
Abstract
This study aims to investigate the role of STING in promoting macrophage apoptosis and regulating macrophage polarization in severe acute pancreatitis (SAP)-associated lung injury in vitro and in vivo. A murine model was established by intraperitoneal injection of caerulein and lipopolysaccharide (LPS). Meanwhile, ANA-1 cells were stimulated with LPS to induce apoptosis in vitro. More primary alveolar macrophages underwent apoptosis and M1 macrophage polarization in the SAP group compared with the control group, which was reversed by inhibiting STING. When ANA-1 cells were induced into M2-type macrophages, the reduction of M1 macrophage markers was accompanied by a decrease of LPS-induced apoptosis. Finally, the inhibitory effect of C-176 on STING ameliorates lung injury and inflammation by adjusting macrophage polarization and rescuing apoptosis. Therefore, inhibiting STING could be a new therapeutic strategy for treating acute pancreatitis-associated lung injury.
Introduction
Acute pancreatitis (AP) is a common digestive disease. With the adjustment of lifestyle, the global incidence is gradually rising, about 34 cases per 100,000 people, of which 20%–30% can develop into severe disease, and the in-hospital mortality rate is about 15% (Iannuzzi et al., 2022; Petrov and Yadav, 2019). Severe cases are combined with multiple organ dysfunction, lung injury is the most common systemic complication (Boxhoorn et al., 2020; Garg and Singh, 2019; Hines and Pandol, 2019; Szatmary et al., 2022; Wang et al., 2022), which increases the frequency of ventilator use and the hospitalization rate in an intensive care unit (Lin et al., 2022), severely endangering patients' life and health.
Alveolar macrophages (AMs) are the central immune cells in the lung, which highly express CD170 (Siglec F) and CD11c, and maintain lung homeostasis together with alveolar epithelial cells. During the occurrence of AP, the damaged pancreatic acini releases trypsin and inflammatory mediators into the blood to activate macrophages. Kupffer cells are stimulated to produce more inflammatory substances and cytokines to reach the lungs through systemic circulation. As the largest number of innate immune cells in the lung, AMs can exhibit functional heterogeneity and plasticity in the disease states (Bissonnette et al., 2020; Guilliams and Svedberg, 2021; Hu and Christman, 2019). It polarizes to M1, producing IL-6 and TNF-α mediating lung injury in AP (Hu et al., 2020a). M1 macrophages highly express inducible nitric oxide synthase (iNOS) and other proinflammatory factors. In contrast, M2 macrophages highly express arginase, CD206, CD163, and other anti-inflammatory cytokines, thereby promoting damage repair. AMs can regulate local immune responses under normal conditions by secreting inflammatory mediators, including TNF-α, IL-1β, IL-6, IL-10, and TGF-β (Hussell and Bell, 2014; Yu et al., 2017). Simultaneously, it has an active physiological role in preventing excessive immunity by phagocytosing pathogens and controlling T cell immune responses.
Moderate apoptosis can eliminate aging, injury, tumors, and irreparable stressor cells (Bertheloot and Latz Efranklin, 2021; Sauler and Bazan, 2019). Secondary necrosis occurs due to excessive apoptosis of airway macrophages. Thus, dangerous signals are released into the airway, aggravating the pathophysiological reaction of the lung (Shotland et al., 2021). Several recent studies have observed that regulating the apoptosis of AMs cells interfered with lung injury and inflammation levels (Lam et al., 2022; Guan et al., 2020; Jiang et al., 2021; Wei et al., 2021; ). Therefore, further exploration of the biological behavior of AMs helps maintain lung homeostasis and provides research ideas for improving lung injury.
STING signaling activation, TBK1 phosphorylation, is triggered to guide IRF-3 activation and control the production of type-1 IFNs. In addition, STING can activate the downstream NF-κB signaling pathway, promote the expression of inflammatory factors, and accelerate the progression of inflammation. In AP, STING is highly expressed in pancreatic tissue and mainly in macrophages. When STING activators are applied, or the sting gene is knocked out, the degree of pancreatitis, lung injury, and intestinal injury in animal models is correspondingly aggravated or alleviated (Hu et al., 2020b; Zhao et al., 2018). It is concluded that the expression level of STING is positively correlated with the severity of pancreatitis-related organ dysfunction. At present, the mechanism of AP-associated lung injury is unknown. Our study attempted to explore the apoptosis mechanism and AMs polarization affecting AP-associated lung injury. Moreover, the protective role of C-176 in anti-apoptosis and macrophage polarization was played by inhibiting STING signaling in vivo and in vitro.
Materials and Methods
Animals
We purchased 6-weeks-old male C57BL/6 wild-type mice weighing 18–22 g from SPF (Beijing) BIOTECHNOLOGY Co., Ltd. They were housed under 12 h light/dark cycle at 25°C in a specific pathogen-free environment. The mice ate common laboratory feed and drank freely. All the animals required adaptive feeding for 1 week before the experiment. The Ethics Committee of Capital Medical University approved all experimental procedures (AEEI-2022-285).
