
Abstract
Type III interferons (IFN-lambdas, IFN-λs) are important antiviral cytokines that can also modulate immune responses by acting through a heterodimeric receptor composed of the specific and limited expressed IFN-λR1 chain and the ubiquitous IL-10R2 chain, which is shared with IL-10 family cytokines. Conflicting data have been reported regarding which cells express the IFN-λR1 subunit and directly respond to IFN-λs. This is, in part, owing to transcript levels of the IFN-λR1 gene, IFNLR1, not always correlating with cell surface protein levels. In this study, we tested a panel of novel monoclonal antibodies (mAbs) that specifically recognize human IFN-λR1. Initially, antigen specificity was confirmed by enzyme-linked immunosorbent assay (ELISA), from which a subset of antibodies was selected for additional flow cytometry and neutralization assays. We further characterized two antibodies based on their strong ELISA binding activity (HLR1 and HLR14) and found only HLR14 could reliably detect cell surface IFN-λR1 protein on a variety of cell lines by flow cytometry. HLR14 could also detect IFN-λR1 protein on certain primary human blood cells, including plasmacytoid dendritic cells and B cells from peripheral blood. Availability of the HLR14 mAb will enable the quantification of IFN-λR1 protein levels on cells and better characterization of the cell specificity of the IFN-λ response.
Introduction
Type III interferons (IFNs) are a family of innate immune cytokines that were originally discovered in 2003 (Kotenko et al., 2003; Sheppard et al., 2003). The IFN-λ gene (IFNL) cluster encodes four distinct proteins designated as IFN-λ1-4, which are functionally related to both type I IFNs and IL-10 family cytokines, including IL-10, IL-22, and IL-26 (Kotenko et al., 2003; Pestka et al., 2004; Prokunina-Olsson et al., 2013; Sheppard et al., 2003). Together, type I and type III IFNs are essential for innate immune responses during viral infections especially at mucosal barriers (Ank et al., 2006; Davidson et al., 2016; Doyle et al., 2006; Egli et al., 2014b).
Type III IFNs differ from type I IFNs in part by signaling through a distinct heterodimeric receptor complex composed of the high-affinity ligand-binding chain, IFN-λR1, and the low-affinity accessory chain, IL-10R2. The IL-10R2 chain is a shared receptor chain in the receptor complexes for several IL-10–related cytokines, including IL-10, IL-22, and IL-26 (Donnelly et al., 2004; Kotenko et al., 2001). In contrast, the IFN-λR1 chain is specific for the IFN-λ proteins, IFN-λ1, -λ2, -λ3, and -λ4 (Hamming et al., 2013; Kotenko et al., 2003).
Unlike type I IFN receptors, IFN-λR1 has a more limited cellular distribution that includes hepatocytes, keratinocytes, and epithelial cells such as respiratory and gastrointestinal epithelial cells (Broggi et al., 2020; Mahlakoiv et al., 2015). In addition, IFN-λR1 is expressed by a subset of leukocytes, including B cells, neutrophils (mice only), and dendritic cells (Broggi et al., 2017; Egli et al., 2014a; Megjugorac et al., 2009; Mordstein et al., 2010; Santer et al., 2020; Sommereyns et al., 2008). Owing to lack of monoclonal antibodies (mAbs) specific for IFN-λR1, cellular and tissue distribution information regarding IFN-λR1 has thus far been primarily based on IFNLR1 mRNA expression data.
Studies of cytokine function rely heavily on the use of reliable antibodies to not only identify cells presenting specific receptors but also to specifically neutralize receptor-driven signaling. Motivated by the lack of available antibodies to not only identify cells displaying IFN-λR1 on the cell surface but also neutralize IFN-λR1–mediated signaling, we generated a panel of mAbs to human IFN-λR1 (hIFN-λR1). These were screened for functional characteristics, including the ability to neutralize IFN-λR1–mediated signaling and to detect and measure the cell surface protein by flow cytometry. Functional characteristics of this panel of mAbs were compared with a commercially available anti-human hIFN-λR1 mAb (PBL clone MMHLR-1 referred to here as HLR1), which was known to neutralize type III IFN-mediated antiviral activity, but had not been tested previously for its ability to detect membrane IFN-λR1 protein.
In this study, we describe several mAbs that can be used to specifically identify human IFN-λR1 by enzyme-linked immunosorbent assay (ELISA) and flow cytometry on a range of cell types. These anti-human IFN-λR1 antibodies can also be used to neutralize type III IFN-mediated signal transduction and antiviral activity efficiently and specifically. These new antibodies open the field for further study of IFN-λR1 biology, which was not possible in the 20 years since the receptor was originally discovered.
Materials and Methods
Ethics statement
Human ethics protocols for blood collection were approved by the University of Manitoba Biomedical Research Ethics Board (No. HS25252). All blood donors gave written informed consent in accordance with the Declaration of Helsinki.
Antigen and immunization of mice for antibody generation
For antibody generation, the extracellular domain (ECD) of hIFN-λR1 (corresponding to residues 21–228 of UniProtKB Q8IU57.1; as given in Fig. 1A) was expressed as a soluble protein in HEK293-EBNA1 cells and purified to 95% homogeneity via the methods previously described (Durocher et al., 2002; Tom et al., 2008). Mice were immunized with purified IFN-λR1-ECD and spleens harvested for hybridoma generation via standard protocols (Ausubel et al., 1987).
