, Stat2 was able to activate a type I IFN antiviral response to clear infection. The same observation was noted in mice lacking Stat2. However, codeletion of Stat1 and Stat2 resulted in a heightened increase of circulating TNF-α levels associated with poor survival and high viral load, indicating a coregulatory effect of STAT1/STAT2 signaling. What surfaced from this study is that STAT2 can restrict infection in vivo and control TNF-α production independently from STAT1. It is worth noting that type III IFNs are also induced by dengue infection (Palma-Ocampo and others 2015; Hsu and others 2016). High levels of IFN-λ are detected in dengue fever patients. Pretreatment of epithelial cells with IFN-λ increased IFN-β production during infection indicating a potential crosstalk between both IFN signaling pathways that share STAT2 to mount a robust antiviral response (Palma-Ocampo and others 2015).
Type I IFNs are pivotal in restricting the replication and spread of lymphocytic choriomeningitis virus (LCMV) during acute infection (Ou and others 2001). In contrast, chronic LCMV infection is controlled by impeding type I IFN signaling (Teijaro and others 2013). Ifnar1-deficient mice lacking either Stat2 or Irf9 survived LCMV infection (Hofer and others 2012). Subsequent studies revealed that Stat1-deficient mice experienced a lethal host response to the infection (Li and others 2014a). These mice presented with elevated serum cytokine and chemokine levels (CCL2, CCL5, IL-5, IL-6), including type I IFN and IFN-γ during LCMV infection. Deletion of Stat2, Irf9, or Ifnar1 in Stat1-deficient mice conferred survival to LCMV infection.
Type III IFN also shows antiviral activity against LCMV before the establishment of long-term infection; however, LCMV-infected cells had reduced expression of Ifnlr1 (Lukacikova and others 2015). These findings point to the proinflammatory and lethal effects of noncanonical activation of type I and type III IFN signaling most likely driven by possible negative regulatory effects of STAT2/IRF9 as opposed to ISGF3, which enables persistent LCMV replication.
In recent years, we have learned that type I IFN-induced activation of STAT2 by phosphorylation on tyrosine (Y)-690 (Y689 in mice) is not the only occurring post-translational modification event. Type I IFN signaling is also impacted by STAT2 being further phosphorylated on serine (S287, S734) (Steen and others 2013, 2016) and threonine (T387 and T404/T403 in mice) (Wang and others 2017b, 2021), both of which impact type I IFN signaling. It is unclear as to whether type III IFNs also induce STAT2 phosphorylation on these same sites. Phosphorylation on S287, S734, and T387 negatively regulates whereas phosphorylation on T404 positively activates type I IFN antiviral responses. In the latter, phosphorylation on T404 was reported to cause disruption of the unphosphorylated STAT1/STAT2 heterodimer in its antiparallel “inactive” conformation to switch to the parallel conformation and facilitate STATs 1 and 2 phosphorylation following type I IFN stimulation.
In vivo studies show that mice carrying Stat2-T403 mutated to alanine (T403A) were highly susceptible to VSV and herpes simplex virus infection, whereas wild-type counterparts were protected. Compared with wild-type mice, Stat2-T403A knockin mice infected with VSV produced higher serum levels of IFN-β as well as proinflammatory mediators, CCL2 and CSF-1, compounded by accumulation of immune cells in the brain. It is unclear whether high IFN-β levels produced during VSV infection increased the protein expression levels of unphosphorylated STAT2-T403A or its stability and the formation of mutant STAT2/IRF9 complex to potentially drive the expression of CCL2, an ISG that can facilitate recruitment of inflammatory monocytes to the site of infection (Conrady and others 2013).
In humans, the discovery of a young child and infant sibling born with homozygous germline STAT2 deficiency who experienced severe viral illness has contributed vital information to the biological significance of STAT2 in antiviral immunity (Hambleton and others 2013). Of the 2, only the older sibling was vaccinated and developed disseminated vaccine-strain measles after routine immunization. In contrast, heterozygous relatives were unaffected. In vitro studies demonstrated that restoration of STAT2 in patient's fibroblasts restored type I IFN response and antiviral state.
After this initial report, more STAT2-deficient individuals have since been identified with similar and/or additional clinical manifestations with varying penetrance (Duncan and Hambleton 2021). For instance, 2 cases were reported with distinct homozygous STAT2 mutations that resulted in complete loss of STAT2 protein and presented with secondary HLH (Alosaimi and others 2019; Gothe and others 2020). HLH is a rare condition characterized by severe systemic hyperinflammation associated with high production of IFN-γ, where viral infection provokes a robust, destructive, and inefficient antiviral response (Rosado and Kim 2013). One patient developed secondary HLH upon infection with meningitis due to vaccine-strain mumps (Alosaimi and others 2019). Treatment with high doses of intravenous immunoglobulin enabled patient recovery. The exact viral trigger that led to HLH in the second patient who developed severe illness following MMR vaccination and subsequently fatal hyperinflammation could not be established (Gothe and others 2020), as there was no evidence of vaccine-strain viral replication.