Establishment of animal model
Intraperitoneal injection of caerulein (CAE) (M9316; AbMole) and lipopolysaccharide (LPS) (L2630; Sigma-Aldrich) was used to induce the lung injury model of severe acute pancreatitis (SAP). The specific process involved dividing the C57BL/6 mice into 3 groups: control (CON), SAP, and C-176 intervention (SAP+C-176) groups, with 6 mice in each of them. The SAP and SAP+C-176 group were given intraperitoneal injections of CAE 100 μg/kg every hour after fasting for 12 h, a total of 7 times. Ten milligrams per kilograms LPS was given immediately after the seventh intraperitoneal CAE injection. The CON group was assigned intraperitoneal injections of the same amount with solvent. SAP+C-176 group was intraperitoneally injected 4 mg/kg of C-176 (HY-112906; MedChemExpress) 6 h before the first CAE injection to inhibit STING. C-176 was reconstituted in DMSO at 10 mg/mL and diluted in 5% Tween-80 (HY-Y1891; MedChem Express) and 5% PEG-400 (HY-Y0873A; MedChem Express). Serum, pancreas, lung, and primary AMs were collected after 24 h of modeling.
Extraction of AMs
The bronchoalveolar lavage was performed using cold and sterile phosphate-buffered saline (PBS) containing 1 mM ethylenediaminetetraacetic acid (Nayak et al., 2018). The 22G intravenous indwelling catheter was attached to the trachea (Fig. 1A). One milliliter liquid was injected each time, drawn back, and collected into a 15 mL centrifuge tube. The procedure was repeated 10 times, and the cell suspension was centrifuged at 800 rpm for 3 min to collect cell precipitates. Finally, the cell precipitation was resuspended in a cell culture medium [Dulbecco's modified Eagle's medium (DMEM) medium with 1 × penicillin-streptomycin and 10% fetal bovine serum (FBS) supplementation] or sterile PBS for subsequent tests.
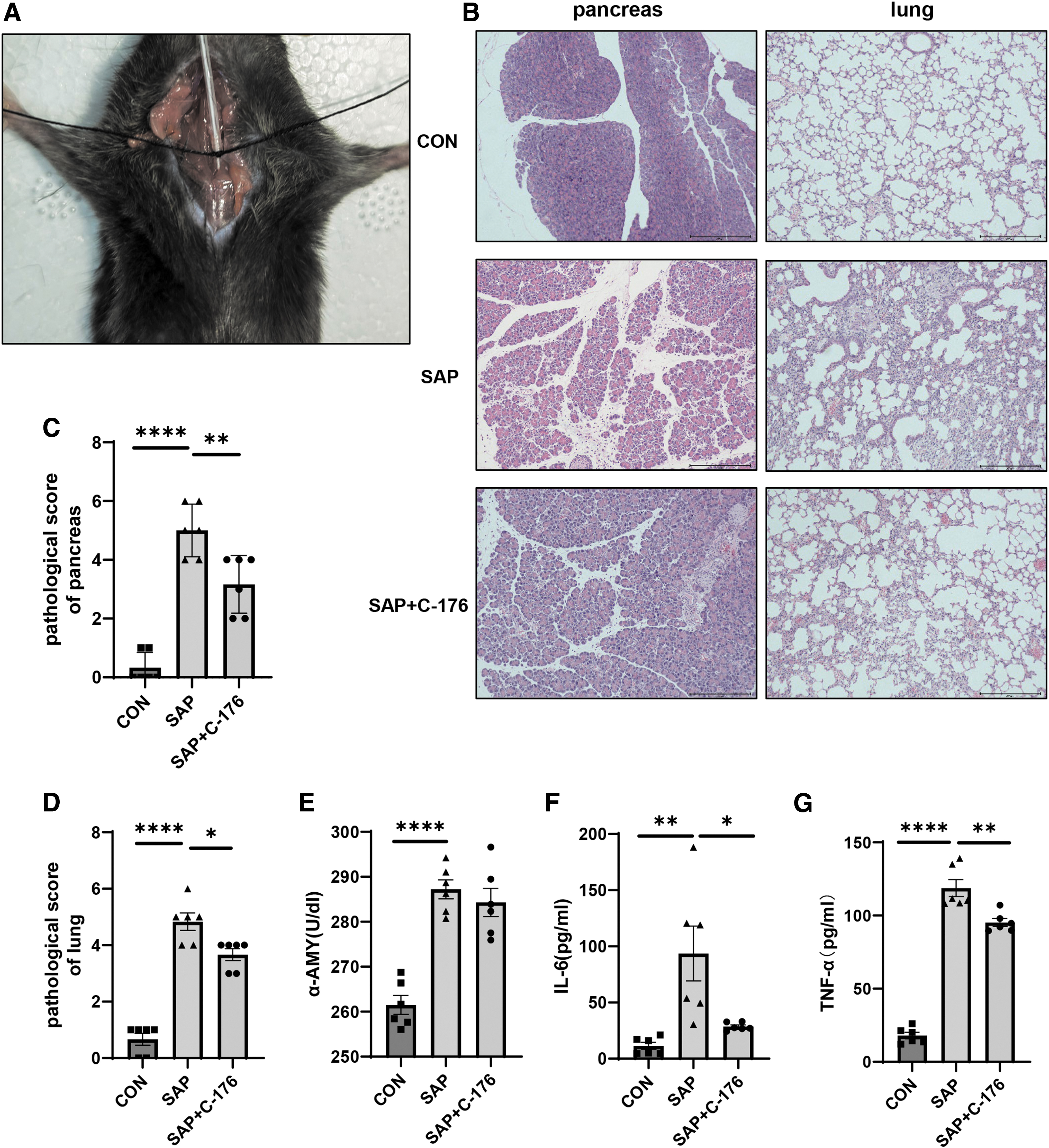
Establishing severe acute pancreatitis-associated lung injury model.