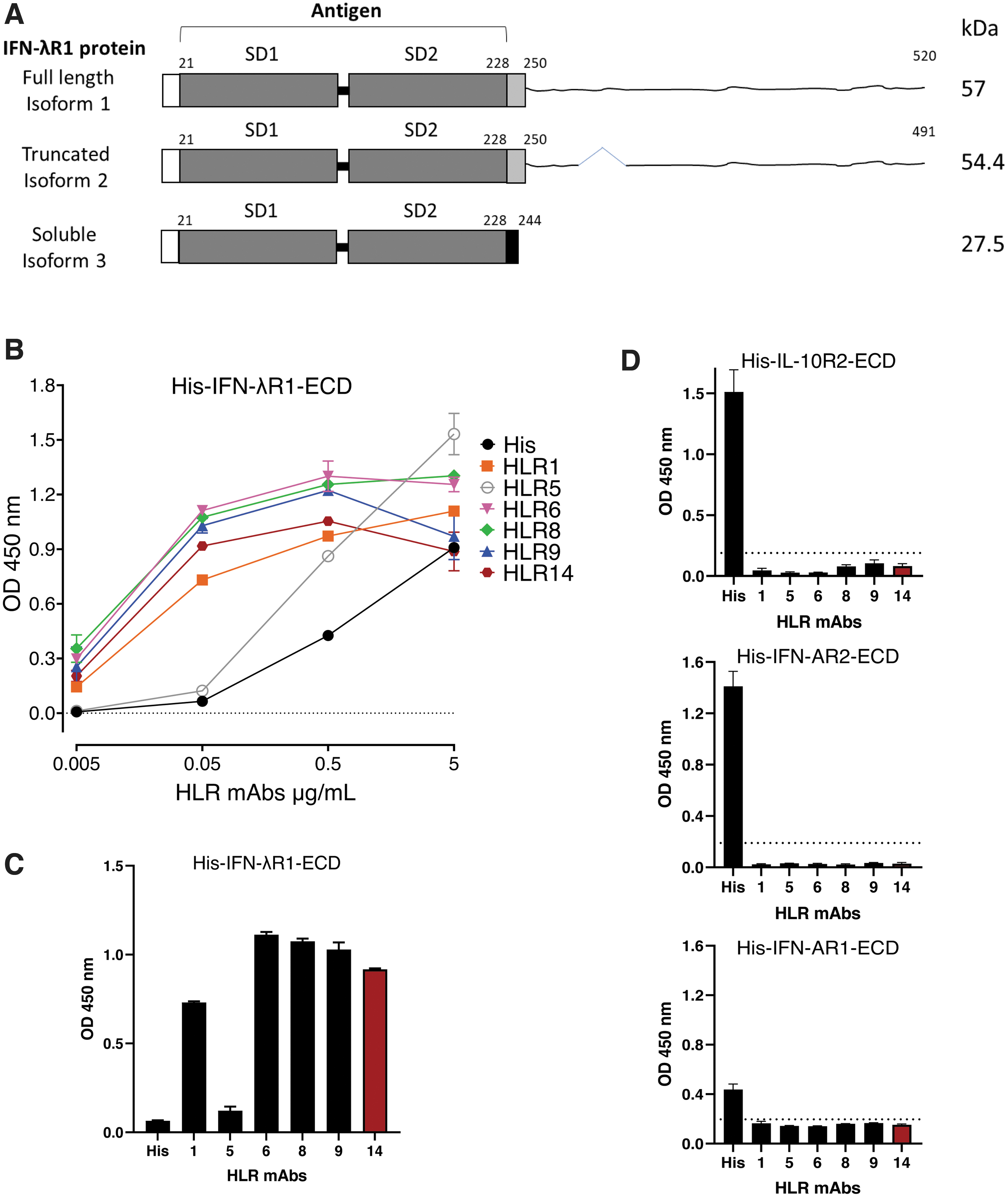
Screen of anti- IFN-λR1 mAbs by ELISA.
Purification of mAbs
Antibodies selected for full characterization were harvested from hybridoma culture supernatants and purified by Protein G affinity chromatography by standard protocols (Grodzki and Berenstein, 2010). Protein concentration was determined by absorbance at OD280 nm and normalized using an extinction coefficient of 1.43. HLR1 refers to commercially available mAb from PBL Assay Science (No. 21885-1). HLR8, 9, and 14 were derived from a common parental clone (2A12), whereas HLR5 and 6 were from independent clones.
Expression and purification of recombinant receptors
IFN-λR1-ECD
For assessment of antibody binding by analytical methods, recombinant human IFN-λR1-ECD was expressed and purified independently of the antigen used for antibody generation as described previously. IFN-λR1-ECD (corresponding to residues 21–228 of NCBI Ref Seq: NP_734464.1) with an N-terminal MGILPSPGMPALLSLVSLLSVLLMGCVA secretion signal and a C-terminal 6xHis tag) was subcloned into a pcDNA5/FRT plasmid and used to stably transform a pFRT HEK293S cell line as per manufacturer's instructions (Invitrogen™ Flp-In™). Following expansion of the HEK293S-interferon lambda receptor line to 1-L culture, the media were harvested and dialyzed into 10 mM Tris pH 8.0, 150 mM NaCl (buffer 1). The protein was then applied to a nickel sepharose column and washed with six column volumes in buffer 1 before elution of bound protein with buffer 1 with 50 mM EDTA. Eluted protein fractions were pooled and loaded onto a S75 16/60 gel filtration column (Cytiva) equilibrated in buffer 1 and purified fractions collected.
IL-10R2-ECD
Recombinant human IL-10R2-ECD (hIL-10R2-ECD) (corresponding to residues 20–220 of UniProtKB deposit Q08334.2) was subcloned into the pHLsec vector and used to transiently transfect 1 L of HEK293T cells. The protein was purified via nickel affinity and size exclusion as described previously for IFN-λR1-ECD.
IFN-AR2-ECD
Recombinant human IFN-AR2-ECD (hIFN-AR2-ECD) [corresponding to residues 28–242 of hIFN-AR2-ECD (GenBank: CAG46616.1)] was amplified from an IFNAR2 clone (Novick et al., 1994) using Pfu polymerase and specific forward (GGTCACCGGTTCATATGATTCGCCTGATTACACAGAT) and reverse primers (AAATGC-ACCCTCCTTCCACCTGGCCAGGAATCAGAATCAGCAGAATCTGCC). The amplified product was cloned into the AgeI and KpnI sites of the mammalian expression vector pHL-Sec (Aricescu et al., 2006). The resultant protein has a GS linker, enterokinase site and 6xHis tag upstream of the C-terminal stop codon. Recombinant IFN-AR2-ECD was purified from cell culture supernatants of transfected HEK293S cells as previously published (de Weerd et al., 2013).
IFN-AR1-ECD
Recombinant human IFN-AR1-ECD (hIFN-AR1-ECD) (corresponding to residues 28–436 of NCBI Ref Seq: NP_000620.2) was amplified from an IFNAR1 clone (Lutfalla et al., 1992) using Pfu polymerase and cloned into a modified version of pFastBac HTa vector (Invitrogen) previously reported.8 The His-tagged expressed protein was purified from insect cell culture by the method previously described (de Weerd et al., 2013).
