
Abstract
Distinct transcriptional isoforms of the interferon lambda receptor 1 (IFNLR1) are expressed in hepatocytes, but whether corresponding full-length and truncated IFNLR1 protein variants have discrete function is unclear. We quantitated IFNLR1 isoforms in liver and blood from individuals with chronic hepatitis C virus (HCV) infection before and after antiviral treatment, hypothesizing their relative expression may differentially change during resolution of virus-induced inflammation. We also expressed FLAG-tagged IFNLR1 variants in stem cell-derived hepatocytes (iHeps) with abrogated endogenous IFNLR1 to evaluate their function. IFNLR1 isoforms decreased in liver and blood during treatment of HCV, but no distinct pattern of decline was observed for any individual isoform. Expression of full-length IFNLR1 enabled lambda interferon (IFNL)-induced expression of antiviral and proinflammatory genes and augmented inhibition of hepatitis B virus (HBV) replication relative to wild-type (WT) iHeps. A noncanonical IFNLR1 variant missing part of the JAK1 binding domain enabled IFNLs to induce antiviral genes but could not support induction of proinflammatory genes or augmented HBV inhibition beyond that observed in WT iHeps with intact endogenous IFNLR1. A secreted IFNLR1 variant had no identified function in iHeps lacking endogenous IFNLR1. Although relative expression of individual IFNLR1 isoforms did not distinctly change during HCV treatment, functional studies in iHeps suggest IFNLR1 variants could function to titrate antiviral versus proinflammatory responses in hepatocytes in the context of viral hepatitis.

Introduction
Lambda interferons (IFNLs) are secreted type II cytokines that signal through the interferon lambda receptor 1 (IFNLR1) and interleukin 10 receptor beta subunit (IL10RB) receptor heterodimer to influence immune and repair responses (Kotenko et al., 2003; Sheppard et al., 2003). IFNLR1 has low endogenous expression that is restricted principally to epithelial cells in the respiratory and gastrointestinal tracts, hepatocytes in the liver, and select immune cells (de Weerd and Nguyen, 2012; Kotenko et al., 2003; Santer et al., 2020; Sheppard et al., 2003; Sommereyns et al., 2008). IFNL binding initiates a JAK-STAT signaling cascade that induces hundreds of interferon-stimulated genes (ISGs), similar to type I IFN signaling (Doyle et al., 2006; Kotenko et al., 2003; Sheppard et al., 2003; Zhou et al., 2007). In contrast to type I IFN signaling, antiviral responses induced by IFNLs are slower in onset, prolonged in duration, less inflammatory, and less sensitive to known negative regulatory mechanisms (Blumer et al., 2017; Jilg et al., 2014; Pervolaraki et al., 2018; Stanifer et al., 2020). Only when IFNLR1 is artificially overexpressed can IFNLs induce a pro-inflammatory response akin to type I IFNs (Evans et al., 2023; Forero et al., 2019; Novotny et al., 2023; Stanifer et al., 2020). Thus, the capacity of IFNLs to combat infection without concomitant damaging inflammation may be mediated by restricted cellular IFNLR1 expression.
RNA-sequencing of primary cells identified multiple transcriptional IFNLR1 isoforms generated by alternative splicing predicted to encode distinct protein variants (Dumoutier et al., 2003; Kotenko et al., 2003; Lauber et al., 2015; Sheppard et al., 2003). Measurement of isoform transcripts has served as a surrogate to evaluate their contribution to pathway regulation as low expression and a lack of specific reagents limit capacity to detect IFNLR1 (Forero et al., 2019; Santer et al., 2020; Witte et al., 2009). IFNLR1 isoform 1 encodes the canonical full-length receptor, IFNLR1 isoform 2 encodes a variant with a truncated intracellular JAK1 binding domain, and IFNLR1 isoform 3 encodes a secreted variant that lacks the transmembrane domain and cytoplasmic tail (Forero et al., 2019; Lauber et al., 2015; Santer et al., 2020; Witte et al., 2009; Zhang et al., 2016). Cellular quantitation of IFNLR1 isoforms identified that a greater ratio of IFNLR1 isoform 1 to isoform 3 correlates with capacity of IFNLs to induce ISGs, whereas cells with a greater ratio of IFNLR1 isoform 3 to isoform 1 are refractory to IFNL stimulation (Forero et al., 2019; Glassman et al., 2022; Lauber et al., 2015; Santer et al., 2020; Witte et al., 2009; Zhang et al., 2016), suggesting IFNLR1 variants may influence IFNL signal transduction (reviewed in Novotny and Meissner, 2024).