The cause was postulated to be either influenza A or vaccination strain of Varicella (Freij and others 2021). In both cases of HLH, STAT2 deficiency was reflected by decreased expression of ISGs upon stimulation with IFN-α. Collectively, these 2 case studies demonstrate the far-reaching detrimental effects of STAT2 deficiency on functionality of the type I IFN response and patients' abilities to combat viral illness (Gothe and others 2022). Based on these findings, one could speculate that type III IFN signaling would also be impaired in individuals with STAT2 deficiency. The emerging trend is that all STAT2-deficient individuals share a susceptibility to viral illness during childhood, which becomes less recurrent when they reach adulthood. Interestingly, no increase in susceptibility to bacterial infections has been reported in STAT2-deficient individuals.
STAT2 in Bacterial Infections
Unlike viral infections, where type I and type III IFNs are protective, both display dual opposing roles in bacterial infection. Depending on the bacterial pathogen, IFNs can be protective or deleterious to the host (Lebreton and others 2011; Cohen and Prince 2013; Boxx and Cheng 2016; Kovarik and others 2016). To elucidate the role of IFN signaling in microbial pathogenesis, mice deficient in Ifnar1, Ifnlr1, and components of the ISGF3 complex are routinely used. However, the phenotypes of STAT2 or IRF9 deficiency may not mirror those of IFNAR1 deficiency. The absence of both STAT2 and IRF9 is rather expected to resemble double IFNAR1/IFNLR1 deficiency, suggesting that both factors have critical roles in bacterial infections that are yet to be fully characterized.
It is widely accepted that type I IFN contributes to the immunopathology of Salmonella Typhimurium, an intracellular Gram-negative enteric pathogen that evades the innate immune system by provoking severe inflammation. In contrast, the role of type III IFN in Salmonella infection is not entirely clear. In vitro studies show IFN-λ treatment safeguards the integrity of epithelial barriers from Salmonella-induced damage, whereas IFN-β provides minimal effect (Odendall and others 2017). Studies in ifnlr1-deficient mice would be needed to determine whether IFN-λ has antibacterial activity in vivo. What is known is that loss of Ifnar1 prolongs host survival to Salmonella infection (Robinson and others 2012). Type I IFN induces cell death of Salmonella-infected macrophages by activating necroptosis, a form of inflammatory cell death. This type of cell death helps to evade the immune response and spread the infection to other organs (McComb and others 2014). Deficiency in RIP3 kinase, a key component of the necroptotic pathway, enhances bacterial clearance.
Similarly, macrophages lacking IFN-a, Stat1, Stat2, or Irf9 were highly resistant to necroptosis. Salmonella infection was reported to drive necroptosis by upregulating the mitochondrial phosphatase Pgam5 that in turn sequestered the transcription factor NRF2 in the cytosol, preventing the expression of antioxidative genes. Ultimately, this led to the production of reactive oxygen species, energy depletion and cell death, which helps the organism evade the immune response (Hos and others 2017). Of note, STAT2 has been reported to increase mitochondrial mass in lipopolysaccharides (LPSs)-stimulated macrophages; such an increase in mitochondrial copy number is needed to support the proinflammatory differentiation of macrophages (Yu and others 2020).
Most recent literature shows that Salmonella flagellin activates the NLRC4 inflammasome to trigger pyroptosis and the synthesis of lysophospholipids to clear early infection (Akhade and others 2020). However, as infection progresses, type I IFN represses NLRC4 and the lysophospholipid enzyme iPLA2. This results in reduced production of lysophospholipids that ultimately downregulates flagellin expression to enable Salmonella to switch to a flagellin-low phenotype to avert immunosurveillance. It is unclear if deletion of STAT1, STAT2, or IRF9 will rescue Salmonella flagellin expression and enhance bacterial clearance. Another study shows that type I IFN remodels lysosome localization and function, which is associated with enhanced Salmonella virulence (Zhang and others 2020).
The expression of a unique group of ISGs (IFITM3, SLC15A3, and CNP) localized to lysosomes after Salmonella infection of intestinal epithelial cells were identified by a combination of CRISPR/Cas9 screen and proteomic profile of lysosomes and found to be important in reducing the pH and protease activity of lysosomes. IFN-dependent lysosome acidification was associated with the expansion of Salmonella-containing vacuoles that is permissive for Salmonella replication, their rupture, and host cell death. Whether this is driven through ISGF3 or STAT2/IRF9 is yet to be determined.
