
Abstract
IFN-γ is recognized as an immunoregulatory cytokine due to its dual role in both accelerating and dampening immunological responses. Accordingly, in the context of tumor immunotherapy, the therapeutic outcome of IFN-γ is contingent upon factors such as dosage and the expression status of downstream signaling molecules. Furthermore, the coadministration of IFN-γ with various immunestimulatory agents, including anticheckpoint inhibitors, chemotherapeutic agents, and herbal-based medicines, may potentially overcome the IFN-γ-related challenges and enhance the response rate. We decipher the mechanisms of tumor cell eradication facilitated by IFN-γ, the last achievements in IFN-γ-mediated tumor immunotherapy across various cancers, and the strategies to address the failure of IFN-γ-based tumor immunotherapy. Unraveling the molecular mechanisms that lead to failure in IFN-γ-based antitumor actions could assist in pinpointing therapeutic agents that target the immune-modulatory features of IFN-γ, thereby increasing the antitumor response rate.

Background
The biologically active form of IFN-γ is composed of two identical 17 kDa polypeptides that are noncovalently linked to form a compact and globular molecule exhibiting a twofold axis of symmetry, with the individual polypeptides interacting in a helical and antiparallel manner. The IFN-γ receptor is comprised of two subunits, IFNGR1 and IFNGR2. Upon ligand binding, Janus kinase (JAK) 1 and JAK2 are recruited and activated, subsequently leading to the activation of interferon regulatory factor (IRF) and signal transducer and activator of transcription 1 (STAT1) (Alspach et al., 2019). It is evident that IFN-γ is produced not only by NK cells, CD8+ T cells, and CD4+ T cells but also by dendritic cells, macrophages, innate lymphoid cells (ILCs), and B cells. ILC1 cells by secreting IFN-γ have the capacity to induce the polarization and activation of CD86+ macrophage, resulting in the inhibition of tumor growth (Zhang et al., 2023). Besides, it has been revealed that B cells generate IFN-γ in response to antigens (Ag) and cytokines such as interleukin-12 (IL-12) and IL-18 (Bao et al., 2014).
IFN-γ is considered to trigger antitumor immune responses by upregulation of MHC molecules, cell cycle arrest, post-proteasomal antigen processing, recruitment of effector cells, and inhibition of angiogenesis by downregulation the pro-angiogenic factors such as vascular endothelial growth factor, basic fibroblast growth factor, and upregulation of antiangiogenic factors, including thrombospondin-1 and interferon-inducible protein 10 (Gupta and Qin, 2003; Textor et al., 2017). Moreover, bystander IFN-γ activity has been documented in human tumors. According to this, neighboring and remote tumor cells, irrespective of the expression of tumor antigens, could be modulated by IFN-γ secreted by tumor-specific CD8+ T cells (Hoekstra et al., 2020). Additionally, a high density of T cells in tumor regions could trigger the apoptosis of tumor lymphatic endothelial cells (LECs) via the IFN-γ signaling pathway. This could reduce the dissemination of tumor lymphatic vessels, consequently diminishing the likelihood of tumor metastasis (Garnier et al., 2022).
Excessive IFN-γ stimulation can also result in various negative effects, such as apoptosis of CD8+ T cells, and the upregulation of inhibitory ligands like programmed death-ligand 1 (PD-L1) on LECs, which consequently inhibits cytotoxic T lymphocyte (CTL) migration to the tumor microenvironment (TME) and dampens the immune response (Zhang et al., 2017). Additionally, IFN-γ can drive the expression of immunosuppressive enzyme, indoleamine 2,3-dioxygenase (IDO), and facilitates the immune surveillance in cancers through genome immunoediting (Banzola et al., 2018). To address these challenges, future directions for IFN-γ-based immunotherapy may include combination approaches with other immunotherapies or targeted therapies to enhance the antitumor effects of IFN-γ. In several cancers, IFN-γ has been detected as an important marker for determining response to immune checkpoint blockade agents (Ayers et al., 2017; Yi et al., 2018). The combination of IFN-γ and a PD-1-blocking agent has been exposed to the potential to augment immunological functioning and may be a critical component in the success of adoptive transfer therapies for pancreatic cancer (Ding et al., 2019). In this review, we delve into the IFN-γ-dependent tumor damage mechanisms, the dichotomous function of IFN-γ in the TME, and the ways to strengthen the effective response to IFN-γ through administration with various immunestimulatory agents.