To explore this possibility, in prior work, we generated HEK293T and induced pluripotent stem cell (iPSC)-derived hepatocyte (iHeps) cell lines with stable expression of individual doxycycline-inducible FLAG-tagged IFNLR1 variants (Evans et al., 2023; Novotny et al., 2023). Each FLAG-IFNLR1 variant supported IFNL-induced antiviral ISG expression, albeit at differing magnitudes, but only variant 1 permitted de novo expression of pro-inflammatory genes similar to type I IFNs and enhanced inhibition of hepatitis B virus (HBV) replication (Evans et al., 2023; Novotny et al., 2023). These data suggested hepatocytes could use balanced expression of IFNLR1 isoforms to promote antiviral responses while also limiting damaging inflammation. As these studies were conducted in cells with intact endogenous IFNLR1, we could not distinguish whether the observed phenotypes related to intrinsic IFNLR1 variant function and/or to modulation of function through endogenously produced IFNLR1.
In the present study, we quantitated IFNLR1 isoform transcripts in whole blood and liver collected from individuals with chronic hepatitis C virus (HCV) infection both before and after a 24-week treatment course with sofosbuvir and ribavirin to further explore their potential role in the context of viral hepatitis. We hypothesized that if IFNLR1 variants function to regulate pathway activity, their relative expression may differentially change during resolution of HCV-induced inflammation. To directly explore function intrinsic to each variant, we generated iHep lines with abrogated endogenous IFNLR1 and inducible expression of each FLAG-IFNLR1 variant and evaluated their influence on IFNL-induced gene expression and HBV replication.
Materials and Methods
HCV samples
Samples were analyzed from the SPARE clinical trial (NCT01441180) in which participants with chronic HCV genotype-1 infection received treatment with 24 weeks of sofosbuvir and ribavirin (Osinusi et al., 2013). Informed consent at enrollment included permission to use samples for future research. The current study was approved by the Institutional Review Board at the Medical University of South Carolina. Gene expression analyses were conducted using RNA from paired pre- and post-treatment liver biopsy (n = 8) and whole blood (n = 42) samples.
Generation of iHep lines
Generation and characterization of iPSCs with abrogated endogenous IFNLR1 (IFNLR1-KO) (Novotny et al., 2023) and constructs with doxycycline-inducible expression of FLAG-tagged IFNLR1 variants (Evans et al., 2023) were previously described. Briefly, to generate iPSCs with abrogated endogenous IFNLR1, exon 4 of IFNLR1 was targeted with CRISPR guide RNA sequence (CCTGGTGCTCACCCAGACGG) and cloned into PX459 pSPCas9(BB)-2A Puro plasmid (#48139; Addgene) (Ran et al., 2013), as previously described (Novotny et al., 2023). The plasmid was transfected into the wild-type (WT) K3 iPSC line using Viafect reagent (Promega). Cells were plated on Matrigel-coated plates and cultured in mTeSR medium (Ludwig et al., 2006a; Ludwig et al., 2006b) supplemented with 40 ng/mL zebrafish bFGF and 1 µg/mL puromycin (Sigma-Aldrich) for 2 days to select for colonies that incorporated the plasmid. Surviving colonies were picked, expanded in medium without puromycin and screened for CRISPR activity. A clone with a −2bp deletion and +1bp insertion, which introduced a premature stop codon and that had high Tracking of Indels by Decomposition and Interference of CRISPR Edits scores was selected for future work (Novotny et al., 2023). IFNLR1-KO iPSCs expressing FLAG-IFNLR1 constructs were generated by electroporation of 30 µg PvuI-linearized plasmid DNA and selection in medium supplemented with 1 µg/mL puromycin. Resultant puromycin-resistant clones (KO-FLAG-Iso1, KO-FLAG-Iso2, and KO-FLAG-Iso3) with FLAG-positive stain (not shown) and a vector-only control (KO-EV) with growth rates comparable to WT iPSCs were selected for experimentation and maintained in medium with 1 µg/mL puromycin. WT and IFNLR1-KO iPSCs were cultured without puromycin.
