
Abstract
Pertussis, caused by Bordetella pertussis, is a resurgent respiratory disease but the molecular mechanisms underlying pathogenesis are poorly understood. We recently showed the importance of type I and type III interferon (IFN) induction and signaling for the development of lung inflammation in B. pertussis-infected mouse models. Classically, these IFNs are induced by signaling through a variety of pattern recognition receptors (PRRs) on host cells. Here, we found that the PRR signaling adaptor molecules MyD88 and TRIF contribute to IFN induction and lung inflammatory pathology during B. pertussis infection. However, the PRRs Toll-like receptors (TLR) 3 and TLR4, which signal through TRIF and MyD88, respectively, played no role in IFN induction. Instead, the DNA-sensing PRRs, TLR9 and STING, were important for induction of type I/III IFN and promotion of inflammatory pathology, indicating that DNA is a major inducer of lung IFN responses in B. pertussis infection. These results increase our understanding of this host–pathogen interaction and identify potential targets for host-directed therapies to reduce B. pertussis-mediated pathology.
Introduction
The recent resurgence in pertussis (whooping cough) is a major public health issue, with hospitalization and mortality in infants and severe prolonged disease in adults (Mattoo and Cherry, 2005). Infection of animal models with Bordetella pertussis, the causative agent of pertussis, results in prolonged inflammation of the airways (Pinto and Merkel, 2017; Scanlon et al., 2014; Zimmerman et al., 2018), but the host and pathogen factors that promote this inflammation are poorly understood.
Previously, we found that type I and type III interferons (IFNs) are induced in the lungs of B. pertussis-infected adult mice and exacerbate lung inflammatory pathology (Ardanuy et al., 2020). Type I IFNs signal through a heterodimeric receptor (IFNAR1/IFNAR2) and JAK/STAT pathways (Pestka et al., 2004). They are important cytokines for antiviral defense and are also involved in pathogenesis and inflammation in various disease models (McNab et al., 2015; Pestka et al., 2004). Type I IFNs also affect outcomes in bacterial infections, providing deleterious effects in some and protective effects in others (Gratz et al., 2011; Martin et al., 2009; Mayer-Barber et al., 2010; O'Connell et al., 2004; Plumlee et al., 2009). The more recently discovered type III IFNs (IFN-λ) induce a similar set of IFN-stimulated genes (ISGs) as type I IFNs (Lazear et al., 2019). IFN-λ signals through a JAK/STAT signaling pathway but utilizes a different heterodimeric receptor, IFNLR1/IL-10Rβ, expressed on epithelial cells and certain immune cells (Donnelly and Kotenko, 2010; Santer et al., 2020; Sommereyns et al., 2008).
Signaling through various pattern recognition receptors (PRRs) can induce type I/type III IFN in mammalian cells (Manes and Nita-Lazar, 2021). PRRs detect conserved microbial patterns, known as pathogen-associated molecular patterns (PAMPs), or host molecules released from damaged cells, known as damage-associated molecular patterns (Li and Wu, 2021). PRRs are located in the plasma membrane, endosomal membrane, or cytosol and include Toll-like receptors (TLRs), intracellular DNA sensors, such as cGAS/STING, and several others. Since B. pertussis is primarily an extracellular pathogen, surface and endosomal PRRs, such as TLR4, TLR7, and TLR9, are more likely to initiate IFN induction in response to infection than cytosolic PRRs (Higgins et al., 2003; Kawasaki and Kawai, 2014).