Detection of serum amylase and cytokine
We centrifuged the blood sample at 3,000 rpm for 15 min to obtain serum. The Ana-1 cells (5 × 104/well) and primary AMs (2 × 105/well) were seeded with 1 mL complete culture medium on a 24-well plate. The supernatant of Ana-1 cells was collected after 48 h of drug stimulation, and the primary macrophages were cultured ex vivo for 24 h to obtain the cell supernatant. α-Amylase Assay Kit (C016–1–1; Nanjing Jiancheng Biological Engineering Research Institute), Mouse IL-6 ELISA MAX™ Deluxe Set (Cat. No. 431304; BioLegend), and Mouse TNF-α ELISA MAX Deluxe Set (Cat. No. 430904; BioLegend) were utilized to determine the concentration of serum amylase and cytokines in the cell supernatant. The experimental procedures were performed based on the operation manual.
Histopathological analysis
We fixed the pancreas and lung tissues in 4% paraformaldehyde solution for 24 h, then dehydrated and embedded them. The paraffin-embedded tissues were cut into 4–5 μm sections and stained using hematoxylin and eosin (HE). It was followed by observing pancreatic edema, inflammation, necrosis, hemorrhage (0–4 points for each), pulmonary tissue edema, alveolar congestion, inflammatory cell infiltration, and atelectasis (0–4 points for each). The pathological scores of all the samples were calculated to assess the tissue damage degree.
Western blotting
The tissues and cells were put into RIPA (R0020; Solarbio) buffer with PMSF, split on ice for half an hour, centrifuged at 10,000–14,000 rpm for 5 min, and the supernatant was retained. BCA Protein Assay Kit (C05-02001; Beijing Biosynthesis Biotechnology Co., Ltd.) helped determine the protein concentration. SDS-PAGE Gel Kit (P1200; Solarbio) was utilized for isolating total protein. Membrane transfer was achieved with a PVDF membrane. We incubated proteins overnight at 4°C using the following primary antibodies: caspase-9 antibody (ab202068; Abcam), cleaved caspase-3 antibody (ab214430; Abcam), Bax antibody (ab32503; Abcam), Bcl-2 antibody (ab182858; Abcam), STING (ab288157; Abcam), Phospho-STING (No.72971; CST), IRF-3 (No. 4302; CST), Phospho-IRF-3 (No. 29047; No. 29047; CST). Then, HRP-conjugated secondary antibody was used for incubation, followed by detecting immune reactive bands using ECL.
Quantitative real-time PCR
HiPure Total RNA Mini Kit (R4111-02; Magen) was utilized to extract total RNA, we used PrimeScript™ RT reagent Kit (Cat No. RR037A; TaKaRa) to undergo reverse transcription, and Hieff® QPCR SYBR Green Master Mix (11201ES03) was utilized for quantitative real-time PCR (RT-qPCR). The comparative cycle threshold (Ct) method (2−ΔΔCT) was used to calculate relative expression, and β-Actin was the internal parameter to assess the target gene expression level. The primer sequences are
iNOS (F: 5′-GTTCTCAGCCCAACAATACAAGA-3′R: 5′-GTGGACGGGTCGATGTCAC-3′)
CD86 (F: 5′-TCAATGGGACTGCATATCTGCC-3′ R: 5′-GCCAAAATACTACCAGCTCACT-3′)
IL-1β (F: 5′-GAAATGCCACCTTTTGACAGTG-3′ R: 5′-TGGATGCTCTCATCAGGACAG-3′)
Arg1 (F: 5′-CTCCAAGCCAAAGTCCTTAGAG-3′ R: 5′-GGAGCTGTCATTAGGGACATCA-3′)
CD206 (F: 5′-CTCTGTTCAGCTATTGGACGC-3′ R: 5′-TGGCACTCCCAAACATAATTTGA-3′)
CD163 (F: 5′-CTGGCGGGTGGTGAAAACA-3′ R: 5′-CAGCCGTTACTGCACACTG-3′)
β-actin (F: 5′-GGCTGTATTCCCCTCCATCG-3′ R: 5′-CCAGTTGGTAACAATGCCATGT-3′)
Cell treatment
Murine macrophage cell line Ana-1 (CTCC-ZHYC-0615) was purchased from MeisenCTCC. The primary culture medium comprises DMEM, 10% FBS, and 1 × penicillin-streptomycin liquid. One microgram per milliliters LPS (L2630; Sigma-Aldrich,) was used to stimulate the cells for 48 h to induce apoptosis. STING expression was inhibited by pretreatment with C-176 5 μM for 12 h before applying LPS. Twenty nanograms per milliliters recombinant mouse IL-4 (P00196; Solarbio) and 20 ng/mL recombinant mouse IL-13 (P02309; Solarbio) were treated with Ana-1 cells inducing the macrophages to differentiate into M2 type for 72 h. Small interfering RNA (si-m-Tmem173; RiboBio) was used to silence endogenous STING expression (sequence: CCATGTCACAGGATGCCAA). Ana-1 cells were transfected with small interfering RNA for 48 h with the help of jetPRIME® in vitro DNA and siRNA transfection reagent (Polyplus, Reference Number 101000046).
Flow cytometry analysis
We used Annexin V-fluorescein isothiocyanate (FITC)/PI Apoptosis Detection Kit (CA1020; Solarbio) to distinguish apoptotic cells. CD170 (Siglec F) Monoclonal Antibody (1RNM44N), Alexa Fluor™ 700, eBioscience™ (56-1702-82; Invitrogen) helped identify primary AMs. We used PE Rat Anti-Mouse CD86 (561963; BD Biosciences) and PerCP/Cyane5.5 anti-mouse CD206 (MMR) Antibody (Cat. No. 141715; BioLegend) to detect the polarization of macrophages. Flow cytometric analysis was performed after incubating on ice in the dark for 15–20 min. All the raw data were saved in the FlowJo software.