The pathogenic role of STAT2 in Salmonella infection was recently evaluated more closely. Similar to Ifnar1-deficient mice (Robinson and others 2012), our group found that Stat2-deficient mice were less susceptible to Salmonella infection and had impaired ISG expression, in contrast to wild-type mice (Wilson and others 2019). Salmonella needs a highly oxygenated environment to expand (Rivera-Chávez and others 2016). We observed that Stat2 deficiency limited the expansion of Salmonella by impairing neutrophil function (defect in generating superoxide anion), thus leading to the establishment of a hostile hypoxic environment in the intestinal lumen, which in turn decreased bacterial burden. Loss of STAT2 also prevented Salmonella from outcompeting the healthy microbiota thereby impeding dysbiosis. Based on these findings, we concluded that STAT2 generates a proinflammatory environment, which allows Salmonella to thrive by increasing luminal oxygenation driven by neutrophils. However, the specific mechanisms by which STAT2 promotes this inflamed state are yet to be elucidated.
Type I IFNs are also detrimental in infection with intracellular Gram-positive Listeria monocytogenes, a foodborne pathogen, by inducing lymphocyte death (Auerbuch and others 2004; O'Connell and others 2004). Induction of apoptosis-associated genes by type I IFN in a STAT1-dependent manner was first proposed as a mechanism to explain susceptibility to infection (Stockinger and others 2002). Subsequent studies showed Listeria infection triggers pyroptosis in macrophages, an inflammatory form of cell death marked by rupture of the plasma membrane, whereby inflammasome activation results in caspase 1/caspase 11 activation, cleavage of pro-IL1-β and pro-IL-18, and activation of pore-forming protein Gasdermin D (Cervantes and others 2008). IFN-γ controls Listeria infection, so a second mechanism was proposed, by which type I IFN antagonizes the antimicrobial effects of IFN-γ in macrophages (Rayamajhi and others 2010). As part of the probacterial effect of type I IFN, Stat2-deficient mice were also found to be more resistant to Listeria infection (Shaabani and others 2021).
Although type III IFN responses have yet to be determined, the possibility that it is detrimental in Listeria infection due to its dependence on STAT2 is plausible. In addition to functioning as a positive activator of type I IFN signaling, it is important to highlight that STAT2 is also a negative regulator of the type I IFN pathway. STAT2 functions as an adaptor molecule to USP18, an ISG, to desensitize cells to IFNs (Arimoto and others 2017). Paradoxically, USP18 expressed in dendritic cells was shown to promote the replication of Staphylococcus aureus and Listeria by inhibiting the production of antimicrobial IFN-γ and TNF-α (Shaabani and others 2021).
Previously, the intestinal microbiota was reported to affect the host transcriptional response to Listeria infection (Archambaud and others 2013). The microbiota inhibited the expression of several microRNAs (miRNAs) that were inversely correlated with the expression of protein-coding genes. The expression of 4 miRNAs, miR-143, miR-148a, miR-200b, and miR-200c, was downregulated in conventional mice upon Listeria infection. Among the 16 top protein-coding genes identified during Listeria infection, germ-free and conventional mice showed upregulation of STAT2 expression. Other ISGs were also identified that are known to be STAT2 dependent. The relationship between these ISGs and miRNAs to the probacterial effects of type I IFN may shed more light into the damaging effects of type I IFN signaling in certain bacterial infections.
Most recently, a link between STAT2 and METTL3 was reported to play a proinflammatory role in the pathogenesis of neonatal bacterial pneumonia (NP), a prevalent cause of neonatal morbidity and mortality (Li and others 2021). Bacterial LPSs induce lung inflammation in neonates (Cui and others 2020). METTL3 catalyzes the methylation of adenosine to N6-methyladenosine (m6A) on mRNA transcripts, an important process in inflammatory responses. In the serum of NP patients and LPS-treated human lung fibroblasts, METTL3 was upregulated, whereas the expression of long noncoding RNA SNHG4 was downregulated. Silencing of METTL3 or overexpression of SNHG4 decreased m6A levels of STAT2 mRNA causing a reduction in STAT2 protein levels and inhibition of LPS-induced production of inflammatory cytokines that were reversed by ectopic STAT2 overexpression. This finding provides insight into the role of STAT2 as a promoter of acute lung inflammation in the setting of bacterial infection.