IFN-γ-Mediated Pathways to Suppress Tumor Cells
IFN-γ primarily utilizes the JAK1/2-STAT1 intracellular pathway to induce transcriptional activation of IFN-γ-inducible genes. The STAT family of transcription factors, comprising seven members, participates in receptor signaling by diverse cytokines and growth factors. Following IFN-γ stimulation, cytoplasmic STAT1a becomes tyrosine-phosphorylated, forming homomeric complexes that translocate into the nucleus and directly bind to palindromic γ-activated sites on IFN-γ-responsive target gene promoters. Hence, STAT1a functions as a vital mediator of transcriptional responses induced by IFN-γ (Saha et al., 2010).
There are three types of regulated cell death including apoptosis, necroptosis, and pyroptosis that assemble together and form PANoptosis cell death (Wang and Kanneganti, 2021). IFN-γ by activating the JAK1/STAT1 pathway leads to the increased expression of apoptosis-inducing molecules, Bax, Bak, caspase-3, and decreased expression of antiapoptotic molecule, Bcl2 (Su et al., 2020). In tumor cells treated with IFN-γ, phosphorylated STAT1/3 (p-STAT1/3) actively promotes the production of reactive oxygen species (ROS), mitochondrial dysfunction, and cell apoptosis. p-STAT1/3 can transport into the mitochondria and initiate the mitochondria respiratory complex and ROS synthesis (Wang et al., 2018). Active IFN-γ signaling has the capacity to instigate apoptosis in cancer cells by activating unfolded protein responses (UPR) and endoplasmic reticulum (ER) stress. IFN-γ accelerated the protein synthesis rate, which leads to upregulation in the UPR-associated molecules via the JAK1/2-STAT1 and phosphatidylinositol 3-kinase (PI3K)-AKT-mTOR signaling pathways. Under conditions of ER stress induced by IFN-γ, autophagy was disturbed through the inhibition of autophagosome-lysosome fusion. Additionally, the IFN-γ pathway induces cell cycle arrest at the G1/S phase and promotes apoptosis by increasing the active form of caspase-3 (Fang et al., 2021).
It was also found that IFN-γ induces cell death via the involvement of ERK pathway. Inhibition of ERK activity by a selective ERK inhibitor ulixertinib inhibited apoptosis in melanoma cells mediated by IFN-γ (Champhekar et al., 2023). It has been similarly observed that blocking the ERK/MAPK pathway leads to suppressing IFN-γ-induced death in A431 epithelial cancer cells via inactivating caspase3 (Burova et al., 2011).
The PANoptosis pathway is being valued in inflammatory and tumor conditions. The pyroptotic cell death can be triggered by activating pore-forming gasdermines through several pathways including inflammasomes or granzymes (Huang et al., 2023). Tumor cells harboring mismatch repair deficiency (MMRD) indicated more susceptibility to chemotherapies and immune checkpoint blockade. One of the main reasons could be the activation of PANoptosis pathways in these types of tumors. IFN-γ has been known as an inducer of these pathways. IFN-γ treatment in colon cell lines with MMRD, elevated the expression of gasdermin E and D (pyroptosis-related molecules) and caspase-3, 7, and 8 (apoptosis-related molecules), leading to a higher rate of cell death compared with cells without MMRD (Li et al., 2024).
The process of necroptotic programmed cell death is significantly enhanced through IFN-γ-mediated signaling. It has been reported that IFN-γ, in synergy with smac inhibitors, promoted the formation of necroptosome complexes consisting of receptor-interacting protein (RIP) 1, RIP3, and mixed lineage kinase domain-like protein (MLKL) in apoptosis-resistant cancer cells (Cekay et al., 2017). Moreover, IFN-γ could potentially instigate the necroptotic death of lung epithelial cells by inducing the activation of MLKL, a key element of necroptosis (Hao et al., 2022).