Enzyme-linked immunosorbent assay
Antibodies were analyzed by direct ELISA for binding to immobilized recombinant His-hIFN-λR1-ECD, His-hIFN-AR2-ECD, His-hIFN-AR1-ECD or His-hIL-10R2-ECD. A 96-well microtiter plate (NUNC MaxiSorp) was coated with 100 μL of 1 μg/mL of protein diluted in 50 mM carbonate/bicarbonate coating buffer pH 9.5 at 4°C for 16 h. After washing with phosphate-buffered saline (PBS) pH 7.4 + 0.05% containing Tween 20 (PBST), plates were blocked with 1% (w/v) bovine serum albumin (BSA) in PBS pH 7.4 at 22°C for 2 h. About 100 μL of anti-hIFN-λR1 antibodies diluted in PBST as specified were added into each well and the plates incubated at 22°C for 2 h before addition of 100 μL of polyclonal rabbit anti-mouse HRP-conjugated IgG (diluted 1:2000 in PBST; Dako). For assessment of binding to hIFN-λR1-ECD, the antibodies were diluted to concentrations of 0.005, 0.05. 0.5, and 5 μg/mL.
For assessment of binding to His-hIFN-AR2-ECD, His-hIFN-AR1-ECD, and His-hIL-10R2-ECD, the antibodies were applied at a concentration of 0.2 μg/mL. Binding was detected by the addition of 100 μL of 3,3′,5,5′-tetramethylbenzidine substrate and incubated for 5 min at room temperature. The reaction was terminated by adding 50 μL of 2 M H2SO4 solution, and immediately assessed at OD450 using a FLUOstar OPTIMA (BMG LABTECH) reader.
Culture of human cells
A549 cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS; Gibco), 1%
Daudi cells were cultured in RPMI-1640 (Gibco) +10% FCS (Corning), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco), 10 mM HEPES (Gibco), and 1 × GlutaMAX (Gibco). Peripheral blood mononuclear cells (PBMCs) were isolated from sodium heparin blood tubes from five different healthy controls as previously described using Lymphoprep density centrifugation (STEMCELL Technologies) (Santer et al., 2020). All cell types were maintained in a humified incubator at 37°C in 5% carbon dioxide (CO2).
Flow cytometric analysis
Cells were detached (Huh7.5, A549, Caco-2) from the culture flask at ∼70%–80% confluency after treatment with accutase (Millipore Sigma), TrypLE™ (Gibco), or taken directly from the flask (Daudi) or tube (PBMCs) and counted so that 200,000 cells (cell lines) or 500,000 cells (PBMCs) were distributed into 1.2 mL library tubes. Accutase was chosen over a traditional 0.05% trypsin enzyme-based detachment method to minimize disruption of IFN-λR1 receptor epitopes. Nonspecific binding was blocked using TruStain FcX (Biolegend) in flow buffer [2% FCS in PBS (Gibco)] for 15 min at room temperature. Without washing off the block, 1.5 μg/mL hIFN-λR1 [HLR-1, HLR5, HLR6, HLR8, HLR9, HLR14, HLR14-bio Ab, or 5 μg/mL of anti-FLAG antibody (Sigma) for Huh7-IFN-λR1-FLAG cells was added to the cells and incubated for 45 min at 4°C, 2 μg/mL of anti-mouse Alexa Fluor 647 secondary antibody (Invitrogen) or 0.33 μg/mL streptavidin–PE (SA-PE (eBioscience)] was added for 40 min, covered with foil at 4°C for detecting unlabeled antibody or biotin-labeled antibody, respectively.
Secondary antibody or SA-PE was added alone to determine background binding and gate setup where background was routinely 0.4%–2%. After washing, 2% paraformaldehyde (Electron Microscopy Sciences) was added to fix cells for 15 min at room temperature in the dark. Samples were analyzed on LSRFortessa (BD Biosciences) or CytoFLEX (Beckman Coulter). FlowJo software (v10, BD Biosciences) was used for data analysis and graph generation. Cell types in PBMCs were identified based on the following markers (all lineage marker antibodies from Biolegend): B cells (CD3-CD56-CD19+), CD4+ T cells (CD3+CD4+CD8-CD56-), plasmacytoid dendritic cells (CD123++CD4+ HLA-DR+), and CD8+ T cells (CD3+CD8+CD4−CD56−). All flow cytometry experiments with PBMCs were completed with freshly isolated cells. Biotin was conjugated to HLR14 using the biotin conjugation kit from Biotium according to the manufacturer's instructions (HLR14-bio Ab).
Neutralization of IFN-mediated gene induction
Huh7.5 cells were plated at 180,000/well in DMEM complete media in a 12-well plate. After 24 h, the cells were preincubated with 5 μg/mL of HLR5, HLR6, HLR8, HLR9, and HLR14 or control mouse IgG1 (Invitrogen) for 60 min in DMEM media. With the antibody still in the well, 10 ng/mL IFN-λ3 (R&D Systems) or Universal IFN-α (R&D Systems) cytokine was added to the cells and incubated for 5 hours in a humidified incubator with 5% CO2 supply at 37°C. After 5 h, the cells were washed with plain PBS. The cells were lysed after using 600 μL TRIzol™ (Invitrogen) and stored at −80°C. RNA was isolated using the Zymo Research mini-RNA isolation kit according to the manufacturer's instructions. cDNA was made immediately after RNA isolation with 300–400 ng of RNA and SuperScript IV VILO mastermix (Invitrogen). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed with power SYBR green mastermix (Invitrogen) using a QuantStudio 3 real-time PCR system (Applied Biosystems). RT-qPCR primer sequences were previously published and purchased from IDT (Santer et al., 2017).
Neutralization of IFN-mediated antiviral protection assay
For comparison of neutralization of IFN-β and IFN-λ3–mediated protection from encephalomyocarditis virus (EMCV) infection, A549 cells were plated at 1.8 × 104 cells per well of a 96-well plate and allowed to adhere for 6 h at 37°C in 5% CO2. Purified anti-hIFN-λR1 mAb (HLR1 or HLR14) was added to wells and serially diluted 2-fold down the plate from a starting concentration of 20 μg/mL and incubated for 30 min at 37°C in 5% CO2. IFN-β (100 IU/mL) or IFN-λ3 (100 ng/mL) was added and the plate incubated for 16 h at 37°C in 5% CO2 before viral infection. EMCV was added as mentioned previously except for 3 days at 37°C in 5% CO2. Live cells remaining after incubation were quantified by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (Stifter et al., 2014). Data are represented as percentage of live cells [mean ± standard error of the mean (SEM)] remaining after the assay from three independent experiments.