Cell fluorescence detection
Ana-1 cells were grown on cell climbing slices plated in 24-well plates to conduct the assay. After induction of apoptosis, the supernatant was removed by aspiration, and cells were rinsed softly in PBS. Annexin V-FITC and propidium iodide (PI) staining solution were introduced and incubated for 10–20 min in the dark at room temperature (20°C–25°C). Then the cells were observed under a fluorescence microscope. The Annexin V-FITC apoptosis detection kit (C1062M; Beyotime) was applied to complete the experimental procedure within 1 h.
Measurement of oxidative stress
We used dihydroethidium (DHE) (S0063; Beyotime) to access reactive oxygen species (ROS) production. Cells were incubated for 30 min in a serum-free cell culture concentration containing 5 μM DHE at 37°C. PBS containing 5 μM DHE was dropped onto fresh frozen sections and incubated in the dark for 30 min. After loading the probe, they were gently washed 3 times with PBS. Then, the DAPI solution was added and incubated in the dark for 5 min before observation under a fluorescence microscope.
Statistical analysis
All the data were represented as means ± standard error of the mean. GraphPad Prism 9.3.0 software was utilized for statistical analysis. Differences between groups were assessed with 1-way ANOVA (nonparametric or mixed) and t-test. P < 0.05 was considered statistically different.
Results
Establishment of SAP-associated lung injury model with CAE and LPS
The animal model was established using intraperitoneal injection of CAE along with LPS. After modeling for 24 h, bronchoalveolar lavage was performed (Fig. 1A). The blood samples and tissues were obtained. We administered C-176 at a dose of 4 mg/kg to mice at 12, 6, and 3 h before modeling and found that mice pretreated with C-176 at 6 h in advance had the lowest lung injury score (Supplementary Fig. S1), so we chose this time point.
In the SAP group, we observed the pathological manifestations of pancreatitis, such as structural disorder of pancreatic lobules, interlobular edema, and inflammatory cell infiltration. However, the above manifestations were alleviated within the SAP+C-176 group (Fig. 1B). Similarly, lung injury could be observed in the SAP group. This was associated with alveolar structure destruction, alveolar septal edema, inflammatory cell infiltration, and hemorrhagic atelectasis after HE staining (Fig. 1B). Significant differences were observed among pathological injury scores between the groups (Fig. 1C, D). In the SAP group, the serum α-amylase was higher than that in the CON group, while it was reduced in the SAP+C-176 group (Fig. 1E). IL-6 and TNF-α are established serum markers of lung injury. The primary AMs were cultured in vitro for 24 h. Then, the cell supernatant was extracted for ELISA, consistent with the pathological injury of lung tissue (Fig. 1F, G).
Apoptosis increases in lung injury with activation of STING signal
Bax is a crucial proapoptotic gene and mutually represses with the anti-apoptotic gene Bcl-2, thereby affecting apoptosis. Apoptosis and the expression of cleaved caspase-3 and cleaved caspase-9 were significantly increased in the lung tissue of mice within the SAP group. Furthermore, the protein expression levels of Bax and Bcl-2 showed an opposite trend (Fig. 2A–C). Simultaneously, the phosphorylation levels of STING and its downstream gene IRF-3 are significantly increased in disease state (Fig. 2H–L), indicating that the STING signaling pathway was activated. After the application of STING inhibitor C-176, the expression level of STING, phosphor-STING, and phosphor-IRF-3 was lower than that of the SAP group, and the apoptosis signal was decreased, with statistically significant differences in protein expression levels among the groups (Fig. 2B–G).