iPSCs were differentiated to iHeps in 48-well microtiter plates using an established 20-day, four-step process, as previously reported (Novotny et al., 2023; Si-Tayeb et al., 2010). Culture medium was supplemented with 1 µg/mL puromycin as appropriate and 10 ng/mL doxycycline (dox) to prevent construct silencing during differentiation (Chang et al., 2014; Peaslee et al., 2021).
Gene expression
Extraction and evaluation of RNA from liver biopsies and whole blood in the SPARE trial has been previously reported (Meissner et al., 2014; Orr et al., 2020). RNA from iPSCs and iHeps was isolated using a Qiagen RNeasy kit (Hilden, Germany) and quantitated by Nanodrop. cDNA was transcribed with a High-capacity cDNA reverse transcription kit (Applied Biosystems, Waltham, MA, USA) and quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was performed with commercially available TaqMan primer-probe sets (Supplementary Table S1) and Fast Advanced Master Mix (Applied Biosystems). Expression of FLAG-IFNLR1 Iso1, -2, and -3 was quantitated using custom primer-probe sets, as described (Novotny et al., 2023). We validated that the FLAG-IFNLR1 isoform construct specific primer-probe sets would not detect endogenous transcripts by sequence comparison prior to use [primer-probe sequences reported in Novotny et al. (2023)]. Expression of antiviral (MX1, ISG15) and pro-inflammatory (CXCL10, IRF1) genes in iHeps was evaluated after 24 h of 100 ng/mL IFNL3 (R&D Systems) treatment with biological and technical duplicates relative to GAPDH.
Protein expression
FLAG-IFNLR1 protein expression in mature iHeps was evaluated by Western blot. After differentiation, dox was excluded from medium for 24 h, followed by +/− dox (100 ng/mL) induction for 24 h. Cells were lysed on ice with RIPA solution (ThermoFisher) supplemented with protease and phosphatase inhibitors. Protein was quantitated by BCA with 5 µg separated on 4%–20% SDS-PAGE gel (BioRad, Hercules, CA, USA). After transfer to nitrocellulose membrane and blocking (EveryBlot buffer, BioRad), blots were probed with murine monoclonal anti-FLAG clone M2 (#F1804, Sigma-Aldrich) then goat anti-mouse IgG conjugated to HRP (#A9044, Sigma-Aldrich). Equivalent protein content was evaluated using polyclonal rabbit anti-GAPDH (#PAB932HU02, Cloud-Clone Corp, Houston, TX, USA) and goat anti-rabbit IgG conjugated to HRP ((#31460, Invitrogen, Carlsbad, CA, USA). Blots were developed with Pierce ECL Western blotting substrate and imaged on Protein Simple FluorChem M.
To evaluate expression of hepatocyte lineage markers α-fetoprotein (AFP) and human nuclear factor 4α (HNF4α), iHeps were fixed (4% paraformaldehyde), permeabilized (0.5% TritonX-100), then blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Cells were stained with antibodies against AFP (#A8452, Sigma-Aldrich) and HNF4α (#sc-8987, Santa Cruz, Dallas, TX, USA) in 1% BSA-PBS overnight at 4°C followed by detection with goat anti-rabbit IgG-AlexaFluor 594 (#A11012, Invitrogen) and goat anti-mouse IgG AlexaFluor 488 (#A11001, Invitrogen). Images were captured with an EVOS Cell Imaging System (ThermoFisher).