In contrast, multiple intracellular bacteria induce type I/III IFN in mammalian cells through nucleic acid or nucleotide sensors, including the AIM 2 inflammasome or the sensor/adaptor proteins cGAS/STING (Barker et al., 2013; Kim et al., 2017; Ori et al., 2017). Detection of unmethylated CpG bacterial DNA by TLR9 in endosomes is a major pathway for induction of type I IFN expression (Kumagai et al., 2008). In contrast, mammalian DNA has few unmethylated CpG sequences, although they are enriched in so-called “CpG islands,” particularly associated with gene promoters (Deaton and Bird, 2011), and self-DNA can be delivered to endosomes and trigger type I IFN production by complexing with antimicrobial peptides, such as LL37 or β-defensins (Lande et al., 2007; Tewary et al., 2013). In addition, mitochondrial DNA released by damaged or stressed host cells can bind and signal through cytosolic and endosomal PRRs (De Gaetano et al., 2021; Xu et al., 2022), another source of self-DNA that can contribute to IFN upregulation.
In this study, we aimed to identify the specific PRRs responsible for upregulation of type I/ type III IFN responses and associated lung inflammation in B. pertussis-infected mice. Our data indicate that these responses are dependent on TLR9 and STING and therefore that DNA is an important inducer of IFN responses in this infection.
Materials and Methods
Bacterial strain
A streptomycin-resistant derivative of B. pertussis Tohama was grown on Bordet–Gengou agar supplemented with 10% defibrinated sheep blood (Lampire, Pipersville, PA) and 200 mg/mL streptomycin at 37°C for 48 h.
Mouse infections
Mouse strains (C57BL/6, C57BL/6-129SF2/J, TRIF KO, TLR3 KO, TLR4 KO, TLR9 KO, STING KO, and MyD88 KO, all from Jackson Lab, Bar Harbor, ME) were used in accordance with an Institutional Animal Care and Use Committee protocol (University of Maryland, Baltimore, MD). Bacterial inocula were prepared in phosphate-buffered saline (PBS) and 2 × 106 colony forming units (CFU) were administered intranasally in 50 μL. C-176 STING antagonist (Med Chem Express, Monmouth Junction, NJ) was administered at 0, 2, 4 days postinoculation (dpi) intraperitoneally at 13 mg/kg per mouse. TLR9 antagonist ODN 2088 (Invivogen, San Diego, CA) was administered at 0, 2, 4 dpi at 2.5 mg/kg intraperitoneally. Following euthanasia, lungs were removed for analysis of bacterial burden, transcript levels, and inflammatory histopathology. In general, groups of mice consisted of four to six animals of both sexes. All experiments were repeated at least once.
RNA isolation and quantitative real-time reverse transcription-polymerase chain reaction
Lung tissue was flash-frozen, and RNA extraction was achieved using the RNeasy Microarray Tissue Kit (QIAGEN, Germantown, MD) following manufacturer's instructions and DNase-treated to remove DNA contamination. cDNA was synthesized using a reverse transcription system (Promega, Madison, WI) following manufacturer's instructions. Quantitative real-time polymerase chain reaction (PCR) was performed with SYBR Green/ROX Master Mix in a 7500 Fast Real-Time PCR system (Applied Biosystems, Waltham, MA). Hypoxanthine phosphoribosyltransferase (hprt) was used as a housekeeping control gene. PCR primers are listed in Supplementary Table S1. Expression was determined by calculating fold changes versus PBS-inoculated animals using the 2-ΔΔCT method.
Inflammatory pathology
Lungs were perfused with PBS, transferred to 10% buffered formalin and hematoxylin and eosin stained at the University of Maryland School of Medicine Pathology Laboratory. Histopathological findings were scored by multiple blinded investigators on a scale of 0–9 as described previously (Scanlon et al., 2014).
Statistical analysis
Data were analyzed using GraphPad Prism software. Student's t-test was used for determining significance between two groups, and two-way analysis of variance was used for determining significance between multiple groups.
Results
Type I/ type III IFN upregulation in B. pertussis-infected mice is MyD88- and TRIF-dependent
MyD88 is a crucial adaptor of numerous TLR signaling pathways, except for the TLR3/4 endosomal pathways that utilize the TRIF adaptor (Kawasaki and Kawai, 2014). B. pertussis infection with our standard dose (2 × 106 CFU) resulted in the death of all MyD88 KO mice by 7 dpi (data not shown). Using a lower dose of B. pertussis (5 × 105 CFU), the bacterial lung burden of MyD88 KO and C57BL/6 (WT) mice at 5 dpi showed no significant difference (Fig. 1A), but induction of lung type I/type III IFN expression was significantly lower in MyD88 KO mice (Fig. 1B), indicating that one or more TLR pathways utilizing the MyD88 adaptor are involved in IFN induction.