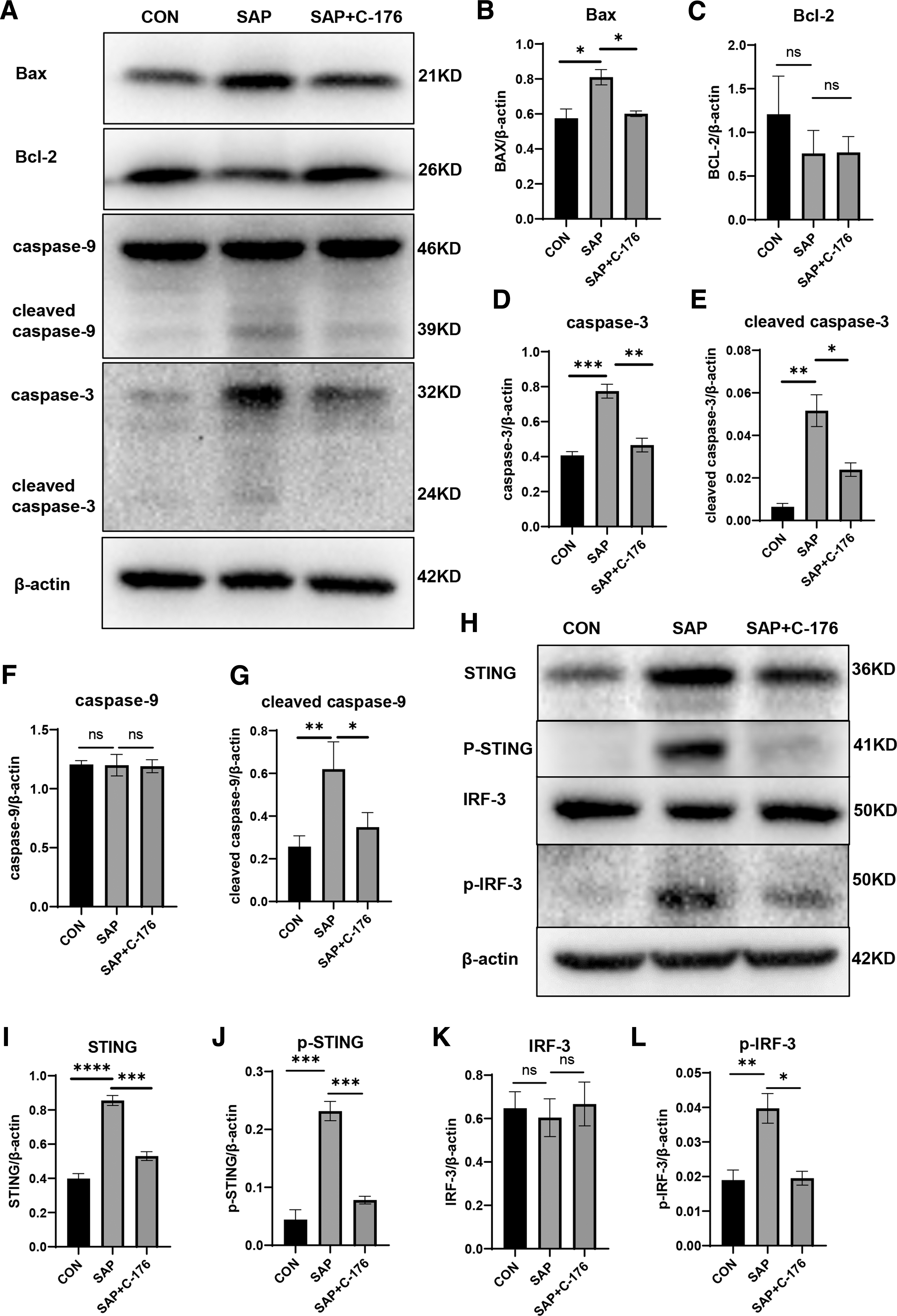
Apoptosis and STING signal activation in lung tissue.
Primary AM apoptosis was accompanied by more M1 macrophage polarization
CD170 was utilized to label primary AMs. It was observed that a satisfactory CD170 positive rate of >95% was obtained from the cells extracted from alveolar lavage (Fig. 3A, C). The AM apoptosis rate in the SAP group was higher by 20.27% compared with the control group. The apoptosis rate significantly decreased to 12.99% after early intervention with C-176, indicating a consistent trend in early (Annexin V-positive) and late (Annexin V-PI double positive) apoptosis (Fig. 3A, D). In addition, the oxidative stress levels of the lung tissue in the SAP group dramatically improved. ROS generation was assessed by DHE staining in the fresh frozen sections of the lung. However, pretreatment with C-176 reduced the ROS level in lung tissues (Supplementary Fig. S2).

Apoptosis and polarization of AMs.
Mitochondria is an essential intracellular energy supply factory, and apoptosis is accompanied by mitochondrial dysfunction and reduced membrane potential. The mitochondria membrane potential of AMs in the SAP group decreased significantly, which rebounded in the SAP+C-176 group (Fig. 3B). CD86 was used to label M1 macrophages. More AMs differentiated toward M1 macrophages among CD170-positive cells in the SAP group and were reversed by C-176 intervention (Fig. 3C, E).
LPS induces apoptosis of murine macrophage ANA-1
Ana-1 cells were stimulated using 1 μg/mL LPS for 48 h, and the total protein was extracted for Western blotting. The results indicated that the STING signal was activated, with increased protein expression levels of apoptotic executor cleaved caspase-3 and initiator-cleaved caspase-9. The expression levels of these 2 apoptosis-related proteins were significantly reduced after administering C-176 to inhibit STING signaling (Fig. 4A–H).