Results
Enzyme-linked immunosorbent assay
A panel of 6 mAbs (Table 1) was initially screened for binding to immobilized recombinant native hIFN-λR1-ECD by ELISA. The antibodies displayed variable binding levels depending on the concentration of antibody protein added. Clone HLR5 showed high levels of binding at 5 and 0.5 μg/mL, but barely detectable binding when diluted to 0.05 μg/mL (Fig. 1B). All other antibody clones were highly reactive in this assay (Fig. 1B, C). We also used direct ELISAs to assess reactivity of the panel of antibodies against recombinant forms of structurally related antigens including hIL-10R2-ECD, hIFN-AR1-ECD, and hIFN-AR2-ECD. Because the absorbance readings recorded for all the HLR antibodies were below those observed for the “no antigen” control wells, these data demonstrate that none of the HLR mAbs showed binding to any of these proteins (Fig. 1D).
Characteristics of the Panel of Mouse Monoclonal Antibodies Assessed
ELISA, enzyme-linked immunosorbent assay.
Flow cytometry
To date, it has been difficult to accurately quantify IFN-λR1 protein levels at the cell surface of human cells using putative anti-IFN-λR1 antibodies from other commercial sources. Based on our results mentioned previously, we selected a subset of our mAbs to assess by flow cytometry, including HLR5 as an example of antibody with low reactivity by ELISA as a control, and HLR1, HLR6, HLR8, HLR9, and HLR14 as promising antibody candidates.
Initially, we used an Huh7 IFNLR1 KO cell line previously shown to display significantly impaired responses to type III IFN stimulation (Jarret et al., 2016) and compared binding with the same cell line reconstituted with FLAG-tagged hIFN-λR1 to demonstrate specificity of the mAbs to hIFN-λR1 on the surface of cells. The results show that whereas HLR5 did not show any detectable binding to the surface of these cells above background levels detected by the secondary antibody alone, HLR1, HLR6, HLR8, HLR9, and HLR14 all showed significant and comparable binding (Fig. 2A). We next sought to confirm the specificity of HLR14 [one of 3 mAbs (HLR8, 9, and 14) derived from the same parental clone] binding using a Huh7 IFNLR1 knockout cell line compared with the FLAG-IFN-λR1 overexpressing line. Our results showed that ∼9.5% of the Huh7 IFNLR1 knockout cells were positive for IFN-λR1 compared with 2.9% of the secondary antibody alone.

IFN-λR1 flow cytometry analyses demonstrated specificity of antibodies.
In contrast, overexpression of FLAG-IFN-λR1 led to >99% IFN-λR1 positivity (Fig. 2B). To confirm that HLR14 was specifically detecting overexpressed FLAG-tagged hIFN-λR1, we compared the binding of HLR14 with that of an anti-FLAG antibody and found that both antibodies detected >99% of the cells (Fig. 2B, right panel).