Our group previously reported that mice lacking Stat2 were highly susceptible to LPS-induced sepsis, as opposed to mice missing components of type I IFN signaling that survived after LPS administration (Alazawi and others 2013). This observation suggested that Stat2 had a protective role. Lethality could not be explained by induction of a cytokine storm as no exaggerated increases were noted in the levels of classical cytokines and chemokines associated with sepsis. Serum levels of TNF-α, MCP-1, and IL-6 were lower than wild-type mice, whereas the levels of IFN-γ were similar between strains. We concluded that the Stat2 deficiency phenotype was due to a distinct mechanism that involved increased cellular transmigration of immune cells. This is in stark contrast to what others have reported regarding type I IFN signaling being responsible for lethality to LPS (Karaghiosoff and others 2003; Kamezaki and others 2004; Bosmann and others 2014) as well as driving TNF-α-induced systemic inflammatory response syndrome (Huys and others 2009) and sepsis in a model of cecal ligation and puncture (CLP) (Dejager and others 2014).
In these models, expression levels of several cytokines and chemokines were drastically reduced in Ifnar1-deficient mice. The latter coincided with reduced trafficking of immune cells in the blood and their migration into tissues. In the model of CLP, antibody-mediated neutralization of IFNAR1 was shown to enhance bacterial clearance by increasing the recruitment of neutrophils (Dejager and others 2014). Therefore, in animal models of sepsis, type I IFNs are detrimental by amplifying the production of proinflammatory mediators that lead to multiorgan failure, although STAT2 has a protective effect.
STAT2 in Superinfection
Superinfection is a secondary infection that occurs following an existing infection and the most common complication of influenza illness. Significant mortality in influenza A virus-infected patients is caused by a secondary bacterial pneumonia infection (Morens and others 2008). S. aureus is the most prominent bacterium in influenza bacterial superinfection. Induction of type I and type III IFNs' antiviral response against influenza enhances susceptibility to bacterial pneumonia infection (Lee and others 2015; Planet and others 2016). Different groups have shown that while influenza virus replicates better in Ifnar1 and ifnlr1-deficient mice, these mice are superior to wild-type mice in clearing Staphylococcus during superinfection (Li and others 2012; Planet and others 2016; Shepardson and others 2016, 2018). One study reported that administration of IFN-λ had the opposite effect with detrimental consequences as it increased bacterial burden due to host immune response to influenza (Rich and others 2019).
Infection by influenza inhibits a Th17-mediated protective response intended to clear bacterial pneumonia infection during influenza and bacterial superinfection (Kudva and others 2011). Bacterial superinfection subsequently antagonizes antiviral type I IFN signaling; STAT1/STAT2 dimerization becomes impaired, causing inhibition of ISG expression and enhanced viral replication (Warnking and others 2015), a finding that can be extended to type III IFN signaling. A recent study compared the outcome of influenza and influenza methicillin-resistant S. aureus superinfection between wild-type and Stat2 null mice (Gopal and others 2018). As expected, influenza-infected Stat2-deficient mice had increased mortality, impaired viral clearance, and severe inflammatory response than wild-type mice. In the context of an influenza bacterial superinfection, Stat2-deficient mice survived and showed bacterial clearance. No discernable differences in perivascular inflammation were noted between wild-type and Stat2-deficient mice as well as in the levels of IL-17, IL-22, and IL-23 cytokines, known to be altered by influenza infection.
RNA-seq analysis revealed that expression levels of IFN-γ, IL-4, and IL-13 were increased in the lungs of Stat2 null mice in comparison to wild-type mice during superinfection. Stat2 null mice also showed elevated expression of chemokines, IFN-γ induced ISGs, and genes representing a dual phenotype of M1- and M2-type macrophages. Additionally, loss of Stat2 signaling was shown to enhance the uptake and killing of bacteria by macrophages. Furthermore, blocking IFN-γ in influenza-infected Stat2-deficient mice before bacterial infection decreased bacterial clearance.
Our most current global health challenge is coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Hu and others 2021). Other viruses that similarly cause dysregulated inflammation and acute respiratory disease are SARS-CoV-1 and MERS-CoV. Optimal and balanced activation of type I and type III IFN-dependent antiviral responses as the first line of defense can subvert SARS-CoV-2 infection. COVID-19 patients in critical condition, however, experience severe inflammation due to the induction of a pulmonary cytokine storm, defective type I IFN secretion, impaired/delayed type I IFN response (inhibiting the JAK/STAT pathway) and production of neutralizing autoantibodies to type I IFN.
All of these were associated with poor outcomes (Zhang and others 2021). SARS-CoV-2 utilizes the angiotensin-converting enzyme 2 (ACE2) as a receptor for virus entry and replication. ACE2 has been described as an ISG because of its induction by all 3 types of IFNs and virus infection (Chua and others 2020; Ziegler and others 2020; Salka and others 2021). However, recent studies invalidate that ACE2 is an ISG. Rather, a spliced variant of ACE2 (dACE2) is an ISG that is nonfunctional and unable to promote infection (Onabajo and others 2020; Blume and others 2021), indicating a protective effect by IFNs.