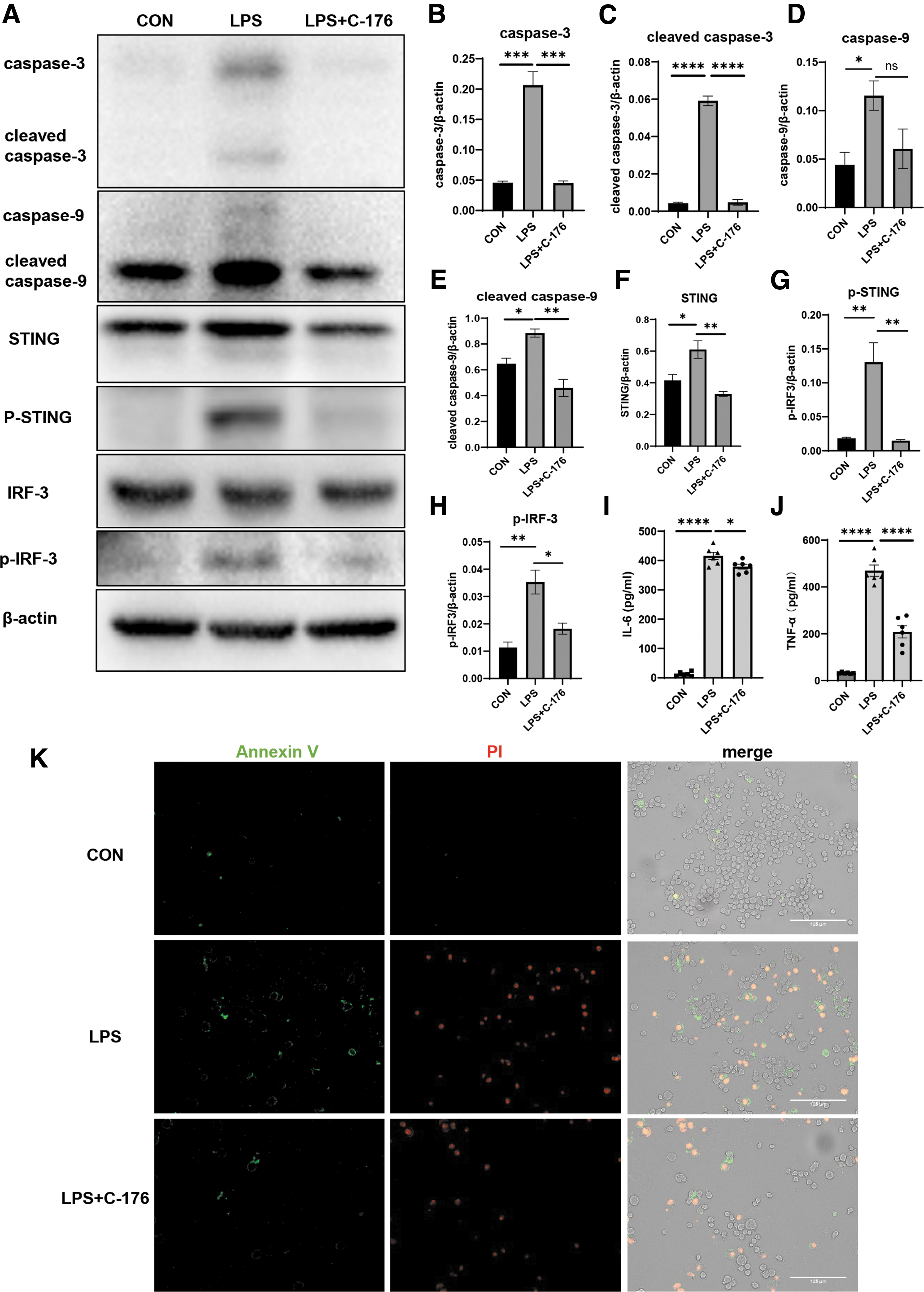
LPS induces apoptosis of ANA-1 cell.
It is visible that the LPS-induced apoptosis of Ana-1 cells ameliorated by inhibiting STING signaling, and they were stained using FITC labeled Annexin V and PI staining solution. The phosphatidylserine on the cell membrane everted to the cell surface and combined with Annexin V to develop green fluorescence during the early stage of apoptosis. PI can stain necrotic cells or other cells that have lost the integrity of the cell membrane during the late stage of apoptosis, showing red fluorescence. The number of green, double green, and red fluorescence-positive cells could elucidate early and overall apoptosis. The number of green fluorescence positive and double positive Ana-1 cells increased significantly after LPS stimulation (Fig. 4K), depicting successful induction of the ANA-1 apoptosis model. The green fluorescence positive and double positive cells decreased after C-176 administration. This suggested apoptosis reduction, consistent with the protein expression level.
Next, Ana-1 cells were transfected with a small interfering RNA to downregulate sting. Flow cytometry showed that siRNA-mediated STING knockdown reduced LPS-induced apoptosis of ANA-1 cells (Supplementary Fig. S3). Subsequently, ELISA was used to determine the concentrations of inflammatory factors within cell supernatants. After LPS induction of apoptosis, secretion of IL-6 and TNF-α elevated. These increased inflammatory factors fell back after inhibiting the STING signaling (Fig. 4I, J). It was consistent with the primary AM culture in vitro. The LPS-induced apoptosis of ANA-1 cells properly mimicked the apoptosis of AMs in the animal model.
Inhibition of STING by C-176 decreases apoptosis and M1 macrophage polarization
After treating with 1 μg/mL LPS for 48 h, the apoptosis rate of Ana-1 cells increased to 26.69%. Moreover, the total apoptosis rate decreased to 18.72% by applying C-176 in advance (Fig. 5A, D). STING inhibition with C-176 reduced LPS-induced apoptosis of Ana-1 cells. Simultaneously, the STING signal inhibition also altered the reduction of mitochondrial membrane potential and controlled the electrochemical potential difference (Fig. 5B). C-176 also induced a significant decrease of intracellular ROS production compared with the SAP group (Supplementary Fig. S4).