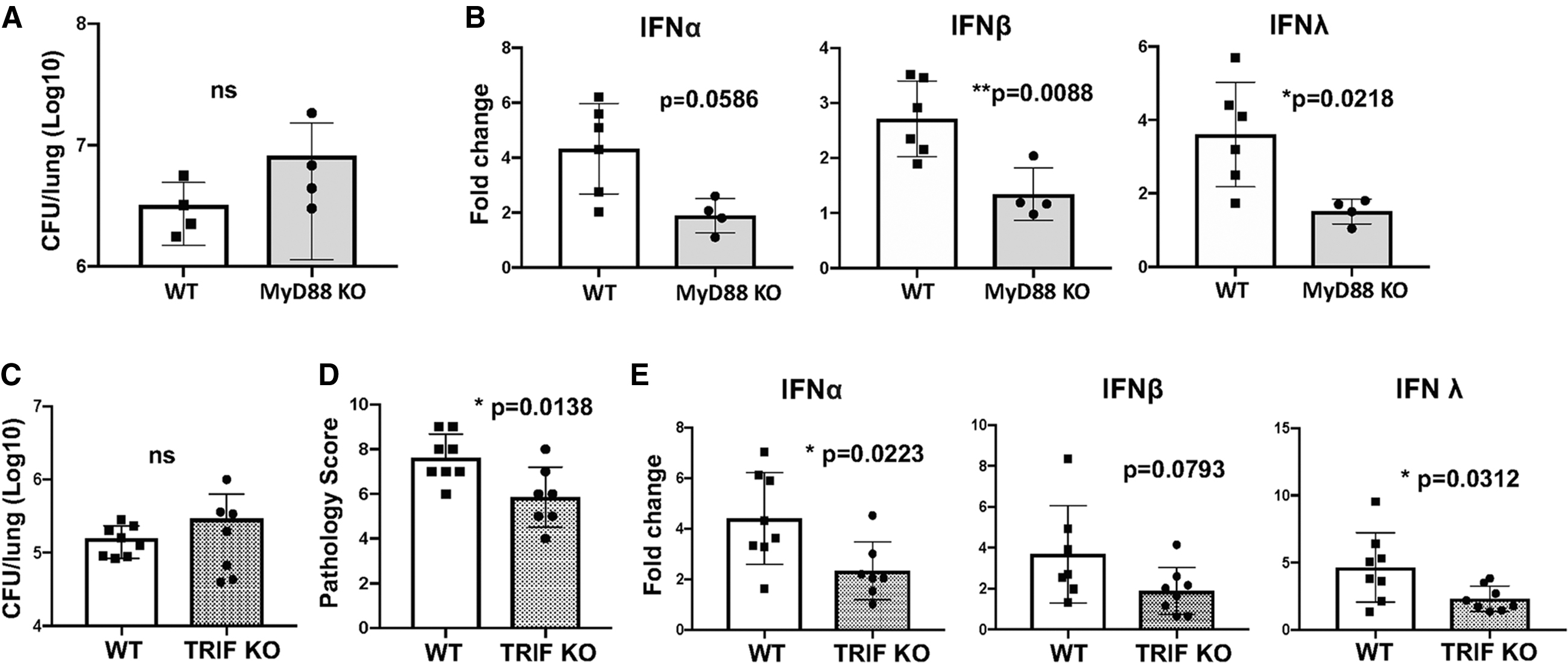
MyD88 KO and TRIF KO mice have reduced levels of induction of type I/type III IFNs in response to Bordetella pertussis infection.