Having demonstrated specificity, we next investigated the ability of a subset of our antibodies to bind endogenous receptors on the surface of several different human cancer cell lines, including Huh7.5 cells (hepatoma), A549 (lung epithelial carcinoma), Caco-2 (colon epithelial carcinoma), and Daudi cells (B cell lymphoblast, Burkitt's lymphoma). We found variable binding characteristics across the different cell types with the highest percentage binding to Huh7.5 cells, closely followed by Caco-2 (Fig. 3A–D). Except for HLR5, all other antibodies tested showed binding to Huh7.5 cells, Caco-2, A549, and Daudi cells, as expected based on these cell types responding to IFN-λ stimulation (Jilg et al., 2014; Kotenko et al., 2003; Santer et al., 2017, 2020; Stanifer et al., 2020). HLR8 and HLR14 showed the highest level of binding on average for each cell type tested (Fig. 3A–D).
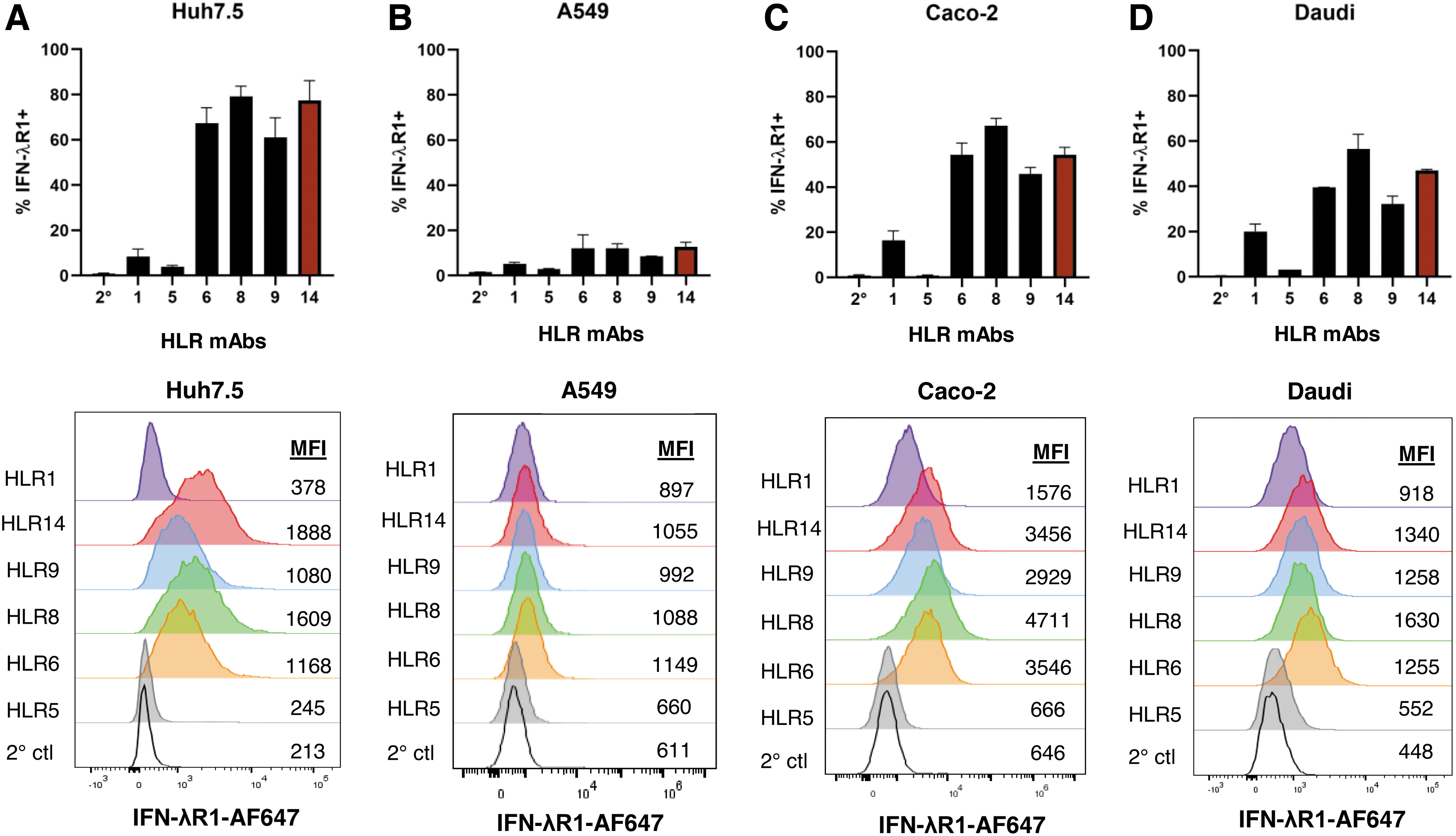
Antibodies show differential binding to surface IFN-λR1 on a variety of cell lines. IFN-λR1 antibodies were screened on Huh7.5
Although A549 cells clearly respond to IFN-λ as shown in later figures in this study or by other groups (Czerkies et al., 2022; Kotenko et al., 2003; Plotnikova et al., 2021), none of the antibodies tested detected >15% IFN-λR1 positivity on A549 cells, with HLR14 detecting the highest levels (by mean %) (Fig. 3B). In all our flow cytometry assays, HLR1 displayed a poor ability to detect IFN-λR1 on the surface of cells despite showing high binding activity to IFN-λR1-ECD by ELISA (Fig. 1B). Additional testing on Caco-2 cells showed that fixing cells first with 2% paraformaldehyde versus at the end of our staining protocol, completely abrogated detection of IFN-λR1 (average 52% among top antibodies down to 1% IFN-λR1+) (Supplementary Fig. S1A). Furthermore, the choice of detachment reagent can also affect receptor epitopes with accutase preferred over trypLE (Supplementary Fig. S1B).
For primary human cells, we assessed binding of our HLR antibodies to immune cells within peripheral blood mononuclear cells (PBMCs) isolated from healthy donors. Conflicting data exist regarding which human blood immune cells express IFN-λR1 (Dickensheets et al., 2013; Kelly et al., 2016; Santer et al., 2020; Witte et al., 2009; Yin et al., 2012). Because HLR14 showed significant binding to all cell lines tested, we prepared biotinylated HLR14 and used it to assess whether hIFN-λR1 can be detected on blood immune cells in multicolor flow cytometry experiments. Similar to our previous work with an IFN-λ3 binding assay (Santer et al., 2017) and previous reports regarding IFNLR1 mRNA expression, we found the highest binding of HLR14 to pDCs and B cells, with lower or no binding to CD8+ or CD4+ T cells (Fig. 4A, B).
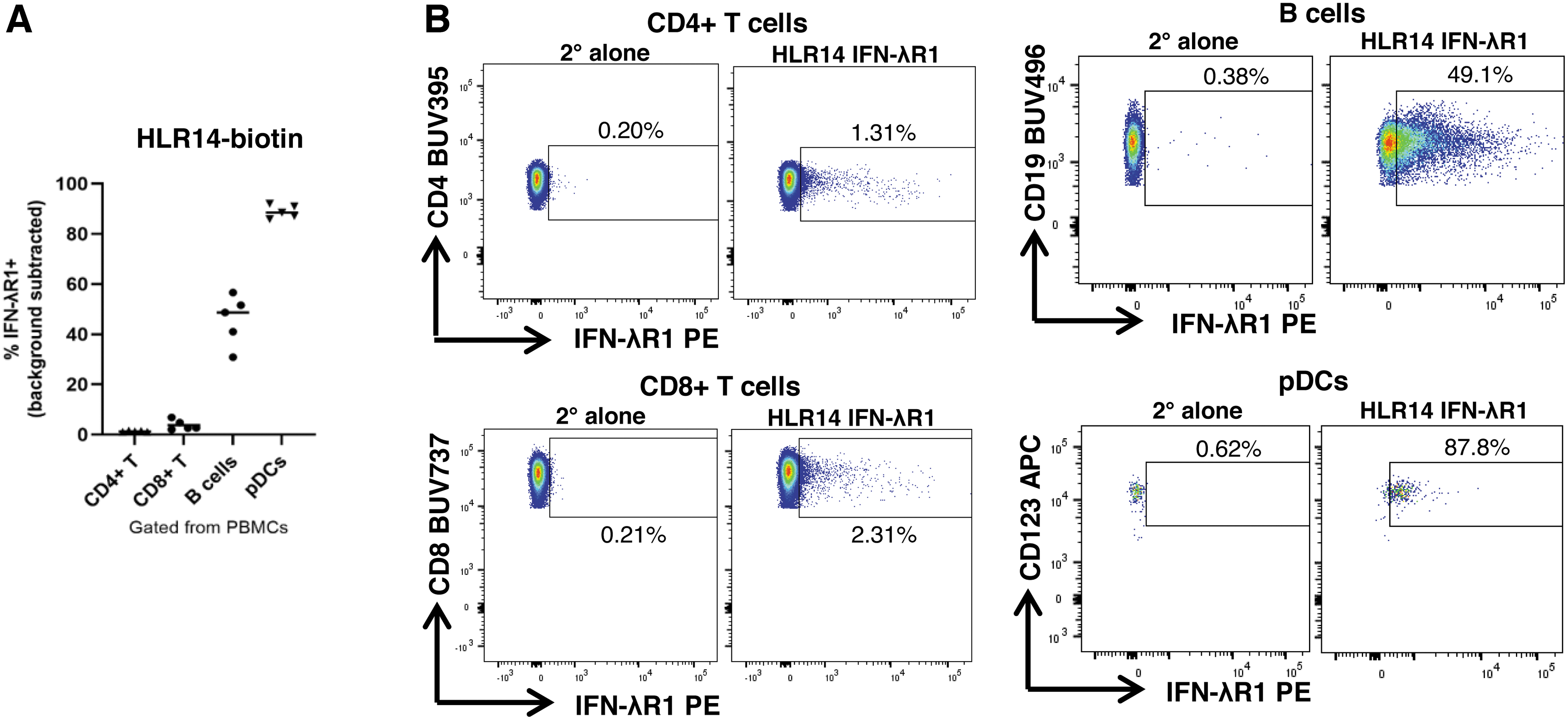
IFN-λR1 mAb HLR14 detects IFN-λR1 protein on the surface of primary human peripheral immune cell subsets.