SARS-CoV-2 replicates poorly in wild-type and immunodeficient SCID mice that lack human ACE2 transgene. Also noted were mice deficient in Ifnar1 presenting with mild lung pathology. However, some of these mice showed an increase in viral titers and peribronchial inflammation, indicating that these mice are not a suitable model to recapitulate the pathogenesis of COVID-19. Recently, a Syrian hamster model of SARS-CoV-2 infection (Chan and others 2020) was utilized to investigate the dual role of STAT2 signaling in the pathogenesis of the disease (Boudewijns and others 2020). Infected wild-type hamsters developed severe lung pathology marked by high infiltration of neutrophils, bronchopneumonia, and edema, all of which resemble the pathology seen in COVID-19 patients (Xu and others 2020). There were no differences in viral RNA levels in the lungs of WT, Stat2-deficient, and IL28ra (also known as ifnlr1)-deficient hamsters.
Nonetheless, when compared with wild-type, Stat2-deficient hamsters showed higher titers of infectious virus in the lung that disseminated to other organs, suggesting STAT2 was critical in controlling SARS-CoV-2 viral replication. In the absence of STAT2, bronchopneumonia and perivascular edema were drastically attenuated unlike that observed with Il28ra deficiency, which could be attributed to the overlapping antiviral effects of type I IFN mediated by STAT2 that remain intact in ifnlr1-deficient mice. Infection of Stat2-deficient hamsters also showed lower baseline expression of antiviral ISGs and inflammatory cytokines (IL-6, IP-10, IFN-λ, and Mx2) with no effect on ACE2 expression. Of note, critically ill COVID-19 patients have reduced IFN responses coupled with an enhanced IL-6 and TNF-α proinflammatory response (Hadjadj and others 2020).
Furthermore, COVID-19 patients have higher rates of coinfection or secondary bacterial infection than influenza patients (Shafran and others 2021). Two studies showed that COVID-19 mortality was associated with elevated expression of both type I and type III IFNs in the lung (Broggi and others 2020; Major and others 2020). These studies also revealed that excessive production of IFN-α, IFN-β, and IFN-λ (more robustly) in the lungs by synthetic viral RNA caused damage to the lung epithelium and hindered repair by inducing p53 to inhibit cell proliferation and differentiation during influenza recovery, which enhances susceptibility to lethal bacterial superinfection. This inherently aggravates severity of disease and clinical outcomes. These findings indicate that STAT2 downstream type I and type III IFN signaling serves a dual function in controlling viral infections at the expense of causing unrestrained inflammation and severe pathologies.
STAT2 as an Inducer of Inflammatory Cytokines in Cancer
IL-6 is a major cytokine involved in the activation of the STAT3 oncogenic pathway that promotes tumor growth and metastasis (Johnson and others 2018). It was recently reported that metastatic colonization of colorectal tumor cells to the liver was reduced in Il6-deficient mice (Toyoshima and others 2019). Without IL-6, CD11c+ dendritic cells that accumulated in the metastatic liver expressed high levels of Ifna and Ifnb. When blocking Ifnar1, metastatic colonization to the liver was restored.
STAT2 is widely recognized for its role in mediating the antitumor activities of type I IFN (Clifford and others 2003; Wang and others 2003; Yue and others 2015). To investigate this closer, we induced tumors in wild-type and Stat2-deficient mice employing chemical models of skin and colorectal cancer (Gamero and others 2010). In the absence of STAT2, mice not only developed fewer tumors but also presented with an attenuated inflammatory transcriptional signature that is found before the onset of cancer. These inflammatory signatures involve the expression of chemokines (Ccl2, Ccl3, Ccl4, Cxcl9, Cxcl10) and cytokines (IL1a, Il1b, and Il6), all of which were reduced. Loss of STAT2 decreased IL-6 secretion and STAT3 activation while reconstitution of STAT2 in an established STAT2-deficient cancer cell line rescued IL-6 production. This finding, thus, uncovered a new association between STAT2 and IL-6 in the setting of skin and colorectal cancer models.
IL-6 is induced weakly by ISGF3 in response to type I IFN, but is significantly enhanced by activators of the NF-κB signaling pathway (Nan and others 2018). Analysis of the IL-6 promoter revealed an ISRE motif needed for activation and occupied by the IRF9/STAT2 complex when present at high levels in concert with the NF-κB subunit p65. STAT2 acts as a bridge whereby IRF9 binds the ISRE complex and the p65 binds the NF-κB DNA element, although STAT2 does not bind to DNA. Consequently, this generates a more robust response that further drives IL-6 transcription. Also, increased unphosphorylated STAT2 protein levels can drive the expression of additional chemokines and cytokines that are dependent on NF-κB. This study also underscored a relationship between STAT2 and lung cancer in which elevated STAT2 mRNA levels were associated with poor clinical outcome. It has yet to be determined if the protumorigenic effects of STAT2 is due to impaired type I and/or type III IFN signaling by the actions of IL-6 or other soluble factors.