The TRIF signaling adaptor is required for both TLR3 and endosomal TLR4 signaling, as well as some cGAS/STING signaling pathways (Wang et al., 2017; Yamamoto et al., 2003). At 7 dpi, lung bacterial burdens in TRIF KO mice showed no significant difference from those in infected WT mice (Fig. 1C). However, lung inflammatory pathology scores and upregulation of type I/III IFN expression levels were significantly lower in TRIF KO mice (Fig. 1D, E), indicating that TRIF is also required for robust IFN upregulation in B. pertussis-infected mice. Induction of proinflammatory cytokine gene expression was also significantly reduced in infected TRIF KO mice (Supplementary Fig. S1A). These data narrow the potential PRRs involved in type I/type III IFN induction to several candidates, including TLR3, TLR4, TLR9, and cGAS-STING.
TLR9 and STING signaling contribute to IFN induction and lung inflammatory pathology during B. pertussis infection
TLR4 senses bacterial LPS and signals from endosomes through TRIF to induce IFN expression (Kawasaki and Kawai, 2014). Despite significantly increased lung bacterial loads in TLR4 KO mice compared to WT mice, lung proinflammatory cytokine induction and inflammatory pathology were lower but no significant differences in induction of IFNs were observed (Fig. 2A–C, Supplementary Fig. S1B). Therefore, TLR4 controls bacterial growth and contributes to lung inflammatory pathology but is not involved in induction of type I/type III IFNs in B. pertussis infection. TLR3 recognizes double-stranded