Assessment of neutralization of IFN-mediated activities
We evaluated the ability of a subset of our HLR antibodies to neutralize functional activities mediated by IFN-λ and/or type I IFNs (IFN-α or IFN-β). HLR1 is already commercially available as an IFN-λR1 neutralizing mAb shown to block IFN-λ–mediated antiviral activities and to neutralize type III IFN-mediated gene induction in T84 (intestinal epithelial) cells (Pervolaraki et al., 2019). Initially, we examined the ability of HLR1 to neutralize IFN-mediated induction of IFIT1, OAS1, and MX1 mRNA in A549 (lung epithelial) cells as a positive control. Our results show that HLR1 completely inhibited induction of gene expression by IFN-λ1, but did not inhibit gene induction mediated by IFN-α or IFN-β confirming its ability to specifically neutralize type III IFN signaling (Supplementary Fig. S2A).
HLR1 also decreased phosphorylated STAT1 (pSTAT1) levels in A549 stimulated with IFN-λ1, but had no effect on pSTAT1 levels with IFN-α, IFN-β, or IFN-γ stimulation (Supplementary Fig. S2B). Next, we expanded our analyses to focus on our new HLR mAbs to determine if HLR5, HLR6, HLR8, HLR9, and HLR14 could neutralize IFN-stimulated gene induction in Huh7.5 (hepatocyte) cells stimulated with IFN-λ3 or IFN-α as a control. Our results show that the HLR mAbs did not inhibit the induction of IFIT1, MX1, or OAS1 genes by IFN-α (Fig. 5A). In contrast, unlike HLR5 and negative control mIgG1, HLR6, HLR8, HLR9, and HLR14 demonstrated potent neutralization of IFN-λ3–mediated gene expression of the 3 IFN stimulated genes (ISGs) by up to 87% (Fig. 5B).
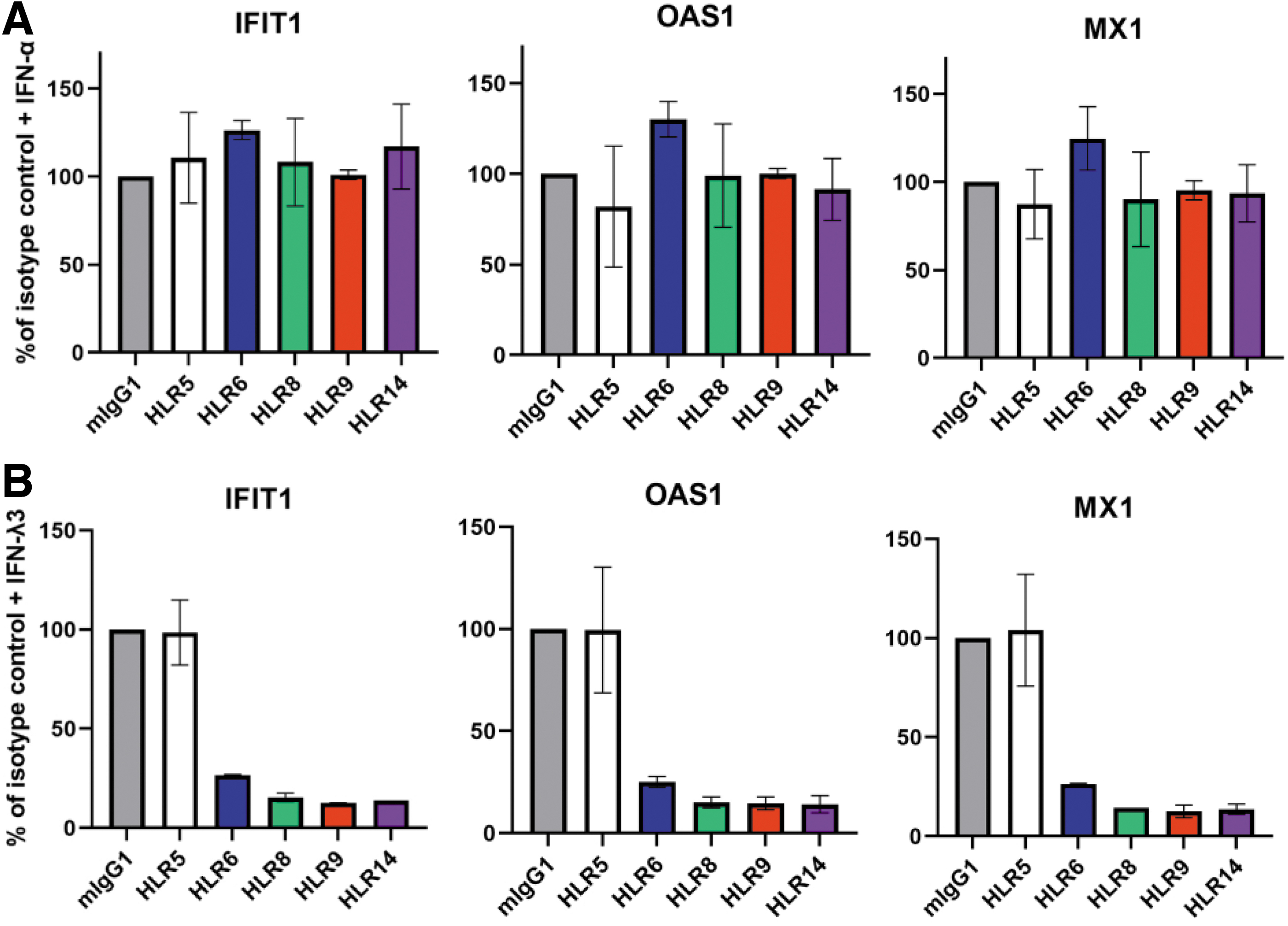
Specific neutralization of IFN-λ signaling by IFN-λR1 antibodies.