Ferroptosis is another programmed cell death propelled by the accumulation of peroxidized lipids, leading to mitochondrial damage. The solute carrier family 7 member 11 (SLC7A11)/glutathione peroxidase 4 (GPX4) axis, acting as a negative regulator of ferroptosis, detoxifies lipid peroxides through the activation of the cysteine/glutathione pathway (Jia et al., 2023). In contrast, erastin, a small molecule compound, positively promoted the ferroptosis events. The potential role of IFN-γ in potentiating ferroptosis inducer agents has been mentioned in several types of cancers. YU et al. reported that IFN-γ-IRF1-STAT pathway, through the downregulation of SLC7A11, amplified the intensity of erastin-induced ferroptotic cell death in adrenocortical adenocarcinoma cells. Erastin significantly repressed the SLC7A11 expression followed by enhancing the activation of IFN-γ-STAT-IRF1 axis (Yu et al., 2022). Lysophosphatidylcholine acyltransferase 3 (LPCAT3) represents another crucial factor in initiating the ferroptosis pathway, through its involvement in the synthesis of unsaturated fatty acids. Tao et al. in their recent research, mentioned the involvement of the IFN-γ-STAT-IRF1 axis in inducing LPCAT3 expression, which ultimately promotes ferroptotic cell death (Tao et al., 2024). A schematic model of IFN-γ involvement in the four types of cell death is illustrated in Figure 1.

IFN-γ-related cell death signaling pathways. IFN-γ, interferon-gamma.
Another prominent characteristic of IFN-γ is its ability to elicit cell senescence by permanently stopping cancer cell proliferation. IFN-γ leads to the activation of cell cycle regulators, such as P16INK4a and P21Cip1, which are known to instigate senescence (Brenner et al., 2020). Simultaneous function of the IFN-γ-STAT1 and TNF/TNFR1 pathway induces the senescence of Tag-positive cancer cells via stimulating the P16INK4a (Braumüller et al., 2013). Moreover, tumor antigen-specific T cell-derived IFN-γ and TNF-α, especially their combination, could potentially provoke senescence in AML cell lines (Hashimoto et al., 2022).
The Observed Potential of IFN-γ in Either Suppressing or Enhancing Tumor Cells
IFN-γ can exert both antitumor and tumorigenesis effects. The impact of IFN-γ therapy varies depending on the dosage, the associated signaling pathways, and tumor type. It has been shown that LD IFN-γ can enhance stem cell-like features in various types of cancers. IFN-γ at low doses of 0.01–0.05 µg/day could provoke AML stem cell stemness by binding to IFNGR1 and stimulating the PI3K/AKT signaling pathway. However, IFN-γ at high doses of 5–10 µg/day could eradicate the tumor stem cells via the JAK1/STAT1 pathway (Xie et al., 2023). In nonsmall cell lung cancer cells, LD IFN-γ improved cancer cells stemness via signaling cascades consist of the intracellular adhesion molecule 1-PI3K-AKT-Notch1 (Song et al., 2019). Likewise, it has been indicated that IFN-γ at low concentrations of 1 and 10 ng/mL increased the metastatic potential of CRC cells, triggered by the elevated expression of metastasis-associated in colon cancer 1. Although these observations were more prominent in the presence of TNF-α (Kobelt et al., 2020).
In melanoma, contrary findings have been achieved. David et al. reported that LD IFN-γ, 100 µg/m2, per week showed tolerable response in all participants and complete response in 12% (Propper et al., 2003). Likewise, Donia et al. reported that the antitumor features of the tumor-infiltrating lymphocytes could be heightened against autologous melanoma cells through low dose (100 IU/mL) treatment of IFN-γ (Donia et al., 2013). Previous trials of IFN-γ at high dose of 0.25–0.5 mg/m2 daily did not achieve the better clinical response, but significantly reduced the WBC count (Creagan et al., 1987; Kopp et al., 1993).