RNA (Kawasaki and Kawai, 2014). B. pertussis-infected TLR3 KO mice showed no significant differences in bacterial loads, lung inflammatory pathology, or IFN expression levels from WT (C57BL/6-129SF2/J) mice (Fig. 2D–F), demonstrating that TLR3 is not involved in these outcomes.
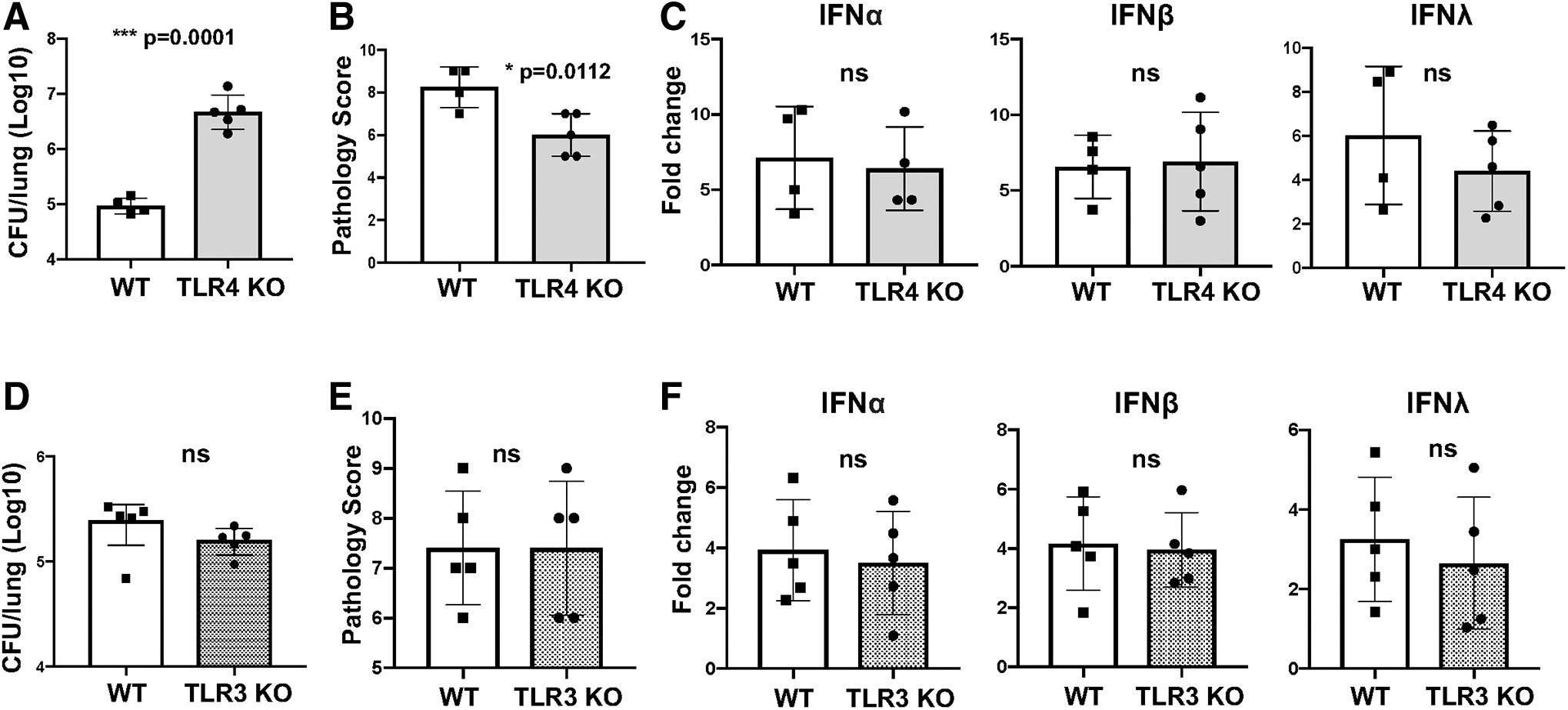
TLR3 and TLR4 do not play a role in induction of type I/type III IFN in response to B. pertussis infection. TLR3 KO, TLR4 KO, C57BL/6 (WT controls for TLR4 KO), or C57BL/6–129SF2/J (WT controls for TLR3 KO) mice (n = 4–5) were infected and euthanized 7 dpi. Lungs were dissected and
Signaling through TLR9 is commonly associated with IFN induction (Ank et al., 2008; Kader et al., 2013; Ma et al., 2015; Mortezagholi et al., 2017; Parker and Prince, 2012). B. pertussis-infected TLR9 KO mice showed no significant difference in lung bacterial burden but had significantly lower lung inflammatory pathology scores and type I and type III IFN expression levels at 7 dpi compared to that in WT mice (Fig. 3A–C). Induction of proinflammatory gene expression was also lower in TLR9 KO mice (Supplementary Fig. S1C). In addition, infected WT mice treated with a TLR9 antagonist (ODN 2088) at 0, 2, and 4 dpi had equivalent bacterial loads, but a similar reduction in lung inflammatory pathology and IFN induction at 7 dpi (Fig. 3D–F). These data indicate that TLR9 signaling is involved in type I/type III IFN induction and promotion of lung inflammation but is not required for controlling B. pertussis infection.