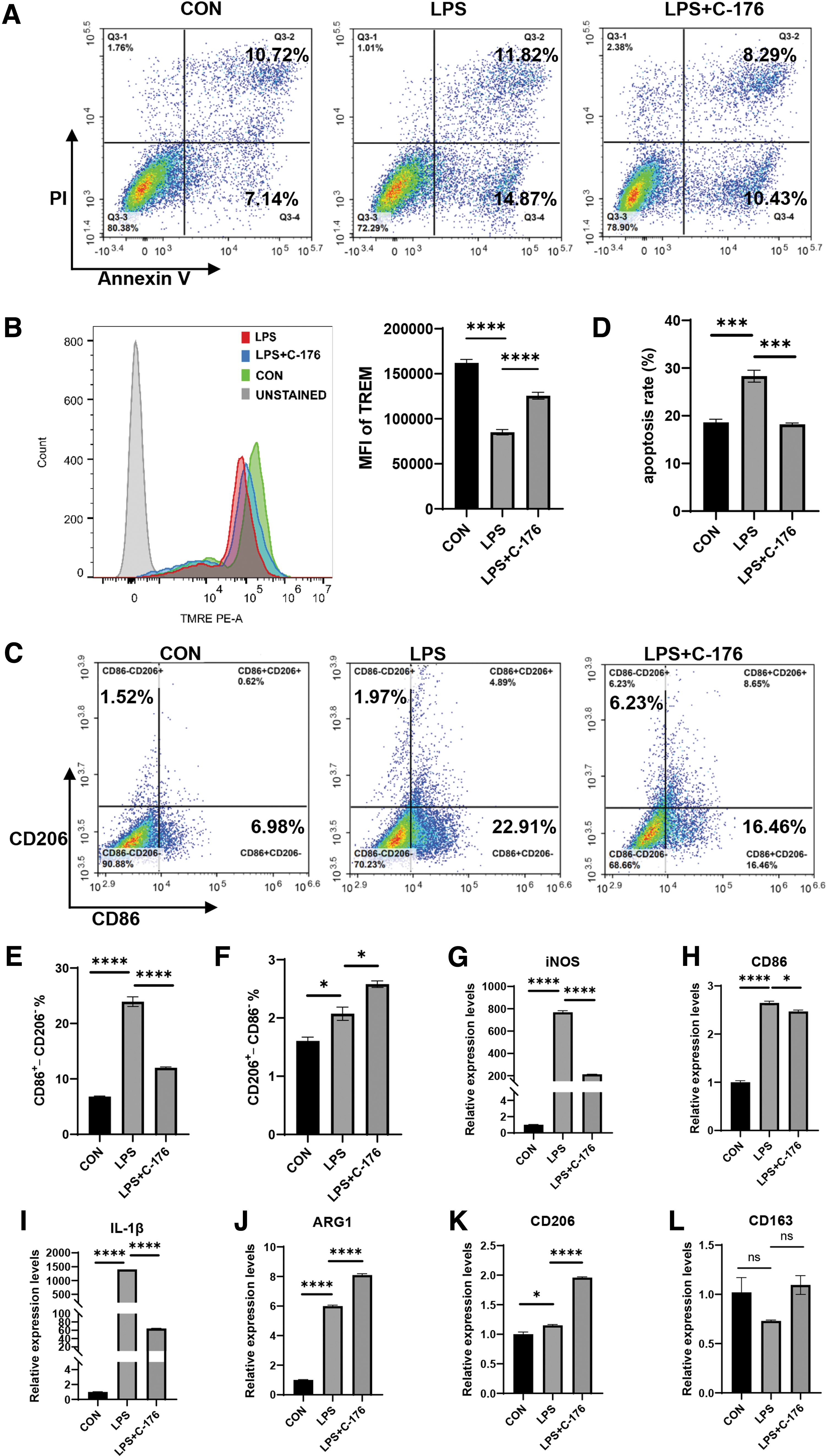
C-176 reduced ANA-1 cell apoptosis and M1 polarization.
LPS-induced apoptosis of ANA-1 was followed by a marked M1 polarization tendency in macrophage polarization, depicted by an increase in CD86+ CD206– cells from 6.68% to 22.91%. However, the M1 macrophage polarization ratio was significantly decreased after the C-176 intervention (Fig. 5C, E). The results were consistent with the in vivo experiments. Additionally, C-176 further elevated the proportion of M2 macrophage polarization, depicted by an increase in CD206+CD86– cells from 1.97% to 6.23%, with statistically significant differences (Fig. 5F). iNOS, CD86, and IL-1β were utilized as M1 macrophage markers. Arg, CD206, and CD163 were the markers of M2 macrophages. RT-qPCR was applied to detect intracellular mRNA transcription levels. C-176 was observed to significantly decrease the LPS-induced elevation of M1-type markers (Fig. 5G–I) while elevating the transcript levels of M2-type markers (Fig. 5J–L). Arg and CD206 transcription increased on administering LPS stimulation, with decreased CD163 transcription.
Differentiation of Ana-1 cell into M2 macrophage reduced LPS-induced apoptosis
The polarization of Ana-1 cells was induced into M2-type macrophages by combining IL-4 and IL-13 and determining the mRNA levels of each macrophage marker 72 h after drug treatment. The increased transcriptional levels of Arg, CD206, and CD163 were accompanied by decreased transcriptional levels of iNOS, CD86, and IL-1β (Fig. 6A–F). This indicated the differentiation of Ana-1 cells into M2 and M1 suppression. Subsequently, LPS was used to induce apoptosis, and the protein expression levels of cleaved caspase-3 and cleaved caspase-9 were reduced after Ana-1 cells differentiated into the M2 type (Fig. 6G–K). Thus, cell apoptosis was alleviated, and LPS-induced apoptosis was ameliorated by modulating Ana-1 polarization.

The polarization of ANA-1 cells to M2 macrophage reduced LPS-induced apoptosis.
Discussion
The current study constructed an animal model of SAP-associated lung injury using mice through intraperitoneal injection of CAE associated with LPS. STING signal was significantly expressed, and AM apoptosis was elevated accompanied by more M1 macrophage polarization within the SAP group. When C-176 was applied, the inhibition of STING signaling controls macrophage polarization to improve apoptosis and alleviate the severity of AP-associated lung injury.