Another considerable issue is dose-limiting toxicities that have prohibited the use of the most effective dosage of IFN-γ. A phase I clinical trial involving patients with gastrointestinal (GI) cancers, adjunctive administration of IFN-γ with FUra/LV was well tolerated at an maximum tolerated dose of 75 µg/m2 and dose-limiting toxicity was observed at a dose of 100 µg/m2 (Schwartzberg et al., 2002). Similarly, patients with ovarian cancer receiving IFN-γ-1b at a dose of 100 µg three times a week plus carboplatin and paclitaxel have indicated severe hematological toxicities (Alberts et al., 2008). Vector-based intratumoral gene expression and combination therapy are strategies that not only mitigate toxicity by modulating the dose–response but also enhance the efficacy of IFN-γ antitumor responses. It has been reported that the local administration of adenoviruses expressing IFN-γ into melanoma lesions in combination with systematic infusion of IL-2 and TIL cells has yielded promising outcomes for patients with melanoma. The tumor response evaluation indicated that out of 13 patients, 3 presented completed regression and 2 achieved a partial response. Moreover, an impressive response rate of 75% was observed in the melanoma cutaneous lesions (Khammari et al., 2015). According to Relation et al., the direct intratumoral administration of the mesenchymal stem cells secreting IFN-γ on the base of a lentivirus-derived gene expression system demonstrated reduced murine neuroblastoma cell growth without any observed toxicity (Relation et al., 2018). The intratumoral injection of adenoviruses carrying IFN-γ gene resulted in a notable inhibition of tumor cell progress, elevated effector T cells, and decreased regulatory T cells, in an animal breast cancer model (Trofimova et al., 2021). The results of available clinical trials on the treatment of IFN-γ (IFN-γ-1b, Actimmune) or the adenoviral vectors expressing IFN-γ, in combination with other antitumor chemotherapeutic or immunestimulatory agents, are indicated in Table 1.
The Last Achievements of the IFN-γ-Based Tumor Immunotherapy in Clinical Trials
ADCC, antibody-dependent cellular cytotoxicity; AE, adverse event; AML, acute myeloid leukemia; BVZ, bevacizumab; CAR-T cells, chimeric antigen receptor T cells; CB, carboplatin; CIK cells, cytokine-induced killer cells; CR, complete response; CRC, colorectal carcinoma; DCR, disease control rate; DLI, donor lymphocyte infusion; DLT, dose-limiting toxicity; DNX2401, oncolytic adenovirus; FUra, fluorouracil; GI, gastrointestinal; IFN-γ, interferon-gamma; IL-2, interleukin-1; irAE, immune-related adverse event; LV, leucovorin; MDS, myelodysplastic syndrome; MR, marginal response; MTD, maximum tolerated dose; Nivo, nivolumab; PAC, paclitaxel; PD, progressive disease; QOD, every other day; ORR, overall objective response; Q3W, every 3 weeks; SC, subcutaneous; SD, stable disease; T, trastuzumab; TG1042, adeniviral particles expressing IFN-γ; TIL, tumor-infiltrating lymphocyte; T.I.W., three times a week; TR, tumor response.
Strategies to Deal with the Immune-Suppressive Effects of IFN-γ
The most common cause of failure of tumor therapy with IFN-γ is the upregulation of PD-L1 on the tumor surface (Zhao et al., 2020; Numata et al., 2022). As a result, relying solely on IFN-γ for controlling tumor growth has not yielded a good efficient response, leading researchers to explore the combination of IFN-γ with other tumor-restricting agents. The PD-L1 expression induced by IFN-γ is primarily regulated by JAK1/JAK2/STAT1 or STAT3 pathway. It should be implied that high PDL1 expression, whether IFN-γ-dependent or not-dependent, could result in a good response rate to PDL1-blocking agents. This point concurs with the report by Lai et al. that strengthening the expression of IFN-γ-related genes such as IRF1/7, which are implicated in the upregulation of PD-L1, could improve the response to anti-PD-L1 therapy (Lai et al., 2018). Likewise, in colorectal cancer cells with IFN-γ-dependent high level of PD-L1, combining the treatment of IFN-γ with an anti-PD1 blocking antibody remarkably enhanced tumor toxicity of T cells (Tang et al., 2024).
The findings by Dong et al. indicate that the sequential therapy of IFN-γ with chimeric antigen receptor T cells augments the antitumor cytotoxic effects by triggering ICAM1/LFA1 interaction in multiple tumors, even in the presence of increased PD-L1 expression (Dong et al., 2021).
Researchers are now exploring alternative agents that could mitigate the expression of PD-L1, such as some chemotherapeutic drugs, tyrosine kinase inhibitors, or natural products to enhance more robustly IFN-γ-antitumor responses.