We also examined protein expression on mature iHeps by flow cytometry. Dox was excluded from culture medium for 24 h, then iHeps were incubated +/− 100 ng/mL dox for 24 h. Cells were collected using TrypLE Select enzyme (Lonza, Greenwood, SC, USA), then stained with Live-or-Die 615/740 fixable viability stain (Biotium, Fremont, CA, USA). After fixation with 2% paraformaldehyde, cells were surface stained for FLAG (#637310, BioLegend, San Diego, CA, USA) or permeabilized with 0.1% TritonX-100, then stained to permit detection of surface and intracellular FLAG. Samples were assayed on a Millipore Guava easyCyte 8HT flow cytometer and data consisting of 20,000 live events analyzed with FlowJo 10.9.0 software (BD Life Sciences).
HBV-iHep infection
HBV viral stock was precipitated from HepG2.2.15 cell supernatant using a PEG Virus Precipitation kit (Abcam, Cambridge, UK) and quantitated by qPCR relative to M-HBsAg plasmid DNA (#103012, Addgene) and an HBV DNA primer-probe set (Huang et al., 2017). To infect iHeps, 1,000 HBV genome equivalents (GEq)/cell were suspended in Hepatocyte culture media (HCM) +oncostatin M (OSM), 10 ng/mL dox, and 4% PEG8000. After 24 h, iHeps were washed 5× with PBS and cultured in medium without PEG8000 with medium exchange every 2 days. After 14 days to propagate infection, diluents in HCM+OSM were applied daily for 8 consecutive days including: (1) medium only, (2) 100 ng/mL dox, (3) 100 ng/mL IFNL3, or (4) 100 ng/mL dox + 100 ng/mL IFNL3. Quantitation of HBV DNA and covalently closed circular DNA (cccDNA) was performed as previously described (Cai et al., 2016; Huang et al., 2017; Novotny et al., 2023).
Statistical analyses
Statistical analyses were performed using GraphPad Prism v9.1.0 software (Boston, MA, USA) with data presented as mean ± the standard error and statistical significance set at P ≤ 0.05.
Results
IFNLR1 is upregulated in the liver of patients with HBV and HCV and may drive inflammation during chronic infection (Duong et al., 2014; Thomas et al., 2012); however, the functional role of IFNLR1 variants in this context is unknown. We previously observed decreased expression of IFNLR1 in liver during treatment of chronic HCV infection (Meissner et al., 2014). Here, we quantitated distinct IFNLR1 isoform transcripts in paired liver biopsies and whole blood collected from individuals before and after 24 weeks of treatment with sofosbuvir and ribavirin (Osinusi et al., 2013), hypothesizing that if IFNLR1 variants influence pathway activity, their relative abundance may change distinctly during resolution of virus-induced inflammation. Contrary to this hypothesis, we observed similar trends of decreased expression for all IFNLR1 variants during treatment (Fig. 1). Canonical IFNLR1 isoform 1 had the highest overall expression and decreased significantly in liver and blood (Fig. 1). Noncanonical IFNLR1 isoforms 2 and 3 had lower overall expression and exhibited similar trends of decreased expression (Fig. 1). No changes in IFNAR1, IFNAR2, or IL10RB expression were observed (Fig. 1). These findings suggest the decline in IFNLR1 was specific and not a reflection of global gene expression changes, but do not suggest pathway regulation through altered rates of IFNLR1 splicing events during resolution of virus-induced inflammation.

Quantitation of IFNLR1 isoforms in liver and whole blood of HCV patients pre- and post-antiviral treatment.
We next directly evaluated the capacity of IFNLR1 protein variants to differentially transduce signal. Despite observing no unique changes in their relative transcriptional abundance in Figure 1, noncanonical IFNLR1 protein variants 2 and 3 could function to temper the phenotype of IFNL-exposed cells if only IFNLR1 variant 1 was expressed. To evaluate function intrinsic to each variant, we generated IFNLR1-KO iPSCs with dox-inducible expression of FLAG-tagged IFNLR1 isoforms (KO-FLAG-Iso1, -Iso2, and -Iso3) or an empty vector control (KO-EV). Each line expressed the stem cell marker OCT4 and could be differentiated into iHeps that lost OCT4 expression and gained expression of hepatocyte genes (ALB, NTCP) (Fig. 2A) and proteins (AFP, HNF4α) (Fig. 2B). Like WT-iHeps expressing FLAG-IFNLR1 variants (Novotny et al., 2023), we observed a low level of construct expression without doxycycline and dox-inducible expression in each line (Fig. 2C, D). A low level of background signal near the threshold of detection was observed for FLAG-Iso1 and FLAG-Iso3 primer-probe sets in lines not expressing these constructs (Fig. 2C). By flow cytometry, we detected FLAG protein on the cell surface and intracellularly on dox-induced KO-FLAG-Iso1 and -Iso2 iHeps and only intracellularly for KO-FLAG-Iso3 iHeps, as expected, with no signal on KO-EV control cells or uninduced cells (Fig. 2E). The proportion of iHeps with FLAG positive stain by flow cytometry correlated with relative band intensity detected by Western blot (Fig. 2D).