Acute lung injury (ALI) is a common systemic complication of AP. It is characterized by progressive hypoxemia and respiratory distress, and severe cases tend to develop into acute respiratory distress syndrome or multiple organ dysfunction syndromes, thus, endangering human health (Gorman et al., 2022; Meyer and Gattinoni, 2021; Pastor et al., 2003). The pathophysiological process of the lung primarily involves oxidative stress injury, inflammatory factor infiltration, cell apoptosis, and immune dysfunction in AP models (Ge et al., 2020; Kong et al., 2021; Zhang et al., 2021a; Zhou et al., 2021). Recently, the role of AMs in lung injury has garnered significant attention (Allard et al., 2019; Qin et al., 2022). The AMs can secrete TNF-α, platelet-activating factor, and IL-6 to initiate an inflammatory response in the early stage of lung injury (Hu et al., 2022; Soni et al., 2016; Zhang et al., 2013; Zoulikha et al., 2022). Additionally, they recruit neutrophils, T cells, and other immune cells to participate in the subsequent pathological reaction. The extracellular vesicles derived from AMs can transport biological information and interact with alveolar epithelial cells and adjacent macrophages, affecting lung injury (Bissonnette et al., 2020; Clements and Idoyaga, 2021; Hu et al., 2021).
Macrophage polarization is a dynamic and continuous biological behavior of the cell. Multiple studies have indicated that the pathological process of acute lung injury is associated with M1 macrophage polarization (Lin and Song, 2020; Wang et al., 2020; Zhang et al., 2021b). MiR-30d-5p in exosomes from neutrophils activates M1 macrophage polarization to regulate sepsis-related ALI (Jiao et al., 2021). M1 macrophage polarization is mediated by ambient particulate matter through the P2A-MTON-p70S6K/4E-BP1 signaling pathway to cause lung injury. Our study indicated that M1-type differentiation of AMs paralleled lung injury and apoptosis. Therefore, the inhibition of STING signaling by C-176 reduces the proportion of M1-type differentiation and the level of AM apoptosis, thus playing a protective role.
Current research on various disease contexts deals with macrophage polarization and cell death modalities. For instance, quercetin regulates the polarization of synovial to M2 macrophages by inhibiting chondrocyte inflammation. Thus, apoptosis plays a protective role in cartilage (Hu et al., 2019b). IL-10 could attenuate hepatic ischemia-reperfusion injury by controlling apoptosis and altering KC polarization (Ye et al., 2020). However, there is no report on whether polarization directly affects macrophage apoptosis. Therefore, the induction experiment of M2 macrophages was added in vitro. IL-4 was combined with IL-13 to induce Ana-1 cell polarization to M2 macrophages. Moreover, RT-qPCR revealed that M1 polarization was inhibited. The apoptosis level in M2-polarized ANA-1 cells decreased when LPS stimulation was administered. Thus, the polarization of macrophages may affect apoptosis to some extent and jointly affect the severity of lung injury in SAP.
STING is mainly activated by the DNA-sensing cGAS signal and is a potential therapeutic target for inflammatory diseases (Barber, 2015; Decout et al., 2021; Motwani and Pesiridis Sfitzgerald, 2019). In 2018, it was first introduced in macrophages activated through AP (Zhao et al., 2018). Then, it was confirmed to be widely involved in various pathophysiological processes, such as ferroptosis, autophagy, and inflammatory response to pancreatic disease (Dai et al., 2020; Song et al., 2021; Zhao et al., 2019). Several studies have found that there is a close relationship between STING signal and cell apoptosis (Hong et al., 2022; Wu et al., 2019). STING-IRF3 promotes hepatocyte injury in non-alcoholic fatty liver disease by mediating inflammation and apoptosis (Qiao et al., 2018). STING disturbs intestinal barrier and aggravates sepsis by increasing intestinal epithelial cell apoptosis (Hu et al., 2019a).
C-176 is a specific inhibitor of STING in mice, utilized in mouse-related animal and cell experiments (Haag et al., 2018). C-176 in our study exerted a protective effect by enhancing macrophage polarization and decreasing apoptosis, consistent with the previous studies (Wu et al., 2022; Yang et al., 2022). Thus, the potential therapeutic value of this signaling molecule has been demonstrated in AP-associated lung injury. However, our study has certain limitations. STING knockout cells and mice would be useful to strengthen our conclusions. Macrophage polarization is a dynamic continuum, and it is impossible to provide the polarization behavior of the whole disease process with more refined polarization types. Therefore, we need to track the biological behavior of macrophages and gather more evidence for identifying organ function injury due to AP.
Conclusions
C-176 exerted protective effects in the LPS-induced apoptosis model of ANA-1 cells in the experiments in vitro. STING signaling inhibition by C-176 ameliorated SAP-associated lung injury severity by controlling AM polarization and decreasing apoptosis, which could be a new therapeutic strategy for treating AP-associated lung injury.
Footnotes
Acknowledgments
We thank all the staff of Clinical Laboratory at Capital Medical University Beijing Obstetrics and Gynecology Hospital for their technological support.
Authors' Contributions
Y.P., Y.L., and C.Y. were involved in the entire conception, study design. Y.P., Y.L., Y.Y., and R.L. completed all experiments and data analysis. Y.P. wrote the manuscript. Y.L., and C.Y. contributed to the project's supervision and the article's revision. All authors read and approved the final manuscript.
Ethical Approval
The Ethics Committee of Capital Medical University approved all experimental procedures (AEEI-2022-285).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (No. 81873943).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4