Inhibition of STAT-3 phosphorylation has emerged as a promising strategy to mitigate PD-L1 expression. STAT-3 is also activated in the MET receptor kinase signaling pathway. Notably, a positive correlation has been observed between MET and PD-L1 expression. This evidence is further substantiated by the findings of Song et al., who demonstrated that melanoma cells treated with the MET tyrosine kinase inhibitor crizotinib exhibit downregulation of IFN-γ-induced PD-L1 expression through the reduction of p-STAT-3 (Song et al., 2023). In an alternative treatment regimen, the combination of IFN-γ (10 ng/mL) with an anti-erbB2 mAb remarkably reduced the viability of breast tumor cells. Although coadministration of an anti-PDL1 mAb in this order caused the most favorable tumor regression response, compared to those without the anti-PDL1 mAb (Nagai et al., 2015).
According to Wu et al., exposure to IFN-γ at a concentration of 5 ng/mL substantially enhanced the PD-L1 expression in the 4T1 breast cell line. A nanoparticle containing alpha-tocopheryl succinate, an antitumor agent extracted from vitamin E, enhanced the antitumor effects, concomitant with the suppression of PD-L1 expression, via attenuating NFKB pathway in the breast cancer cells (Wu et al., 2020).
Some herbal antitumor compounds have been identified as inhibitors of IFN-γ-dependent PD-L1 expression. Myricetin, belonging to the flavonol class of flavonoids, has been characterized to inhibit the upregulation of PD-L1 by interfering with the JAK-STAT-IRF1 pathway (Chen et al., 2022). Licochalcone A (LCA) is also a flavonoid agent that exhibits a broad spectrum of antitumor characteristics, including antiproliferation, antiangiogenesis, and antioxidative properties (Liu et al., 2021). In recent years, the capacity of LCA to constrain PDL-1 expression has undergone assessment. Yuan et al. provided evidence that LCA could impede the IFN-γ-stimulated PD-L1 expression by interfering with its translation process in lung cancer cells (Yuan et al., 2021). Both apigenin, a flavonoid, and curcumin, a polyphenolic compound, have demonstrated the capacity to attenuate STATA1 activation, thereby blocking IFN-γ-induced PD-L1 expression and enhancing T cells-mediated antitumor responses (Coombs et al., 2016). Berberine, an alkaloid compound has also indicated an immune-enhancing role by reducing IFN-γ-dependent PD-L1 expression in hepatocellular carcinoma (HCC) cells and thereby rendering HCC cells more susceptible to NK cells (Wang et al., 2022). In a recent research, it has been discovered that sokotrasterol sulfate (SKS), a marine compound extracted from Topsentia sp. Spange, possesses the capability to suppress the IFN-γ-stimulated PD-L1 expression without altering the expression of constitutive PD-L1 in pancreatic, lung, and ovarian cell lines. SKS directly inhibits the active form of JAK, which results in the suppression of STAT activation and PD-L1 induction (Wang et al., 2024).
Similar effects have been noted in chemical compounds. The mechanistic potential of propranolol, a beta blocker, in mitigating tumor burden and metastasis has been related to the reduced level of both IFN-γ in activated immune cells and PD-L1 expression in cancer cells. Moreover, combination therapy of propranolol with an anti-PD-L1 inhibitor improved the antitumor immunity (Falcinelli et al., 2023). Nimesulide, a nonsteroidal anti-inflammatory agent, has the capability to inhibit the cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE2) pathway and exhibits anti-inflammatory and tumorigenic effects. It has been shown that nimesulide could reduce high PD-L1 expression incited by IFN-γ and improve the antitumor responses of T cells in breast cancer cells, independently of COX-2. The underlying mechanism remains to be elucidated (Liang et al., 2009). BRAF-MEK pathway also affected the PD-L1 level by influencing the IFN-γ pathway-related molecules. Accordingly, in melanoma cells with prevalent BRAF-V600E mutation, treatment with vemurafenib, a selective BRAF inhibitor, showed the capacity to modulate the level of PD-L1 protein by interfering with both STAT1 activation and proteins involved in the translation process (Górniak et al., 2020).
The positive effect of IFN-γ has also been observed in the context of anti-CTLA-4 inhibitors. Genomic defects affecting IFN-γ pathway genes have been associated with reduced responsiveness to anti-CTLA4 treatment (Gao et al., 2016). Additionally, anti-CTLA-4 therapy indicated an elevated number of ICOS+ CD4+ T/FOXP3+ CD4+ T cells accompanied by an enhanced expression of IFN-γ level in both tumoral and normal prostate tissues (Chen et al., 2009; Lukhele et al., 2019).