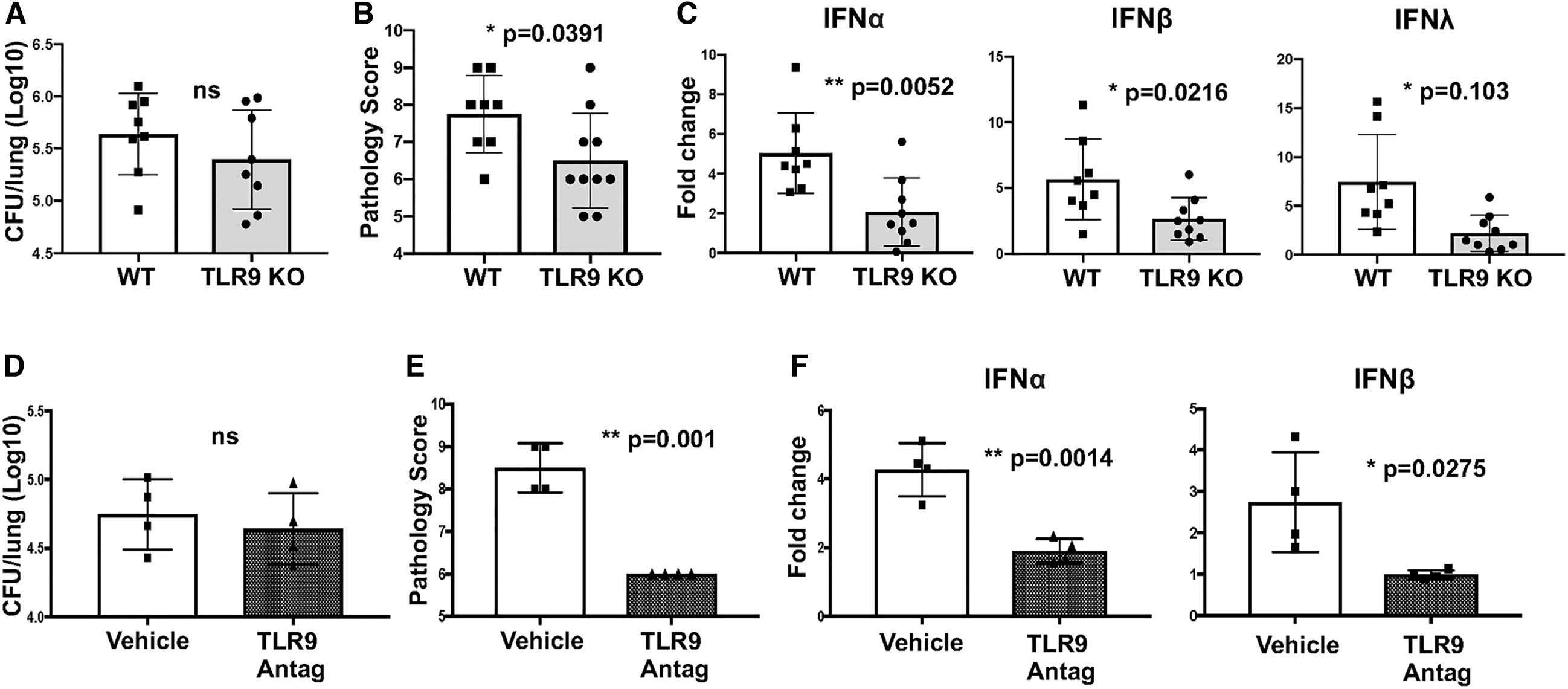
TLR9 plays a significant role in induction of type I/III IFN in response to B. pertussis infection.
Another PRR potentially involved in IFN induction is the cGAS/STING cytosolic DNA sensor (Cai et al., 2014; Marinho et al., 2017). Although B. pertussis is considered primarily an extracellular pathogen, PAMPs from phagosomal bacteria can be translocated to the cytosol and signal through STING and other cytosolic PRRs (Ragland and Kagan, 2021). B. pertussis-infected STING KO mice showed no significant difference from WT mice in lung bacterial burdens but had significantly lower levels of lung inflammatory pathology, IFN expression levels, and induction of proinflammatory genes (Fig. 4A–C, Supplementary Fig. S1D). WT-infected mice treated with the selective STING antagonist C-176 (Haag et al., 2018) also had lower lung inflammatory pathology and induction of IFNβ, but not of the other IFNs, compared to control mice (Fig. 4D–F). These data demonstrate that both TLR9 and STING are important PRRs for induction of type I/type III IFN responses to B. pertussis infection therefore that DNA is a major inducer of these responses.