Characterization of IFNLR1-KO IFNLR1-variant expressing iHeps.
To evaluate intrinsic capacity of IFNLR1 variants to signal absent expression of endogenous IFNLR1, we treated KO-FLAG-Iso1/2/3 iHeps with IFNL3 and quantitated expression of antiviral and proinflammatory genes. Like WT-FLAG-Iso1 iHeps (Novotny et al., 2023), IFNL3-treated KO-FLAG-Iso1 iHeps expressed both antiviral (MX1, ISG15) and proinflammatory (CXCL10, IRF1) genes in dox-induced conditions (Fig. 3A, B). In contrast, IFNL3-treated WT-iHeps expressed antiviral but not proinflammatory genes, while no genes were induced in KO- and KO-EV iHeps. Previously, we observed that dox-induced and -uninduced WT-FLAG-Iso1 iHeps supported comparably high expression of antiviral and proinflammatory genes, implying low levels of transcript from a leaky tet-on promoter were sufficient to allow a maximal response to IFNL3 (Novotny et al., 2023). In KO-FLAG-Iso1 iHeps, we observed no antiviral or proinflammatory gene expression in dox-uninduced conditions (Fig. 3A, B), implying the amount of FLAG-Iso1 produced by leaky expression was inadequate to support signaling without protein from endogenous IFNLR1.

IFNLR1 variants differentially support induction of antiviral versus proinflammatory genes after IFNL3 stimulation. iHeps were incubated with 100 ng/mL doxycycline (+dox) to induce expression of FLAG-Isoform constructs or maintained in medium (−dox) for 24 h then either mock-treated (−IFNL3) or stimulated with 100 ng/mL IFNL3 (+IFNL3) for an additional 24 h. Expression of antiviral (MX1, ISG15) and proinflammatory (CXCL10, IRF1) genes relative to GAPDH was quantitated by qRT-PCR. *P ≤ 0.05 versus KO-EV cell line or as indicated by Student’s t-test. Biological replicates were assayed in technical duplicate and data are representative of two independent experiments. IFNL, Lambda interferon; ISG, interferon-stimulated gene.
IFNL3-treated, dox-induced KO-FLAG-Iso2 iHeps expressed antiviral genes, indicating this variant intrinsically supports signaling independent of endogenous IFNLR1, however, this variant did not support proinflammatory gene expression (Fig. 3A, B). No genes were induced in IFNL3-treated KO-FLAG-Iso3 iHeps (Fig. 3A, B), suggesting this variant depends on protein from endogenous IFNLR1 to influence signaling.
We next evaluated how each IFNLR1 variant influenced IFNL3-mediated inhibition of HBV replication (Fig. 4A). HBV replication kinetics prior to IFNL3 treatment were comparable in all lines (Fig. 4B). In IFNL3-treated WT-iHeps, HBV declined in culture supernatant and cells (Fig. 4C, D), while no decline was observed in KO- and KO-EV iHeps (Fig. 4C, D). KO-FLAG-Iso1 iHeps had accelerated HBV decline and lower HBV DNA and cccDNA at the end of treatment only in dox-induced conditions (Fig. 4C, D). KO-FLAG-Iso2 iHeps exhibited viral decline comparable to WT-iHeps, while IFNL3-treated KO-FLAG-Iso3 iHeps exhibited no viral decline (Fig. 4C, D).