IDO-1 is an immune-suppressive and tolerogenic enzyme by activating the tryptophan-kynurenine-aryl-hydrocarbon receptor (AhR) axis, causing effector T cells apoptosis, T-regulatory cells induction, and upregulation of the inhibitory molecules (Schramme et al., 2020). IFN-γ is the potent inducer of IDO-1, as well as its downstream signaling molecules. IFN-γ also has the capacity to induce dormancy in tumor cells, a temporary cellular quiescence, which is one of the leading causes of tumor recurrence. Evidence suggests that IFN-γ through activating the IDO-Kyn-AhR pathway leads to the upregulation of P27, a downregulator of the transition of cells from G0 to G1, thereby causing dormancy in cancer stem cells (Qiu et al., 2020). Besides, p27 impeded phosphorylated STAT1 translocation from the cytoplasm into the nucleus. Knocking down p27, AhR, or IDO-1 results in the stimulation of the IFN-γ/p-STAT1 pathway subsequently triggers apoptosis of reactivated tumor cells (Liu et al., 2017). The two well-recognized IDO-1 blocker agents, including epacadostat and indoximod, do as a selective competitor of IDO-1 by blocking tryptophan binding to IDO-1 and a tryptophan mimetic, respectively. Exposure to DC encompassing IFN-γ-derived high IDO-1 level with epacadostat boosted the functional activity of DC to enhance tumor-specific T cells response (Jochems et al., 2016). In clinical trials, the combination of epacadostat or indoximod with anti-PD-1 or anti-CTLA4 mAbs demonstrated an enhanced response rate when compared to using immune checkpoint inhibitors alone (Zakharia et al., 2021; Ziogas et al., 2023).
Additionally, the empirical data indicated the impact of some naturally and chemically based compounds in inhibiting IFN-γ-induced IDO-1 expression. The study by Chen et al. presented that in the presence of IFN-γ, Myricetin treatment impaired the protein expression of IDO-1 in multiple tumor cell lines (Chen et al., 2022). In the study by Yaun et al., it was highlighted that LCA, alongside PDL-1, has the potential to inhibit several IFN-γ-induced proteins, including IDO-1 (Yuan et al., 2021). Galanal, which is a phytochemical compound derived from the buds of myoga flower, showed inhibitory potential in blocking high IDO-1 activity. The underlying mechanism functioned by inhibiting the IFN-γ signaling pathway, especially through interference with the formation of phosphorylated STAT-1 (Yamamoto et al., 2014).
Carbidopa, an FDA-approved drug for Parkinson’s disease, has been recognized as a selective suppressor of IDO-1, in both the presence and absence of IFN-γ. According to that, Carbidopa is an activator of AhR, the anti-IDO-1 potential of Carbidopa could be mediated by AhR stimulation. As it was observed, AhR through its capacity for ubiquitination possesses the potential to modulate the IDO-1 function along with decreased tumor growth in pancreatic cancer cells (Korac et al., 2022). Similarly, fludarabine, as a purine analogue and chemotherapeutic agent, repressed the induction of IDO-1 protein by activating proteasomal degradation pathway following exposure to IFN-γ (Hanafi et al., 2014). Sodium butyrate acts as a histone deacetylase inhibitor presenting antiproliferative and apoptotic features in multiple cancers (Louis et al., 2004; Xie et al., 2016). In the study conducted by He et al., sodium butyrate was found to modulate the IFN-γ-stimulated IDO-1 expression in nasopharyngeal cancer cells by restricting STAT1 action through inducing STAT1 acetylation, blocking its phosphorylation, and reducing its nuclear translocation (He et al., 2013). A demonstration of inhibitors blocking the expression of IFN-γ-dependent PD-L1 and IDO-1 is depicted in Figure 2.