STAT2 in Asthma
Asthma is a complex inflammatory disease characterized by narrowing of the airways of the lungs, eosinophilic inflammation, mucus hypersecretion, increased Th2 cytokines (IL-4, IL-5, and IL-13), elevated IgE production, shortness of breath, and wheezing (Hamid and Tulic 2009). The development and exacerbation of the disease is influenced by lifestyle, genetics, and environmental factors. Research has shown genetic variants of STAT6, STAT3, and STAT4 being strongly associated with this respiratory condition (Litonjua and others 2005; Korman and others 2008; Qian and others 2014). A more recent study further implicates the STAT family in progression of asthma by identifying a STAT2-related polymorphism directly linked to asthma susceptibility (Hsieh and others 2009). Proportions of genotypes at a single nucleotide polymorphism site in the STAT2 gene (rs2066807) were observed for both asthma and control patients.
Results indicated that the distribution of the genotypes CC/CG/GG were significantly different when these 2 groups were compared. Based upon statistical analysis, the study predicts that STAT2*C-associated variants could be correlated with increased asthma susceptibility. In this same study, TLR4 and CD40-related polymorphisms were found not to contribute to increased susceptibility to asthma. In the future, identification of STAT2 polymorphisms in the exons or promoter region will enable expansion of a database of markers and help with predicting disease susceptibility.
Type I and type III IFN levels are found increased in patients with asthma (da Silva and others 2017). Both types of IFNs have been shown to inhibit the development of Th2 cells and secretion of Th2 cytokines, which suppress allergic responses that in turn attenuate lung inflammation (Jordan and others 2007; Huber and others 2010). Intranasal delivery of IFN-λ1 was found to decrease severity of airway inflammation in an experimental model of ovalbumin-induced asthma (Li and others 2014b). A recent study looked at asthmatic children's peripheral blood mononuclear cells, which had elevated IFN-λ and STAT2 expression (Krug and others 2021). These cells were noted to have a degree of protection against rhinovirus infections. One study reported that 80%–85% of asthma exacerbations in children are associated with upper respiratory viral infections and rhinovirus being the most common (Johnston and others 1995).
Therefore, a decrease in type I and type III IFN response during viral infection in asthmatics can have detrimental effects in promoting asthma exacerbation by the inability of IFNs to restrict Th2 cytokine secretion. Collectively, these studies imply a protective anti-inflammatory effect of STAT2 and identifies a possible target for the induction of antiviral responses in asthmatic children.
Glucocorticoids are a first-line treatment in the prevention and symptomatic control of asthma. One study looked at the association of glucocorticoid usage among asthmatic children and rhinovirus infection. The study showed reduced type I IFN signaling among glucocorticoid-treated children and an association with rhinovirus replication (Marcellini and others 2021). Stimulation of rhinovirus-infected Beas 2b epithelial cells with IFN-β in the presence of the glucocorticoid, fluticasone propionate, decreased mRNA expression of ISGs. This reduction in ISG expression was due to impaired STAT1 and STAT2 tyrosine phosphorylation, thus preventing an antiviral response and enabling rhinovirus replication. This finding helps to explain why long-term use of inhaled corticosteroids in patients with asthma increases the risk of respiratory infections. Additionally, these data further solidify the role STAT2 plays in this process.
STAT2 in Inflammatory Bowel Disease
Inflammatory Bowel Disease (IBD) is a chronic intestinal condition characterized by inflammation of the digestive tract. IBD occurs in 2 forms: Crohn's Disease (CD) and Ulcerative Colitis (UC) (Friedrich and others 2019). Type I and type III IFNs have been studied in models of acute and chronic colitis and are considered a double-edged sword as they can reduce or intensify the severity of inflammation (Rauch and others 2014; McElrath and others 2021; Wallace and others 2021; Xu and others 2021).
A growing body of evidence shows that patients with active IBD displaying a high IFN response signature are poor responders to anti-TNF-α drugs (Andreou and others 2020; Mavragani and others 2020). In addition, IFN-λ is increased in the serum of CD patients with active disease (Günther and others 2019). Clinical trials of type I IFN therapy have produced mixed results (Musch and others 2002; Pena Rossi and others 2009); therefore, the beneficial effects of type I IFN therapy in treating IBD remain controversial. IFN-λ has not yet been tested to treat IBD but given the detrimental and beneficial effects seen in mouse models, like those with type I IFN, extreme caution is urged.