Based on the reliability of HLR14 in our flow cytometry assays (Figs. 3 and 4) and in neutralization of gene induction (Fig. 5B), we compared the ability of this mAb to neutralize IFN-λ3–mediated antiviral activity alongside the commercial mAb, HLR1. The type III IFNs are well known for their ability to protect lung epithelial cells (A549s) from the cytopathic effect of EMCV infection (Kotenko et al., 2003). Therefore, we assessed whether HLR1 and HLR14 can neutralize antiviral protection mediated by IFN-λ3 or the type I IFN, IFN-β in A549 cells. Our results show that both HLR1 and HLR14 neutralized the antiviral activity of IFN-λ3, but not that of the type I IFN, IFN-β (Fig. 6). These findings are consistent with our observations that the HLR antibodies show specificity for binding to hIFN-λR1 and not to the related type I IFN receptors (e.g., IFN-αR1 or IFN-αR2) (Fig. 1D).
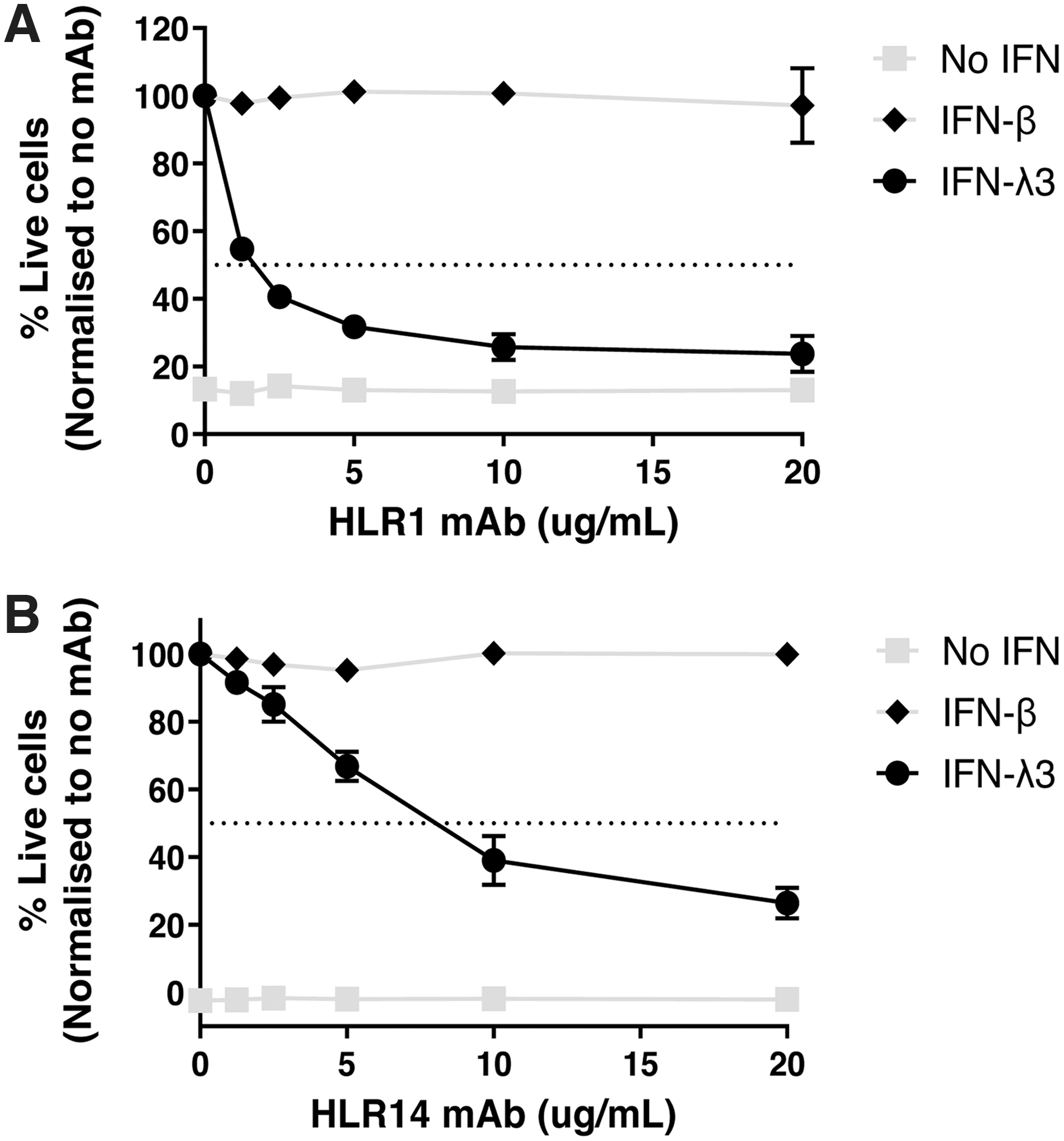
Dose–response of antibody-mediated neutralization of IFN-λ3–driven antiviral protection. A549 cells were pretreated with anti-IFN-λR1 HLR1
Our results do however demonstrate that HLR1 was more efficient than HLR14 at neutralizing IFN-λ3–mediated antiviral activity compared with HLR14, with the latter showing ∼50% neutralization capacity at ∼8 μg/mL compared with HLR1 at ∼1.5 μg/mL (Fig. 6). Although HLR1 is a more potent neutralizing mAb for cell culture assays, our data demonstrate that HLR14 is a more suitable mAb for quantifying hIFN-λR1 protein levels on various cell types by flow cytometry.