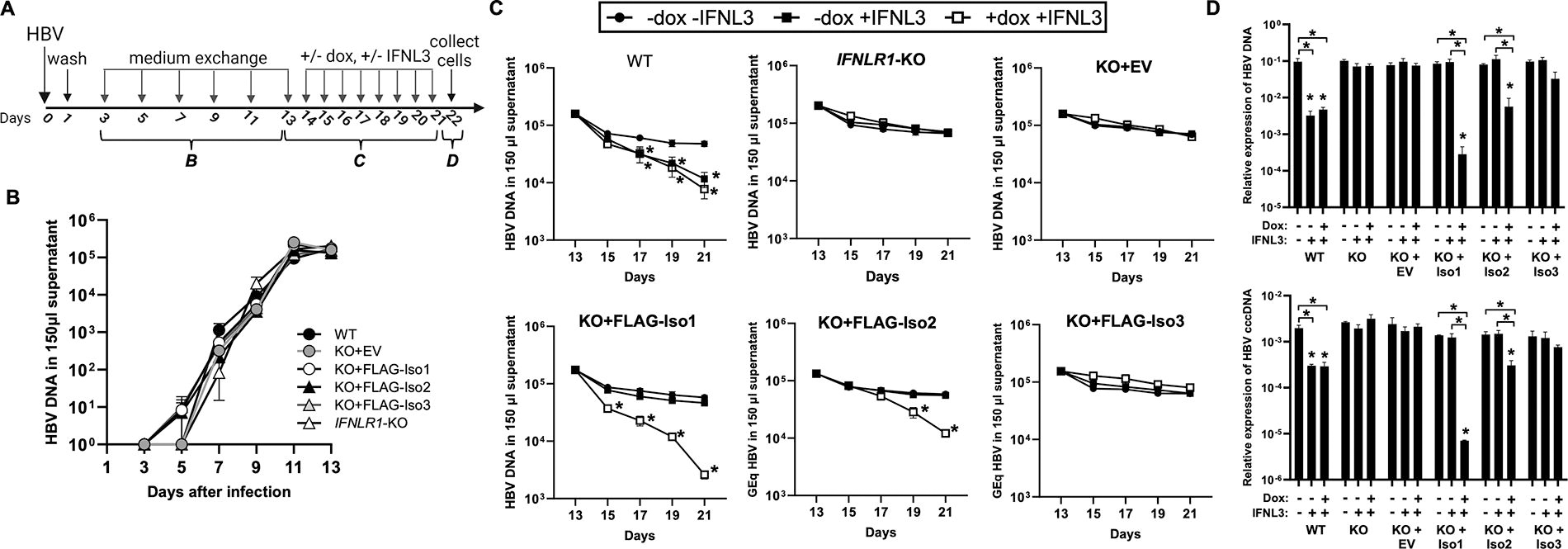
Influence of IFNLR1 variants on IFNL3-mediated inhibition of HBV replication.
Discussion
Using iHeps with abrogated expression of endogenous IFNLR1, we demonstrate unique and intrinsic function of IFNLR1 variants encoded by transcriptional IFNLR1 isoforms that were identified in primary human cells (Dumoutier et al., 2003; Kotenko et al., 2003; Lauber et al., 2015; Sheppard et al., 2003). Full-length canonical IFNLR1 variant 1 enabled expression of both antiviral and proinflammatory genes and increased IFNL-mediated inhibition of HBV replication. IFNLR1 variant 2, missing a portion of the JAK1 binding domain, functioned intrinsically to support induction of antiviral genes, but did not support expression of proinflammatory genes and only partially supported IFNL-mediated inhibition of HBV replication. Secreted IFNLR1 variant 3 had no identified function in iHeps with abrogated endogenous IFNLR1, suggesting this variant has no intrinsic signaling capacity and requires canonical IFNLR1 expression to modulate signaling. While no distinct differences were observed in relative expression of IFNLR1 isoforms in liver and whole blood during HCV treatment (Fig. 1), the distinct signaling phenotypes identified in iHeps suggest these variants could function to regulate pathway activity and balance the antiviral and proinflammatory effects of IFNL signaling.