Today, hardly anything is known about STAT2 in IBD and no genetic association to UC or CD has been reported. Current knowledge regarding the role of STAT2 in IBD is limited to flow cytometric analysis of different STATs in lamina propria lymphocytes from healthy individuals and patients with active disease in which intracellular levels of STAT2 were decreased in both UC and CD patients (Mudter and others 2005). As the sample size of this study was small, more studies that use a larger cohort of patients would be required to elucidate the activation status of STAT2. In another study, IRF9 was found to be proinflammatory, in a type I and type III IFN independent noncanonical fashion, by forming a complex with STAT1 in an acute model of dextran sodium sulfate (DSS)-induced colitis (Rauch and others 2015). From this study, it was implied that STAT2, as part of the ISGF3 complex, is protective against acute colitis.
In a mouse model of CD, in which Caspase 8 is deleted in the intestinal epithelium (Casp8ΔIEC ), mice spontaneously developed inflammatory lesions in the terminal ileum (Günther and others 2011). Administration of DSS resulted in colonic inflammation and necroptosis of Paneth cells, which produce antimicrobial peptides.
Treatment of Casp8ΔIEC intestinal organoids with either IFN-β- or TNF-α-induced cell death (Stolzer and others 2021). This same study investigated the different contributions of STAT1 and STAT2 in this model. In vivo, STAT1 played no role in inducing inflammation and instead, partially contributed to the death of Paneth cells. In contrast, Paneth cell death occurred independently of STAT2. However, unlike STAT1, STAT2 contributed to the severity of inflammation. In a different study, expression of IFN-λ in Casp8ΔIEC mice was lethal by promoting massive epithelial cell death and loss of immune homeostasis independently of TNF-α through STAT1 signaling (Günther and others 2019). However, the potential contribution of STAT2 in this context is still unclear. These findings highlighted the distinctive, nonoverlapping roles that STAT1 and STAT2 serve in intestinal inflammation.
STAT2 in Type I Interferonopathy
Unrestrained activation of type I IFN signaling pathway can result in a group of disorders defined as type I interferonopathy (Crow and Stetson 2021). Patients with type I interferonopathy often show signs of severe auto-inflammation such as cerebral calcifications and skin ulcerations, in addition to upregulation of IFN-α and aberrant induction of ISGs. Recent studies have implicated STAT2 in the pathogenesis of type I interferonopathies by demonstrating that STAT2 mutations that directly impact its interaction with USP18 interfere with the role of STAT2 as a key negative regulator of the type I IFN pathway.
Two independent studies identified children who died due to excessive type I IFN activity and severe inflammation caused by a homozygous germline missense mutation in STAT2. In the first study, a lethal germline homozygous variant of STAT2 at position c.442CC>T was identified in 2 children (Duncan and others 2019). This mutation resulted in an amino substitution at arginine 148 with tryptophan (R148W). As a result, this mutation affected the recruitment of USP18 to IFNAR2, causing desensitization to type I IFN stimulation. Dysfunction of the negative regulatory system, thus, caused prolonged tyrosine phosphorylation, nuclear localization of STAT2, heightened ISG expression, and hyperinflammation. In the second study, arginine at position 148 was replaced by glutamine (R148Q) at the site where USP18 binds (Gruber and others 2020).
This mutation does not interfere with ISGF3 activity, nor does it inhibit the interaction between USP18 and R148Q STAT2; however, it entirely impairs the ability of STAT2 to mediate USP18 trafficking to the receptor to turn off type I IFN signaling. Whether type III IFN contributes to this phenotype is unclear. Altogether, this contributed to the development of severe type I interferonopathy in the patient. Research that furthers our understanding of type I interferonopathies early in their development will prove valuable to efforts toward therapeutic treatments.
STAT2 in Psoriasis
Psoriasis is a chronic inflammatory skin disease that commonly results in the formation of lesional plaques, which appear as red, silvery, scaly patches on the skin. Pathogenesis of psoriasis is marked by the penetration and infiltration of immune cells, namely Th1 and Th17 cells, into superficial layers of the skin (Nograles and others 2008). While previous research had demonstrated increased expression of STAT1 and STAT3 in psoriatic skin, recent studies point to the involvement of STAT2 in the pathogenesis of the disease. Genome-wide association studies show STAT2 as a psoriasis susceptibility gene (Gupta and others 2014; Yin and others 2015; Fodil and others 2016). A recent study revealed activation of STAT2 signaling in skin lesions of patients with psoriasis (Johansen and others 2017). Paired skin biopsies taken from psoriasis patients with lesional and nonlesional skin as well as skin from healthy individuals were analyzed for STAT2 mRNA levels.