Discussion
Type III interferons (IFN-lambdas, IFN-λ) were discovered in 2003 (Kotenko et al., 2003; Sheppard et al., 2003) and since their discovery multiple groups have uncovered roles for these cytokines in regulating immunity with an emphasis on inducing strong antiviral immunity. Meanwhile, the study of the high-affinity IFN-λ receptor, IFN-λR1, has been limited by the lack of availability of reliable and specific antibodies against this protein. IFN-λR1 expression appears to be limited to epithelial cells and certain immune cell types, and most information has been gained via indirect methods such as assessment of IFNLR1 mRNA levels (Goel et al., 2020; Santer et al., 2020; Witte et al., 2009). To formulate a more in-depth understanding of IFN-λ biology, it is necessary to accurately identify cell types displaying IFN-λR1 protein by more direct means.
Hence, for this project, we generated 6 novel mAb clones and compared them with a commercially available antibody for IFN-λR1 (HLR1). We found that the commercial clone, HLR1, is a potent neutralizer, but could not detect IFN-λR1 on cell lines by flow cytometry, unless the receptor was overexpressed, whereas one of the novel clones, HLR14, was a good neutralizer and effectively detected surface IFN-λR1 protein on a variety of cell types by flow cytometry.
We tested these antibodies using several different techniques. First, we assessed the ability of all 6 antibodies to bind specifically to IFN-λR1, but not type I IFN receptors or IL-10RB, protein using an ELISA, demonstrating minimal cross-reactivity to structurally related proteins. The top HLR mAbs were not able to detect denatured IFN-λR1 in western blots (data not shown) or if cells were fixed first before staining in flow cytometry. Therefore, the HLR antibodies are likely best suited for detecting native IFN-λR1. IFN-λR1 has multiple splice variants in human cells (Evans et al., 2023; Santer et al., 2020; Sheppard et al., 2003), including a soluble form that lacks the transmembrane domain.
The IFN-λR1 ECD amino acid sequence used for immunization to generate the HLR mAbs and to detect and measure binding by ELISA is identical in each of the three splice variants. Because soluble IFN-λR1 antagonizes IFN-λ responses in cells, it will be important to fully characterize where and when it is produced in the human body (Evans et al., 2023; Santer et al., 2020). Although we did not directly measure the ability of the HLR mAbs to detect endogenous soluble IFN-λR1, they could be assessed in future studies.
Although several of the HLR mAbs could detect IFN-λR1 (HLR6, 8, 9, 14), we chose to fully characterize HLR14 as a representative clone of the 3 top antibodies derived from the same parental clone. Our results demonstrated HLR14 can detect surface IFN-λR1 protein on all cell lines tested, each chosen based on their ability to respond to IFN-λ stimulation. Despite displaying the lowest detectable levels of IFN-λR1 by flow cytometry, the lung epithelial cell line (A549) is known to be responsive to both type I IFN (Budd et al., 1984) and type III IFNs (Czerkies et al., 2022; Kotenko et al., 2003; Plotnikova et al., 2021).
A549 cells were found to have the lowest IFNLR1 mRNA of the 4 cell lines examined in our study [Human Protein Atlas,
Antibody specificity was also confirmed by examining ISG induction in Huh7.5 and A549 cells, where the top IFN-λR1 antibodies selectively inhibited IFIT1, OAS1, and MX1 mRNA induction by IFN-λs, without affecting ISG induction by the type I IFNs, IFN-α and IFN-β. Both these antibodies also efficiently neutralized the ability of type III IFNs, but not type I IFNs, to protect cells from the cytopathic effect of viral infection, an observation in line with the limited receptor cross-reactivity displayed by these antibodies by ELISA.
Altogether, these results expand the IFN-λ field to finally be able to properly quantify IFN-λR1 levels by flow cytometry and use the same antibody to neutralize IFN-λ–mediated signaling. We identified and characterized HLR14 as a novel anti-IFN-λR1 mAb that can be used to detect and quantify cell surface expression of IFN-λR1. Availability of this antibody should help to facilitate future studies to further identify and characterize type III IFN target cells across the human system.
Footnotes
Acknowledgments
The authors thank Dr. Christine Zhang at the University of Manitoba Flow Cytometry core for her support and also thank staff at the University of Alberta Faculty of Medicine and Dentistry Flow Cytometry Facility, which receives financial support from the Faculty of Medicine & Dentistry and Canada Foundation for Innovation (CFI) awards to contributing investigators. The authors also thank Dr. Faruk Sheikh (Food and Drug Administration) for his assistance with the STAT1 western blot analysis.
Authors' Contributions
Conceptualization: N.A.W., P.J.H., D.M.S.
Data curation: N.A.W., O.O., X.L., A.Y.M., A.I., J.P.V., S.S.L., M.S., N.P., H.D., T.B.L., R.P.D., D.M.S.
Formal analysis: N.A.W., O.O., X.L., A.I., N.P., H.D., R.P.D., D.M.S.
Funding acquisition: D.L.T., T.B.L., R.P.D., P.J.H., D.M.S.
Project administration: R.P.D., P.J.H., D.M.S.
Supervision: N.A.W., M.S., T.B.L., R.P.D., P.J.H., D.M.S.
Writing (original draft): N.A.W., O.O., X.L., R.P.D., D.M.S.
Writing (review and editing): all authors.
Disclaimer
This article reflects the views and opinions of the authors and should not be construed as representing the views or policies of the FDA (H.D., R.P.D.).
Author Disclosure Statement
During the preparation of the immunogen, monoclonal antibodies, and execution of certain of the experiments, N.P., M.S., and T.B.L. were employed by PBL Assay Science, which commercializes or may commercialize certain of the anti-IFN-λR1 mAbs. M.S. is a current employee of PBL Assay Science.
Funding Information
This work was supported by funding from the University of Manitoba, a Research Manitoba New Investigator Operating Grant, CFI award (No. JELF 41799) and Natural Sciences and Engineering Research Council of Canada Discovery Grant (No. RGPIN-2022-04133) to D.M.S., Australian Research Council Discovery Project (No. DP210103122) to N.A.W. and P.J.H. and Li Ka Shing Institute of Virology funding to D.L.T. This work was also supported by the Victorian Government's Operational Infrastructure Support Program and National Health and Medical Research Council (P.J.H.). O.O. and X.L. were supported by University of Manitoba Rady Faculty of Health Sciences graduate studentships.
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.