The outcome of IFNL signaling, which evolved to confer immune defense at mucosal surfaces and barrier sites, can be either protective or deleterious to the host dependent on the context examined (Dowling and Forero, 2022; Johnson and Carbonetti, 2023; Piperakis et al., 2024; Stanifer and Boulant, 2024; Ye et al., 2019). IFNLs can divergently either facilitate repair and limit inflammation (Broggi et al., 2017; Davidson et al., 2016; Galani et al., 2017) or exacerbate inflammation, tissue damage, and risk of secondary infections (Antos et al., 2024; Kumar et al., 2024; Major et al., 2020; Piperakis et al., 2024), contingent on the timing of IFNL exposure during infection. Restricted expression of canonical IFNLR1 limits the inflammatory impact of IFNL exposure (Evans et al., 2023; Forero et al., 2019; Novotny et al., 2023), which may be one mechanism cells use to induce antiviral effect while restricting harmful inflammation. Our data identify that IFNLR1 variant 2 supports expression of antiviral genes but not proinflammatory genes (Fig. 3), suggesting expression of this variant could be another mechanism cells use to titrate pathway activity in the setting of infection and repair. As this variant is missing a portion of the cytoplasmic tail responsible for JAK1 binding and stability (Ferrao et al., 2016; Glassman et al., 2022; Zhang et al., 2016), differential affinity for JAK1 and/or use of alternative signaling mediators could account for the observed differences in downstream gene expression.
In prior work, we unexpectedly observed maximal augmentation of antiviral and proinflammatory gene expression and inhibition of HBV replication in dox-uninduced WT-FLAG-Iso1 iHeps (Novotny et al., 2023). This implied FLAG-Iso1 protein produced from a leaky tet-on promoter was adequate to fully sensitize the pathway to the proinflammatory effects of IFNL3 exposure when endogenous IFNLR1 was intact. In contrast, we observed no changes in gene expression or anti-HBV effect in similarly treated KO-FLAG-Iso1 iHeps despite comparable leakiness of transcript expression from the tet-on promoter (Figs. 2–4). Thus, protein produced from endogenous IFNLR1 in WT-iHeps, which may include all 3 variants, is inadequate to support maximal IFNL-induced expression of proinflammatory genes and augment inhibition of HBV replication, suggesting the receptor signaling complex is poised to gain these attributes with addition of low amounts of exogenously-expressed FLAG-Iso1. In contrast, in iHeps that lack protein from endogenous IFNLR1, higher levels of FLAG-Iso1 in dox-induced conditions are required to support IFNL signaling. Future work will be needed to further decipher the mechanisms by which protein made from endogenous IFNLR1 influences the phenotypes imparted by FLAG-IFNLR1 variants.
Secreted IFNLR1 variant 3 in iHeps with abrogated endogenous IFNLR1 imparted no discernible phenotype distinct from KO-EV iHeps (Figs. 3 and 4), suggesting this variant functions by modulating signaling through canonical IFNLR1, consistent with prior reports (Santer et al., 2020). Further work will be needed to understand the mechanisms by which FLAG-Iso3 enhances antiviral ISG expression in iHeps with intact endogenous IFNLR1 (Novotny et al., 2023).
These data are intriguing in light of growing insight that the IFNL receptor complex may utilize noncanonical mechanisms to signal in cells. For unclear reasons, IFNL signaling is intact in genetically TYK2-deficient persons and persons treated with TYK2 inhibitors (Fuchs et al., 2016; Kreins et al., 2015; Schnepf et al., 2021), which is inconsistent with the paradigm of canonical IFNL signaling wherein JAK1 bound to IFNLR1 and TYK2 bound to IL10RB function to initiate a JAK-STAT signaling cascade (Doyle et al., 2006; Kotenko et al., 2003; Sheppard et al., 2003; Zhou et al., 2007). Recent work using synthetic IFNLR1 heterodimers suggests the IFNLR1 signaling complex may utilize multimeric receptor complexes or a subunit other than IL10RB to function (Lin et al., 2024; Mesev et al., 2023). As such, it is intriguing to conceptualize a potential role for IFNLR1 variants 2 and/or 3 in contributing to noncanonical signaling complexes.