Only lesional psoriatic skin displayed elevated STAT2 mRNA expression when compared with normal and nonlesional skin. This increase in STAT2 expression also matched at the protein level. Psoriatic skin lesions not only had elevated STAT2 protein but also displayed activated STAT2. Curiously, no changes in STAT2 expression were detected in atopic dermatitis, a different inflammatory skin condition. This study also reported that human keratinocytes responding to type I IFN stimulation produced the chemokines CCL5 and CXCL11, both of which required STAT2 but not STAT1 for their induction. These findings imply that STAT2 is involved in the recruitment of immune cells into the epidermal and dermal layers of the skin. In a different study, T cell lines generated from the skin of psoriasis patients showed increased sensitivity to type I IFN when compared with T cells from healthy subjects (Eriksen and others 2005).
The level and kinetics of STAT2 activation by IFN-β were elevated and prolonged when compared with STAT1 activation. Another study profiled transcriptomic changes in psoriatic keratinocytes isolated from paired lesional and nonlesional skin of psoriasis patients (Pasquali and others 2019). A subset of upregulated genes identified in psoriatic lesions contained binding sites for STAT2. The role of type III IFN in the pathogenesis of psoriasis is not entirely clear. Psoriatic lesions display elevated IFN-λ associated with an ISG signature (Wolk and others 2013) and psoriatic patients have elevated serum levels of IFN-λ (Cardoso and others 2016). Treatment of human keratinocytes with type III IFN induced chemokines CXCL10 and CXCL11 (Witte and others 2016). Of note, induction of CXCL11 is mediated by STAT2 in response to type I IFN. This hints at a shared response between these 2 cytokines and their overlapping contribution to the pathogenesis of psoriasis.
Current knowledge pertaining to psoriasis pathogenesis also encompasses our understanding of risk loci and variants that confer risk for psoriasis and psoriatic arthritis (PsA). PsA is a chronic inflammatory condition closely linked to psoriasis (Rahmati and others 2020). To date, all risk loci known to be associated with psoriasis also confer risk for PsA. Thus, identifying risk loci that distinguish risk for PsA from psoriasis can significantly aid in the development of treatments and processes by which patients at high risk for each inflammatory condition are identified. These data point to STAT2 as having an active deleterious role in psoriasis.
Concluding Remarks
Over the years, multiple studies have reaffirmed the essential role of STAT2 in activating the transcriptional response to type I and type III IFNs. It is becoming increasingly clear that STAT2 has distinct functions in pathogenic infections, autoimmune and autoinflammatory diseases as depicted in Fig. 2. STAT2 is protective against viral infections while deleterious in bacterial infections. Loss of STAT2 can lead to hyperinflammation and tissue injury in the setting of viral illness due to the failed attempt of the host to clear the virus. Severe inflammation is also observed in certain bacterial infections and in superinfections; however, in this case, STAT2 activates a damaging transcriptional inflammatory program. Similarly, severe inflammation is noted in patients with persistent type I and type III IFN signaling as observed in multiple human diseases (IBD, psoriasis, type I interferonopathies).

STAT2 plays a dual role as a transcription factor involved in promoting proinflammatory and anti-inflammatory activities. Unrestrained STAT2 signaling due to uncontrolled infection, tissue injury, or heightened IFN-I production can lead to detrimental and severe inflammation. Shown are the various diseases to which STAT2 can be associated as either protective or pathogenic or both.
It is important to consider that STAT2 is multifaceted: initially, it assumes the role of an activator and later, functions as a suppressor of type I IFN signaling. This became apparent with the identification of individuals born with a primary immunodeficiency disorder characterized by a STAT2 deficiency.
During childhood, they experienced recurrent viral infections and others born with a lethal homozygous STAT2 variant (R148W/Q), which cannot restrict type I IFN signaling and led to severe inflammation. In that regard, STAT2 can be viewed as an immune rheostat. However, whether the inflammatory effects of STAT2 are driven solely by a secondary transcriptional response mediated by unphosphorylated STAT2/IRF9 complex and if this operates independently of type I or type III signaling, is still unknown. Already, STAT2 has been shown to promote the expression of protumorigenic cytokines, IL-6 and TNF-α. This may aid in explaining the contribution of STAT2 in promoting cancer. Nevertheless, mechanistic studies are warranted to understand mechanistically how STAT2 promotes hyperinflammation and whether this feature is regulated by post-translational modifications and association with other proteins. Insight into these inquiries will prompt the development of treatments that target key factors in a myriad of inflammatory diseases assumed to be linked to STAT2.
Footnotes
Acknowledgments
The authors thank Kennedy Darling and Nataliya Pryimych for their critical reading of this article. Figures were created with Biorender.com.
Authors' Contributions
A.M.G. conceptualized, wrote, and approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by grants to A.M.G. from the Department of Defense (CA170751) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R21 AR078350-01A1).