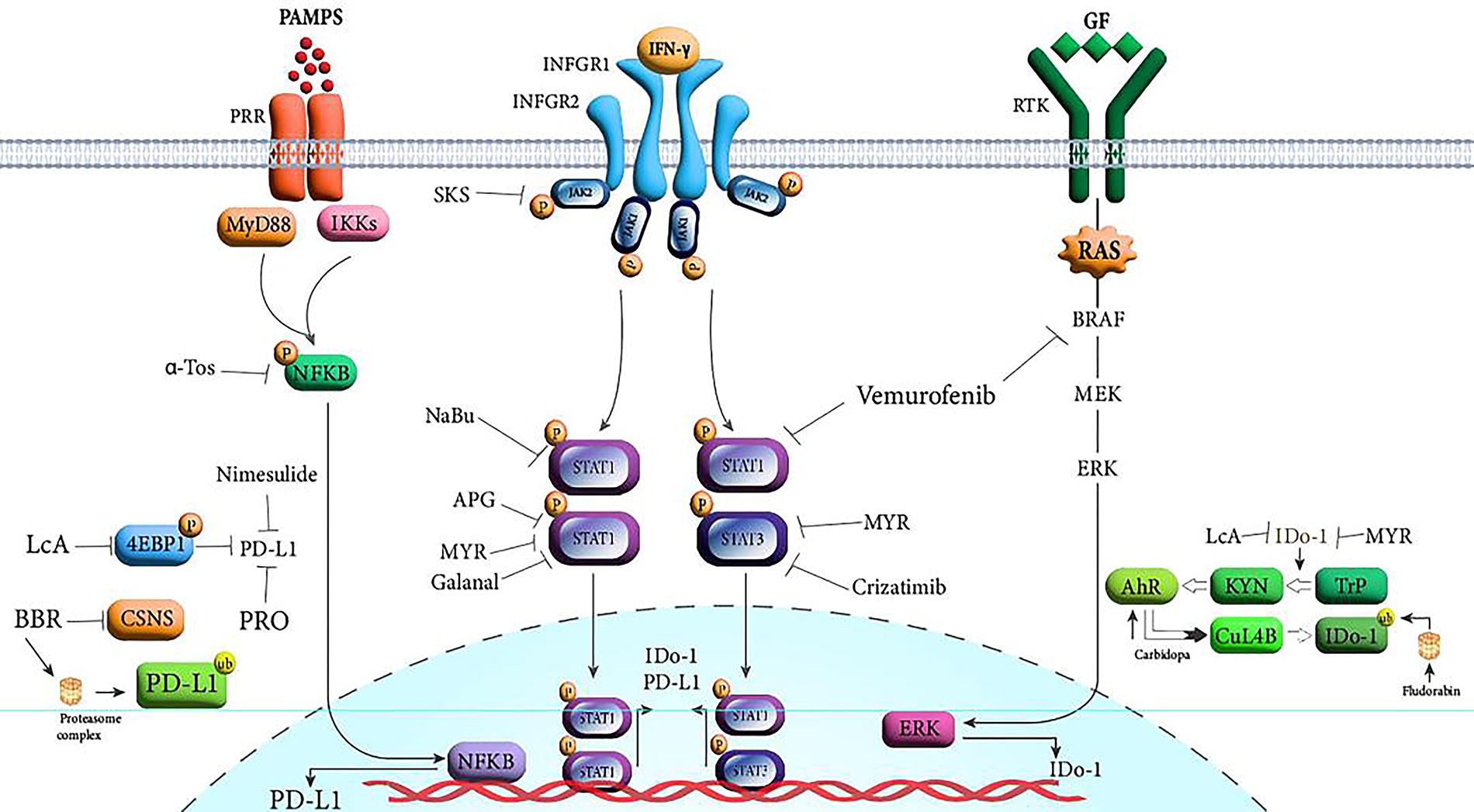
Inhibitory agents blocking the IFN-γ-dependent PD-L1 and IDO-1 expression. α-TOS, alpha-tocopheryl succinate; APG, apigenin; BBR, berberine; GF, growth factor; IFN-γ, interferon-gamma; LCA, licochalcone A; MYR, myricetin; NaBu, sodium butyrate; PAMPS, pathogen-associated molecular patterns; PRO, propranolol; PRR, pattern recognition receptor; RTK, receptor tyrosine kinase; SKS, sokotrastrol sulfate.
Another noteworthy finding pertains to the certain oncolytic viruses, particularly the herpes simplex virus (HSV), which has been shown to induce high levels of IFN-γ secretion within the TME. Intratumor administration of HSV engineered to carry IL-12, IL-18, and anti-PD-1 has demonstrated potent antitumor responses driven by elevated IFN-γ production while minimizing the side effects associated with systemic therapies (Xie et al., 2022). It appears that combining this approach with therapies to modulate IFN-γ-derived immune-inhibitory molecules may achieve optimal antitumor cytotoxic IFN-γ responses.