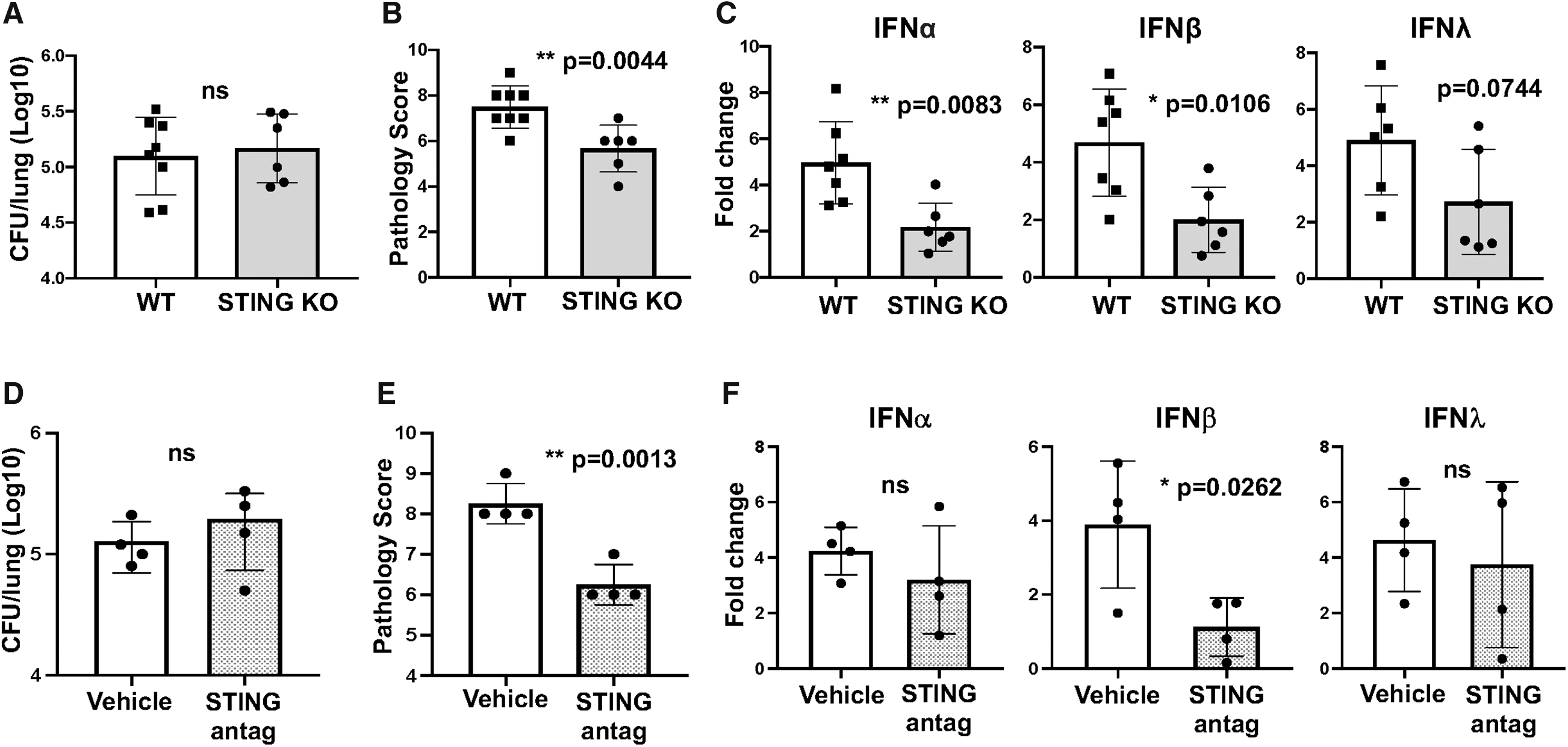
STING KO mice have reduced levels of lung inflammatory pathology and induction of type I/type III IFN in response to B. pertussis infection.
Discussion
B. pertussis infection induces significant lung inflammation in animal models, and we previously showed the involvement of type I/type III IFNs in this inflammation in mice (Ardanuy et al., 2020). Here we found that the signaling adaptor molecules MyD88 and TRIF and the DNA-sensing PRRs TLR9 and STING are involved in the induction of type I/type III IFNs and lung inflammatory pathology during infection, whereas TLR3 and TLR4, other non-DNA binding PRRs dependent on TRIF or MyD88, are not involved.