This work also allows speculation to explain the outcomes observed when exogenous pegylated-IFNL was tested as a therapeutic for treatment of chronic HBV infection (Chan et al., 2016), which was predicted to be an effective therapy based on in vitro and animal models (reviewed in Novotny et al., 2021). The cohort of participants who received pegylated-IFNL had greater early on-treatment viral decline as compared to the cohort receiving a pegylated type I IFN (Chan et al., 2016). However, the IFNL cohort had more ALT liver enzyme flares during treatment and the antiviral effect of IFNLs plateaued during treatment and was not sustained post-treatment (Chan et al., 2016). Interestingly, detailed immune analysis of a subset of IFNL responders and nonresponders identified a correlation of immune response with restoration of markers of anti-HBV specific adaptive immunity (Phillips et al., 2017). It is intriguing to speculate that inter-individual differences in expression of canonical IFNLR1 variant 1 relative to noncanonical variants 2 and/or 3 on hepatocytes or immune cells, given the disparate signaling events that are supported by each variant, could have correlated with either ALT flares or immune responsiveness. Unfortunately, no samples were available to test this hypothesis.
Important limitations of this work merit discussion. For the analysis conducted with HCV clinical samples, total RNA from whole liver biopsies or whole blood was used to quantitate IFNLR1 isoform expression. As such, significant differential changes in relative expression of IFNLR1 isoforms on certain cell types, particularly cell types that comprise a minority of liver of blood, would not have been detected in this assay. Second, the in vitro work relied on detection of IFNLR1 transcripts and overexpression of FLAG-tagged IFNLR1 variants. Thus, extrapolation of the physiological relevance of the disparate signaling outcomes observed in the setting of overexpressed proteins should be interpreted with some caution, as an excess of receptor can form nonsignaling complexes that can cloud interpretation of functional assay readouts. Finally, the in vitro iHep and HBV replication model used to test IFNLR1 variant function consisted only of hepatocyte-like cells and does not mimic the complexity of chronic viral hepatitis infection wherein changes in the immune function of hepatocytes and immune cells occur over the course of decades.
In conclusion, this mechanistic work in iHeps with abrogated endogenous IFNLR1 identified distinct and unique function intrinsic to IFNLR1 variants encoded by IFNLR1 transcriptional isoforms, suggesting these isoforms could be a mechanism cells use to balance antiviral and proinflammatory sequelae of IFNL signaling. Prior IFNLR1 studies conducted in epithelial cells identified an influence of cellular polarization and spatial orientation with cellular distribution of IFNLR1 and IFNL responsiveness (Bhushal et al., 2017; Metz-Zumaran et al., 2024), thus it will be informative in future work to utilize WT- and IFNLR1-KO iHep lines expressing FLAG-tagged IFNLR1 variants to examine receptor localization and signaling within more architecturally complex cellular systems, such as liver organoids derived from iPSCs. With increasing use of inhibitors that target JAK-STAT signaling pathways to treat cancer and autoimmunity (Adas et al., 2022; Levine and Hubbard, 2022; Rusinol and Puig, 2023) and which impart a risk of infection, it will also be informative to evaluate how these inhibitors impact IFNL signaling and whether IFNLR1 variants are differentially impacted.
Footnotes
Acknowledgments
The authors acknowledge Paige Lamprecht-McGinnis and Troy Dearth for help with iHep reagents and technical assistance (Medical University of South Carolina). The graphical abstract was created using BioRender software.
Authors’ Contributions
Conceptualization: L.A.N. and E.G.M. Methodology: L.A.N., C.S.K., and E.G.M. Investigation: L.A.N., C.S.K., and E.G.M. Writing—original draft: L.A.N. and E.G.M. Writing—review and editing: L.A.N., C.S.K., and E.G.M. All authors have read and agreed to the submitted version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
E.G.M. received the funding from the National Institute of Allergy and Infectious Diseases (K08 AI121348), the National Institute of General Medical Sciences (P20 GM130457), the National Institute of Diabetes and Digestive and Kidney Diseases (Analytic Cell Models Core of the MUSC Digestive Disease Research Core Center, NIH P30 DK123704), and the South Carolina Clinical & Translational Research Institute (NCATS: UL1 TR001450).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.