Resistance Mechanisms of Cancer cells to IFN-γ
The inherent instability of the genomic and epigenomic landscape in tumor cells affects gene expression profiles, which profoundly impacts the IFN-γ responses. Especially, genes involved in chromatin modifications, DNA repairmen, and cell cycle progression significantly influence the response to the IFN-γ’s antitumor response. High-throughput knockout techniques and transcriptome analysis in various IFN-γ sensitive and resistant tumor cells revealed elevated expression of double-strand break (DSB)-related DNA repair genes. In addition, inhibiting DSB repair genes has synergistically enhanced the cytotoxic responses of IFN-γ in resistant tumor cells (Han et al., 2023). Moreover, tumor cells induce loss-of-function mutations or impair gene expression in genes related to IFN-γ signaling, including JAK1/2, IFGR, and STAT1. A defective IFN-γ signaling pathway is associated with an impaired response to immune checkpoint blockades, thus highlighting the critical role of this pathway in modulating the TME (Gao et al., 2016; Hugo et al., 2017).
Conclusion
IFN-γ plays a non-negligible role in eradicating tumor cells, as the susceptibility of tumor cells to the IFN-γ determines the effectiveness of T cell therapies. The prominent role of IFN-γ lies in its involvement in triggering various mechanisms of tumor cells suppression, including apoptosis, necroptosis, ferroptosis, and senescence. Despite the prominent role of IFN-γ in tumor immunity, it has not yielded desired outcome in human research. In order to enhance the remedy efficiency while maintaining minimal toxicity, the common trend is to combine IFN-γ with approved antitumor agents such as the conventional chemotherapeutic agents, T cells, GMCSF cytokine, and anti-PD1 antibodies. A portion of these trials were prematurely terminated due to suboptimal response rate and issues with tolerability, and others are currently in progress. Besides, the delivery of the IFN-γ gene via safe, tumor-specific vectors has been observed to mitigate severe adverse effects and enhance response rates, especially following dose escalation and repeated injection. To bolster the IFN-γ therapy, the subsequent considerations should also be taken into account.
Chronic exposure to IFN-γ results in the activation of immune-modulating molecules. The direct intervention toward these regulatory molecules, in combination with conventional treatment, has demonstrated favorable effects in managing cancer. The augmented expression of PD-L1 and IDO-1, mediated by IFN-γ, can be construed as an optimistic occurrence as it amplifies the response ratio to their inhibitors and expedites the efficacy of antineoplastic T cells. Thus, assessing the PD-L1 and IDO-1 expression levels in tumors can help identify patients who are more likely to respond to IFN-γ-based therapies and guide treatment decisions. Presently, besides anti-PD-L1 and IDO-1 antibodies, a vast variety of chemical, natural, and organic compounds aimed at restricting IFN-γ-triggered PD-L1 or IDO-1 expression has been subjected to rigorous evaluation. It is more beneficial to select inhibitors that block PD-L1 or IDO-1, which are exclusively overexpressed in an IFN-γ-dependent manner. In this context, by impeding the absolute suppression of immune-regulatory molecules, the hemostasis of the immune system could be significantly preserved.
It should also be noted that the dysregulation of downstream signaling molecules and IFN-γ-inducible genes potentially influences the response to IFN-γ treatment. The genetic predisposition can considerably influence the outcome of IFN-γ therapy in inducing stimulatory or inhibitory immune responses. Besides, evaluating the existence of IFN-γ-resistance mechanisms is essential for determining therapeutic decision-making. Ongoing research is exploring novel biomarkers and molecular signatures to improve patient selection and treatment response prediction in IFN-γ-based therapies.
Footnotes
Authors’ Contributions
All authors contributed to the study conception and design. The first draft of the article was written by M.R.A. The final version of the article was prepared by N.H. The figures were designed by F.S.A., and the article was revised by E.F.S. All authors commented on previous versions of the article and approved the final article.
Data Availability Statement
Because of the type of current study, datasets have just been collected from various recent studies, and no datasets were generated.
Author Disclosure Statement
This study was performed in the absence of any financial and nonfinancial conflicts of interest.
Funding Information
No funding was received for this article.