We previously found that increased expression of TREM-1, a receptor associated with inflammatory response amplification (Bosco et al., 2016), is TLR9-dependent in B. pertussis-infected WT mice (Gallop et al., 2021), but otherwise there are no previous reports of the involvement of TLR9 or STING in B. pertussis infection, although TLR9 and STING agonists added to acellular pertussis vaccine were found to enhance bacterial control in mouse models (Allen et al., 2018; Auderset et al., 2019). However, TLR9 and STING induce type I IFN responses and affect bacterial loads in other infections (Koppe et al., 2012; Parker and Prince, 2012; Parker et al., 2011).
Our results indicate that DNA is the major inducer of type I/type III IFNs during B. pertussis infection, although the source of the DNA is unclear. While killed B. pertussis or biofilm growth are potential bacterial sources (Bosch et al., 2006), host cells represent another potential source. Typically, self-DNA released following cell death is degraded by macrophages (Kawane et al., 2014) but can also be delivered to the endosome and stimulate production of type I IFNs by complexing with antimicrobial peptides such as LL37 and β-defensins (Lande et al., 2007; Tewary et al., 2013). Since infection with B. pertussis induces upregulation of antimicrobial peptides in the airways (Elahi et al., 2006), self-DNA is a potential stimulator of IFN responses and lung inflammation. TLR9 responses to self-DNA have been observed in autoimmune diseases such as systemic lupus erythematosus (Arneth, 2019) as well as in infections where large numbers of cells are killed (Stanbery et al., 2020; Tian et al., 2007).
In addition, mitochondrial DNA released by damaged host cells can signal through cytosolic and endosomal PRRs (De Gaetano et al., 2021). Neutrophil extracellular traps (Conceicao-Silva et al., 2021) represent yet another source of IFN-inducing host DNA. However, we found that antibody-mediated depletion of neutrophils had no effect on lung IFN induction or inflammation in B. pertussis-infected mice (Ardanuy and Carbonetti, unpublished data). If self-DNA is the predominant inducer of type I/type III IFN during B. pertussis infection, it is presumably derived from damaged cells and tissues in the early phase of inflammation, which is independent of type I/type III IFNs [since they are not induced until several days into the infection (Ardanuy et al., 2020)] and dependent on other PRRs such as TLR4. We previously found that induction of lung proinflammatory cytokines in B. pertussis-infected mice occurs earlier than type I/type III IFN induction (Andreasen et al., 2009; Ardanuy et al., 2020).
DNA-mediated induction of IFNs and stimulation of increased inflammation would therefore represent a positive feedback effect that may account for some of the deleterious respiratory effects in pertussis. Although certain aspects of the inflammatory response would contribute to control of bacterial infection, such as recruitment of immune cells that ultimately clear the infection, our results indicate that DNA-mediated induction of IFN responses does not affect bacterial loads within the first week of infection, since TLR9 KO and STING KO mice show no differences in bacterial loads from WT mice.
In summary, our results demonstrate that the DNA-sensing PRRs TLR9 and STING are the major inducers of type I/type III IFN responses in the lungs of B. pertussis-infected mice. Since these IFN responses exacerbate lung inflammation, this indicates the potential for TLR9 or STING antagonist compounds as therapeutic options in pertussis infections, in conjunction with antibiotic treatment to clear the infection. DNase I treatment, as used in cystic fibrosis patients (Aitken, 1993), represents another therapeutic option, although this assumes that IFN-inducing DNA is present in the airways. It may be that individuals who are well into the paroxysmal stage of pertussis disease will receive little benefit from reducing inflammatory responses, but individuals postexposure in an outbreak situation with early symptoms may benefit from such treatment in combination with antibiotic therapy.
Footnotes
Acknowledgment
The authors thank Danisha Gallop for animal care and the Carbonetti laboratory members for helpful feedback on this project.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Institutes of Health grants R01-AI141372 (to N.H.C.), F31-AI136377 (to J.A.), and T32-AI095190 (which supported J.A.).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1